
Abstract
To describe the clinical spectrum and prognosis of atypical tumefactive demyelinating lesions (TDLs), which were confirmed by pathology. A total of 11 patients were diagnosed with atypical TDLs confirmed by brain biopsy and surgery between January 2006 and December 2017. The clinical spectrum and prognosis in these patients were analyzed. The patients’ ages ranged from 29 to 62 years, with a mean age of 48.9 years; 72.7% were males. The Expanded Disability Status Scale (EDSS) of the patients with first onset was 2.36. Most of the patients started with limb numbness and weakness (45.5%) or alalia (27.2%). The mean time from symptom onset to biopsy or surgery was 12.9 days (3–30 days). Most of the patients had solitary lesions (72.7%), supratentorial lesions (90.9%, particularly predominant in the frontal, temporal, and parietal lobes), moderate edema (63.6%), mild mass effect (54.5%), and patchy lesions (54.5%). Among them, three patients were positive for myelin basic protein (MBP) and one patient was positive for myelin oligodendrocyte glycoprotein (MOG). The patients were followed up for an average of 6.9 years (2–14 years), and recurrent TDLs were observed in 2 patients. Except for the 2 patients who relapsed, only 1 of the 9 patients died; the other 8 patients improved or maintained the status quo (the EDSS scores were lower or unchanged). The patients did not have any serious nervous system injury at onset, and the main presentation included extremity weakness, headache or dizziness, and alalia. The most common form was patchy on MRI enhancement. Cerebrospinal fluid and demyelination test can be an indicator of TDLs, and seizures may be a poor prognostic indicator. Most atypical TDLs have monophasic courses and good outcomes. The effect of neurosurgery alone was good in our group, and the effect of surgery on atypical TDLs can be further studied.
Similar content being viewed by others

Introduction
Tumefactive demyelinating lesions (TDLs) can also be called “demyelinating pseudotumors”, “tumor-like demyelinating lesions (TDLs),” “tumefactive demyelinating lesions,” or “tumefactive multiple sclerosis lesions”. TDLs are tumors or lump-like white matter space-occupying lesions, generally larger than 2 cm1. TDLs generally have small edema and mass effects. It is difficult to differentiate TDLs from brain tumors, but most TDLs have a high degree of specificity, especially in radiographic features. Most of the TDLs showed incomplete annular enhancement, and the open side faced the cortex, with dilated veins centrally. MRI of TDLs are multiple lesions with high minimal apparent diffusion coefficient values2. TDLs require serial MRI scans, and some continuous evolutions will be found. However, a brain tumor will show stability in an MRI scan3. However, there are certain TDL lesions that are atypical on imaging and difficult to distinguish from tumors or the TDLs not responding well to immunotherapy, which are called atypical tumefactive demyelinating lesions.
The prevalence of TDLs has been reported to be 1–2/1000 for multiple sclerosis (MS)1,4,5,6,7. TDLs have been associated with multiple sclerosis (MS) and neuromyelitis optica spectrum disorders (NMODS). However, patients with TDLs could not fully meet the diagnostic criteria of MS and NMODS; therefore, TDLs may be an independent diagnosis1,6,8,9.Some reports about TDLs found that the conversion rate to MS or recurrence fluctuated between 10%8 and 70%10; this difference is huge. It is difficult to assess the prognosis of patients with TDLs. The possible risk factors of relapse include age, enhancement pattern, and the presence of other typical MS lesions7,9.
Some patients were not suspected of having TDLs before brain biopsy and neurosurgery; they may be suspected of having brain tumors. The primary aim of our study was to evaluate the clinical characteristics of pathologically confirmed demyelinating lesions that were easily confused with brain tumors (which we call atypical tumefactive demyelinating lesions) and to identify the risk factors of poor prognosis.
Method
The present study was a retrospective review of patients presenting with TDLs as their first clinical event. We identified patients who underwent brain biopsy or neurosurgery at Tianjin Huanhu Hospital between January 2006 and December 2017 and received a final neuropathological diagnosis of TDLs. Patients who could not be followed up were excluded. Cases diagnosed with neoplasms, ADEM (acute disseminated encephalomyelitis), NMOSD, infection, vascular, and other fatal diseases were excluded.
The Tianjin Huanhu Hospital Ethics Committee on Human Research approved the study protocol for this retrospective review.
We recorded the following details: previous history, age, sex, race, CSF analysis results if it could be obtained, radiographic spectrum, clinical manifestations, treatment, and all the patients were followed up. Clinical information was obtained by board-certified neurologists, and the neuroimages were obtained by board-certified radiologists. The certified neuroradiologist who evaluated the radiographic material was blinded to the clinical data. We recorded the following neuroimaging details: lesion location, lesion number, degree of mass effect (mild, (sulcal effacement); moderate (ventricular compression); or severe (midline, sub-falcine, or uncal herniation)), degree of edema (mild: < 1 cm from the lesion; moderate:1–3 cm from the lesion; severe: > 3 cm from the lesion)11, and enhancement pattern (nodular, patchy, radiating, linear, and complete rim).
Stereotactic puncture biopsy and exploratory craniotomy were performed by a professional neurosurgeon. Informed consent have been obtained from all subjects and/or their legal guardians. The samples were obtained from the lesions with significant enhancement on MRI. All the cases diagnosed as TDLs during pathological examination were identified by 2 professional pathologists. All the cases underwent routine sections, special stains, and immunohistochemical studies.
The follow-up was conducted through personal interviews, examinations, telephone contacts, and family contacts. The EDSS were assessed at first onset and during follow-up in patients with TDLs.
Ethical approval
All human studies have been approved by the appropriate ethics committee and have therefore been performed in accordance with the ethical standards laid down in the 1964 Declaration of Helsinki and its later amendments.
Statistical analysis
We performed descriptive statistics as the statistical analysis.
Result
As shown in Table 1. Eleven patients were diagnosed with TDLs by brain biopsy or neurosurgery. The ages of the patients ranged from 29 to 62 years, with a mean age of 48.9 years; 72.7% were males. The average EDSS of the patients at first onset was 2.36. Among these patients, there were five patients whose EDSS was 1, three patients whose ESSS was 3, three patients whose EDSS was 4, and all of the patients’ EDSS was ≤ 4. Regarding clinical symptoms (chief complaint on admission), there were five patients with headache or dizziness, seven patients with numbness or weakness of the limb, four patients with alalia, one patient with epilepsy, one patient with memory deterioration, and one patient with eye movement disorders. Five patients had a history of hypertension, two had a history of diabetes, one had a history of colonitis, one had a history of cholangiolithiasis, one had a history of duodenal ulcer, and one had a history of viral encephalitis. In conclusion, most of the patients with atypical TDLs were middle-aged (mean age 48.9 years), did not have any serious nervous system injury (EDSS ≤ 4), most started with limb numbness and weakness (45.5%) or alalia (27.2%). The mean time from symptom onset to biopsy or surgery was 12.9 days (3–30 days).
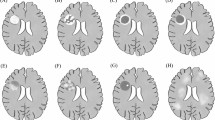
In terms of imaging, there were eight patients with solitary lesions (72.7%) and three patients with multifocal lesions (27.3%). Only one (9.1%) of the 11 patients had sub-tentorial lesions, three patients (27.2%) had right hemispheric lesions, six patients (54.5%) had left hemispheric lesions, and 1 patients (9.1%) had bilateral hemispheric lesions. Frontal lobes (5 cases, 45.5%), temporal lobes (3 cases, 27.3%), and parietal lobes (3 cases, 27.3%) were the most prevalent lesions. Mild edema surrounded the lesions in three cases (27.3%), moderate edema surrounded the lesions in 7 patients (63.6%). Six patients had mild mass effects (54.5%), four patients (36.4%) had a moderate mass effect. Enhancement following the administration of gadolinium was observed. There were six patients whose lesions were patchy (Fig. 1a) in MRI enhancement (54.5%), no patients with incomplete rim lesions, one patients with linear lesions (Fig. 1b), two patients with complete rim lesions(Fig. 1c), one patient with nodular lesions (Fig. 1d) and two patient with no enhancement of the lesion. No patients had spinal lesions. In conclusion, most of the patients had solitary lesions (72.7%), supratentorial lesions (90.9%), in particular, the frontal, temporal, and parietal lobes were predominant), moderate edema (63.6%), mild mass effect (54.5%), and patchy lesions (54.5%).

Representative examples of different enhancement patterns: (a) patchy (b) linear (c) Complete rim (d) nodular.
Most of the patients were considered to have intracranial tumors before surgery; therefore, few cerebrospinal fluid analyses and related demyelination tests were performed before surgery. Only four patients underwent lumbar puncture and related demyelination test. Among them, three patients were positive for MBP, one patient was negative for MBP, and one patient was positive for MOG.
Of the 10 patients who underwent frozen pathology, two patients were considered as having brain tumor in frozen pathology, seven patients could not be determined as having TDLs or tumors, and only one patient was considered as having TDLs in frozen pathology. Five cases were considered as tumors before surgery, five cases could not be determined as having tumors or TDLs and the symptoms and imaging findings did not improve after immunotherapy, one case whose cerebrospinal fluid was positive for MOG that was considered as having TDLs, but the symptoms worsened after hormone therapy; therefore, surgery was performed.
Regarding the treatment, five patients underwent exploratory craniotomy and hormone therapy; one patient underwent exploratory craniotomy, hormone therapy, and immune globulin therapy; 4 patients underwent exploratory craniotomy with complete removal of the lesions in the following MRI examination; therefore, these 4 patients were not given hormone or immunoglobulin therapy. One patient underwent brain biopsy and hormone therapy. The patients were followed up for an average of 6.9 years (2–14 years), two patients relapsed, and one of them died after 2 years. Of the other 9 patients, one died after 2 years, and the other 8 patients recovered very well (the EDSS scores decreased or did not change) and did not develop new lesions over time. The average EDSS of the 8 patients was 1.25 (range, 0–3). Among the 8 patients, 4 patients underwent only neurosurgery, two underwent exploratory craniotomy and hormone therapy, one patient underwent exploratory craniotomy, hormone therapy, and immunoglobulin globulin therapy, and one patient underwent brain biopsy and hormone therapy.
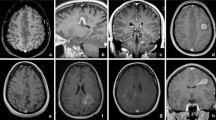
There were two epileptic patients, one of who was a 29-year-old male with 10 days of paroxysmal convulsion and 10 days of dizziness. Head MRI showed a space-occupying lesion on the left frontal parietal (Fig. 2) with irregular morphology and linear enhancement. Pathological findings showed that a large number of foam cells were deposited in the local brain tissue of the test material, with myelin loss, axonal retention, peripheral glial cell proliferation with small vascular hyperplasia, lumen dilation, and focal vascular lymphatic sleeve formation, which was considered as demyelinating pseudotumor.

Male, 29 years old, 10 days of paroxysmal convulsion and 10 days of dizziness. (a) T1 sagittal position: left frontoparietal long T1 signal; (b) T2 axial position: left frontoparietal long T2 signal; c Flair axial position: left frontoparietal high signal ;d enhanced NMR axial position: linear enhancement.
The other patient was a 57-year-old male patient with dizziness and memory loss for more than 1 month. MRI of the head (Fig. 3) showed that the right frontal lobe, corpus callosum, basal ganglia, and periventricular lesions with irregular morphology, unclear boundary, and insignificant peripheral edema. MRI enhancement of the head shows patchy enhancement. Pathological examination showed a large number of foam cell deposits in brain tissue, scattered lymphocytes and plasma cells in infiltration and perivascular cuff cluster, accompanied by a large number of astrocyte proliferation, which was considered demyelinating disease. The symptoms improved after hormone therapy. One year later, the patient developed increased limb weakness and recurrent seizures, and the MRI showed abnormal signals in brachium pontis (Fig. 3). He was diagnosed as MS. After immunoglobulin and hormone therapy, symptoms worsened. The patient's family gave up treatment and died two years later.

Male, 57 years old, dizziness and memory loss for more than 1 month (a–d). The patient relapse after 1 year, weakness of left limbs and seizures (e–g). (a) T1 sagittal position: right frontal lobe, corpus callosum, basal ganglia, and periventricular long T1 signal; (b) T2 axial position: right frontal lobe, corpus callosum, basal ganglia, and periventricular long T2 signal; (c) Flair axial position: right frontal lobe, corpus callosum, basal ganglia, and periventricular high signal; (d) enhanced MRI axial position: patchy enhancement. The patient relapse after 1 year (e–g). (e) T2 axial position: right brachium pontis long T2 signal; (f) Flair axial position: right brachium pontis high signal; (g) enhanced MRI axial position: none enhancement.
Recurrent TDLs were observed in two patients. One patient relapsed 3 years after the exploratory craniotomy and hormone therapy. The recurrent lesion in this patient was located in the pons, different from the first lesion. After immune globulin therapy, the patient recovered very well within 3 years of follow-up. The other patient relapsed 2 months after the exploratory craniotomy and hormone therapy. The recurrent lesion in this patient was located in brachium pontis, different from the first lesion. Immune globulin therapy was administered. The patient died after 2 years. The lesions of the patients who relapsed were solitary. There were no enhancement in both two recurrent patients. Except for the two patients who relapsed, only one patient of the 9 patients died, and the other 8 patients improved or maintained the status quo (the EDSS scores were lower or did not change). Three patients maintained the status quo and the other five patients improved. Among the 8 patients, four patients underwent exploratory craniotomy and other immunotherapies, while 4 patients underwent only exploratory craniotomy.
Postoperative paraffin pathological findings showed discrete border, different degrees of demyelination in the lesion area (Luxol-fast-blue, LFB) (Fig. 4a) ,macrophage infiltration (CD68) (Fig. 4b), Creutzfeldt cells(Fig. 4c), perivascular lymphocytic inflammation(Fig. 4d), preserved axons (neurofilament protein immunohistochemistry, NF) (Fig. 4e) and reactive astrocytes (glial fibrillary acidic protein, GFAP) (Fig. 4f). A final diagnosis of TDLs was made in all the cases. The myelin loss in the areas of macrophage infiltration and the preservation of axons in the areas of demyelination are supportive histological findings in TDLs.

Pathologic features (a) Discrete border and demyelination (b) Discrete border and Macrophage infiltrate (c) Creutzfeldt cells (red arrows) (d) Perivascular lymphocytic inflammation (e) Preserved axons (f) Reactive astrocytes.
Discussion
TDLs were first described in 1979 by van der Velden et al.12 It has characteristic clinical and radiographic features, sizes ˃2 cm , incomplete annular enhancement, small edema and mass effects, and experienced neurologists can diagnose typical TDLs in most cases. However, there are no consensus criteria for the clear definition of TDLs, and the 2-cm cut-off for the definition of TDLs is an arbitrary one. One report found that when the TDLs were grouped into < 2 cm and > 2 cm, there was no difference in clinical features and MRI manifestations13. In our study, we included 11 patients, of which 5 patients (45.5%) were considered to have tumors before the surgery, and for 5 patients (45.5%), it was not sure whether they were tumors or TDLs and had poor response to immunotherapy, and one (9.1%) case was considered as TDLs, but the symptoms worsened after hormone therapy. In all the cases, it was difficult to establish the diagnosis of TDLs or tumors; therefore, a biopsy or surgery was advisable. Consequently, these cases were histopathologically proven to be a demyelinating disease. This suggests that it is difficult to completely differentiate TDL from a tumor, and pathological biopsy is still the gold standard for the diagnosis of TDLs. Misdiagnosis can result in unwarranted and overly aggressive therapy; therefore, it is critical to be aware of this serious diagnostic pitfall. TDLs have no biomarker or indicator, and the prognostic value of TDLs is uncertain. Therefore, we summarized the clinical and radiological features and prognostic factors of atypical TDLs and hoped to find clues as to why some patients developed TDLs.
TDLs is estimated to have an annual incidence of 0.3/100,00014. The figure is likely an underestimation because of under-reporting, and the true incidence is unknown. TDLs can occur at any age (2–71 years)8,15,16, but persons in their 20 s and 30 s were more frequently affected1,17. In our study, the ages of the patients ranged from 29 to 62 years, with a mean age of 48.9 years. The mean age was higher than for previous studies due to the number of atypical cases and the small number of cases. Female predominance has been reported in previous reports17,18,19. However, research including 168 cases of pathologically confirmed tumefactive MS observed no gender predilection, which is consistent with our results1.
The clinical manifestations of TDLs vary depending on the location and size. TDLs generally have an acute onset and gradually aggravate. In our study, the patients did not have serious nervous system injury (EDSS ≤ 4) at onset; the duration from onset of symptoms to biopsy or surgery was mostly within 30 days, and the main presentation included weakness of the limbs, headache or dizziness, and alalia. Cognitive impairments and seizures have also been reported1,20; one patient presented with a seizure and one patient with cognitive impairments in our study. There was also one patient with eye movement disorders. These findings suggest that the disease is insidious and that these clinical manifestations are indistinguishable from tumors. Our research did not involve children, but some studies found that TDLs have a relatively acute onset (mostly less than 1 weeks) in children, and symptoms are severe, including headache and vomiting because of intracranial hypertension15,16. The most common lesions of TDLs are located in the subcortical hemispheric white matter and corpus callosum8,21,22,23. TDLs lack a mass effect and less perifocal edema21, but the mass and perifocal edema increases with larger lesions and in a more acute onsets(< 3 weeks) 1,19,24.TDLs are generally thought to be solitary lesions8,21. There are various patterns such as nodular‑,closed‑ring‑, open‑ring‑, and flame‑shaped enhancements that are noted in strengthen MRI25.The patterns of incomplete, C-shaped, or “open-ring” enhancements are considered to be typical TDLs imaging findings. In particular, the open-ring sign was present in 70% of the demyelinating disease26. The open portions of the open-ring face the cortical gray matter or basal ganglia and the portions of enhancement face the white matter27.The lesions of enhancement are regarded as demyelination and the lesions that are not enhanced are regarded as chronic inflammation28. In our study, all the cases appeared as hypointensities on T1-weighted MRI and hyperintensities on T2-weighted MRI. The lesions in most of the patients were solitary, supratentorial, and unilateral (there was no obvious distinction between left and right hemispheric) lesions, and the frontal, temporal, and parietal lobes were the most prevalent locations of the lesions, which is consistent with the results of some studies1,19.
Most of the patients in our study had moderate edema and mild mass effect; that is, neither the edema nor the mass effect was serious. The characteristics mentioned above are consistent with the literature reports. However, the forms of MRI enhancement in atypical TDLs were inconsistent with the literature, and the most common form was patchy enhancement, which was confused with tumors. There was no incomplete rim in the MRI enhancement. The reason would be that the typical TDLs with incomplete rims were diagnosed and had avoided surgery. The atypical TDLs without incomplete rims are easily confused with tumors. TDLs could also occur in the spinal cord, and cervical lesions appear to be the most commonly occurring spinal lesion29,30,31,32. The cases in our study did not include spinal lesions.
Sometimes, conventional MRI imaging was unable to discriminate between demyelination and neoplastic processes. Special imaging examinations such as magnetic resonance spectroscopy (MRS), MRI perfusion, and positron emission tomography (PET) are helpful for the identification of TDLs. In MRS, TDLs have decreased N-acetylaspartate peaks/Cr ratios and increased Cho/Cr33. A study found that dilated veins drain towards distended subependymal veins in MRI perfusion studies34. The mean regional cerebral blood volume (rCBV) in TDLs was lower than the mean rCBV in the contralateral normal-appearing white matter and substantially less than in gliomas34,35. In PET-CT, TDLs have lower metabolic activity than neoplasms36. However, advanced MRI techniques can lead to equivocal results because there is a broad overlap between the TDL and tumors. Unfortunately, only two of our patients underwent functional MRI. The magnetic resonance venogram (MRV) of one patient was normal and the MRS of the other suggested a high possibility of TDLs, but a glioma could not be excluded. As the sample size was small, we could not summarize the role of the special imaging examination. Further research is required to clarify this is needed.
Typical TDL are open loop enhancement and vertical sign (linear enhancement perpendicular to the lateral ventricle). Our atypical TDL are all non-open loop enhancement, and there is no vertical sign. Most of our atypical TDL enhancement were patchy and nodular reinforcement, and some of them had no significant enhancement changes. If the imaging findings are confused with tumors, we recommend further examination, for example, magnetic resonance perfusion examination. Although the enhancement of TDLs is obvious, it often presents equal or low perfusion due to the absence of tumor neovascularization, while neoplastic lesions, such as glioblastoma, often present obvious high perfusion. MRS Examination is also helpful in the diagnosis of TDLs.
The TDLs were dynamic, consistent with the disease progression37. Most TDLs lesions showed an excellent response to hormone therapy, especially on diffusion-weighted imaging (DWI), and there was a substantial decrease in size or disappearance of the lesions after 6–8 weeks, which is in contrast to the more stable findings in patients with tumors8,38,39.Therefore, the dynamic changes of the image is a feature that differentiates TDLs from tumors.
The patients with TDLs had slightly elevated concentrations of cerebrospinal fluid (CSF) protein or mild CSF pleocytosis40. The myelin basic protein (MBP) may play a role in TDLs41,42. One study found that oligoclonal band positivity was less frequent in patients with TDLs onset19. The importance of anti-MOG testing in NMO spectrum disorders and other demyelinating disease phenotypes, including TDLs, has since been highlighted in emerging research43. Some reports showed that TDLs with MOG antibody positivity had MS-like histopathological features and had a good response to immunotherapy44. Some studies found that patients with NMOSD who are AQP4-IgG-seropositive or myelin oligodendrocyte glycoprotein (MOG)-IgG-seropositive can develop TDLs45,46,47. One study found that the conversion of TDL to aquaporin-4 AQP4-NMOSD is increased47. In our study, 3 patients had MBP positivity and one patient had both MBP and MOG antibody positivity. One patient with MBP positivity evolved into MS 4 years later. The patient with both MBP and MOG antibody positivity did not develop MS or NMOSD. In our study, although the sample size was small, the cerebrospinal fluid and demyelination tests could be an indicator of TDL. The absence of oligoclonal bands or other markers of intrathecal inflammation cannot exclude the demyelination disease48. More studies on the demyelination test will be needed.
The histologic features, particularly those observed on intraoperative frozen sections, can be deceptive and difficult to interpret, and they often lead to the misdiagnoses of TDLs as astrocytomas because it is difficult to distinguish macrophages from swollen astrocytes. As seen in this study, only one of the frozen sections suggested a diagnosis of TDLs. However, the CD68 marker can distinguish between macrophages and swollen astrocytes. In our study, for most of the patients (7/10, 70%) in the intraoperative frozen section, it could not be determined whether they were TDLs or tumors, and some of them were diagnosed as tumors (2/10, 20%) in the intraoperative frozen section; only one of them (1/10, 10%) was diagnosed as TDLs in the intraoperative frozen section, while postoperative paraffin section confirmed the TDLs. The accuracy of frozen pathology in diagnosing TDLs is relatively low. Therefore, in the process of craniotomy exploration, our patients underwent total resection without affecting the neurological function as far as possible. Brain biopsy may sometimes lead to misdiagnosis. Due to sampling bias of the tissue biopsied, misdiagnosis can occur after pathological confirmation. Biopsy samples may be insufficient or nonrepresentative of tissue compounds. The size, quality, and site of the biopsy, fixation conditions, and other factors can affect the accuracy of the pathological results. Experienced interpretations of neuropathologists and neurologists are critical. In our study, the patient who was diagnosed with TDL only before surgery underwent brain biopsy, while the others underwent exploratory craniotomy. During the operation, all the lesions were removed as far as possible.
The treatment of TDL is based on the findings from case reports and series, and there are no recognized recommendations. TDLs respond well to steroid hormones49, corticosteroids are the usual first-line treatment, as in conventional MS19. If patients on hormonotherapy recover incompletely or worsen, plasma exchange (PLEX) is an appropriate next-line therapy50. If the symptoms of TDLs worsen and the lesions enlarged after hormonotherapy and PLEX, immunosuppressive therapy should be started without hesitation17. If brainstem herniation seems to be imminent due to increased intracranial pressure, decompressive craniectomy should be strongly considered51. If the patients with TDLs are subsequently diagnosed with MS or NOSDM, the standard immunotherapies for those conditions should be considered. We had four treatments: exploratory craniotomy only, exploratory craniotomy combined with hormone therapy, exploratory craniotomy combined with hormone therapy and immunoglobulin therapy, brain biopsy, and hormone therapy. No patient received PLEX. The 4 patients who underwent exploratory craniotomy recovered very well (the EDSS scores decreased or did not change) and did not develop new lesions over time. The patient who underwent exploratory craniotomy combined with hormone therapy and immunoglobulin therapy as well as the patient who underwent brain biopsy and hormone therapy recovered well and did not develop new lesions over time. However, among the 5 patients who underwent exploratory craniotomy combined with hormone therapy, 2 patients relapsed and one of them died after 2 years. The lesions of the patients who experienced relapse were both located in the pons. One patient was diagnosed with TDLs after the relapse and had a good prognosis. Another patient was diagnosed with MS and died 2 years later. The patient died due to a worsening of the disease with seizures and complications of lung infection. One patient died 2 years after the exploratory craniotomy and hormone therapy. Due to the large lesion, frequent epileptic seizures, and economic reasons, the gamma globulin treatment was not applied. The 2 patients who died both had epileptic seizures; therefore, the seizures may be a poor prognostic indicator.
The lesions in two patients with epilepsy were close to the cortex, had space-occupying effects and edema, and were prone to epilepsy. Pathological findings showed foam cell deposition, disappearance of myelin sheath, retention of axons, scattered infiltration of lymphocytes and plasma cells and perivascular cuff aggregation, accompanied by massive proliferation of astrocytes, all of which were considered demyelinating disease.
One of the two patients complained of epilepsy, while the other was admitted with dizziness and memory loss and later recurrent limb weakness and seizures, which were not isolated seizures. Epilepsy is rare in TDLs. In a case series study, epilepsy was associated with hemiplegia in 2 out of 18 patients with TDLs52.In another study, epilepsy occurred in 6% of patients with TDLs1. An Indian child was also reported to have suffered epilepsy as well as hemiblindness and mild hemiplegia53. There has also been a report of patient with TDLs presenting with epilepsy as an isolated manifestation54. Therefore, epilepsy is rare in TDLs patients, most of which are combined with other signs of neurological impairment. However, TDLs characterized by isolated seizures epilepsy are relatively rare.
The recurrence rate of TDLs has been extremely variable (10–70%) 1,7,10,19,48,55.The risk of relapse or imaging progression between patients with solitary TDL and patients with multiple lesions is similar48. The recurrence rate of TDLs in our group was 16.7% (2/12). In our group, one patient recurred as MS; therefore, the TDLs were associated with MS or NOMDS. Some TDLs have a preexisting diagnosis of MS17,56,57,58,59,60. Some studies have suggested that TDLs may represent either a unique form of demyelinating disease8 or a variant of MS61. TDLs are sometimes regarded as Shilder's disease or Marburg's variants61. TDLs can occur as part of other demyelinating diseases: NMOSD or acute disseminated encephalomyelitis (ADEM)1,62. One report found that most patients followed the typical course of MS, while a small subset developed relapsing TDLs1. Therefore, it may also be necessary to further determine the relationship between TDLs, MS, or NMOSD.
There are no definitive diagnostic criteria for TDLs. Some of the TDLs have no pathognomonic imaging or clinical manifestations. Currently, biopsy cannot be replaced by imaging in the diagnosis of TDLs. If the radiology is uncertain and the clinical features are atypical, we can attempt to use steroids or plasma exchange for treatment. A response to the treatment is particularly suggestive of an inflammatory demyelinating lesion. We suggest to avoid unnecessary surgical treatment and to choose drug control first. However, when surgery is unavoidable, total surgical resection without affecting the neurological function is recommended. Postoperative immunotherapy will be used according to the situation. In patients with mild or improved symptoms, it can be considered not to use immunotherapy because the lesion has been completely excised during the surgery. But this idea is still controversial. Although The effect of neurosurgery alone was good in our group. But in our study, half of the patients who underwent surgery had tumors considered preoperatively, and half were patients who did not respond well to immunotherapy (hormone or hormone combined with propyl bulb). So, this may have something to do with our selection bias. In our group, the effect of surgical excision alone was still good. Among the 5 patients who underwent exploratory craniotomy combined with hormone therapy, 2 died with a poor prognosis, which may be related to the aggravation of the disease, and it is not necessarily related to the use of hormones. The samples size is small; therefore, large sample studies are still needed to confirm this. The effect of neurosurgery alone was good; therefore, the effect of surgery on TDLs can be further studied.
Most TDLs may have a possible benign course, and they are not associated with poorer outcomes1,8,63. Some reports suggested that patients with TDLs often had a monophasic and benign course16,64. The prognosis of patients with TDLs is relatively more favorable than expected in Taiwan. Most of the patients were females (11/12), and all the patients had relapsing-remitting10 .One report found that patients presenting with isolated TDLs might have a better long-term prognosis than patients with conventional MS6,65, which was consistent with our results. Patients who experienced recurrence as MS died in our study. Most of the other patients had a good outcome.
Limitations of our study include the relatively small sample size, incomplete inspection, and some missing data, given that this was a retrospective review. Children with TDLs were not included in our study. Our study did not include spinal lesions. Most of the patients were unable to undergo an imaging examination. The strength of our study is that we included pathologically proven cases.
In the future, we need further large-scale studies comparing suspected TDLs cases that did not undergo pathological examination with the patients who underwent surgery, in order to conclude whether the surgery caused damage or was helpful for the treatment of patients. It may also be necessary to further determine the relationship between TDLs, MS, or NMOSD. This article serves as a starting point.
Conclusion
There was no gender predilection of the atypical TDLs in our group. The patients did not have serious nervous system injury at onset, and the main presentation included weakness of the limbs, headache or dizziness, and alalia. Most of the lesions were supratentorial lesions, and the lesions were most prevalent in the frontal, temporal, and parietal lobes. Neither edema nor mass effect was serious in the cases in this study. The most common form was patchy lesions on MRI enhancement in atypical TDLs. Special imaging examinations are helpful for the identification of TDLs. Cerebrospinal fluid and demyelination test can be an indicator of TDLs, and seizure may be a poor prognostic indicator. The intraoperative frozen section can be deceptive. Most TDLs have monophasic courses and good outcomes. The effect of neurosurgery alone was good in our group, and the effect of surgery on TDLs can be further studied. Is total excision without affecting the neurological function a better option when surgery is unavoidable? This needs to be verified by a large number of further studies. It is necessary to follow-up the patients diagnosed with TDLs by brain biopsy and neurosurgery.
Data availability
The data that support the findings of this study are available from the corresponding author [YW], upon reasonable request.
References
Lucchinetti, C. F. et al. Clinical and radiographic spectrum of pathologically confirmed tumefactive multiple sclerosis. Brain 131, 1759–1775 (2008).
Mabray, M. C. et al. Performance of apparent diffusion coefficient values and conventional mri features in differentiating tumefactive demyelinating lesions from primary brain neoplasms. AJR Am. J. Roentgenol. 205, 1075–1085 (2015).
Kalus, S., Muzio, B. D. & Gaillard, F. Demyelination preceding a diagnosis of central nervous system lymphoma. J. Clin. Neurosci. 24, 146–148 (2016).
Jeong, I. H., Kim, S. H., Hyun, J. W., Joung, A. R. & Kim, H. J. Tumefactive demyelinating lesions as a first clinical event: Clinical, imaging, and follow-up observations. J. Neurol. Sci. 358, 118–124 (2015).
Hardy, T. A., Tobin, W. O. & Lucchinetti, C. F. Exploring the overlap between multiple sclerosis, tumefactive demyelination and Balos concentric sclerosis. Mult. Scler. 22, 986–992 (2016).
Wattamwar, P. R., Baheti, N. N., Kesavadas, C., Nair, M. & Radhakrishnan, A. Evolution and long term outcome in patients presenting with large demyelinating lesions as their first clinical event. J. Neurol. Sci. 297, 29–35 (2010).
Kilic, A. K., Kurne, A. T., Oguz, K. K., Soylemezoglu, F. & Karabudak, R. Mass lesions in the brain: Tumor or multiple sclerosis? clinical and imaging characteristics and course from a single reference center. Turk. Neurosurg. 23, 728–735 (2013).
Kepes, J. J. Large focal tumor-like demyelinating lesions of the brain: Intermediate entity between multiple sclerosis and acute disseminated encephalomyelitis? A study of 31 patients. Ann. Neurol. 33, 18–27 (1993).
Wallner-Blazek, M., Rovira, A., Fillipp, M. & Rocca, M. A. Atypical idiopathic inflammatory demyelinating lesions: prognostic implications and relation to multiple sclerosis. J. Neurol. 260, 2016–2022 (2013).
Yi-Chun, K., Wang, K. C., Yuan, W. H., Ching-Piao, T. & Steven, J. Tumefactive multiple sclerosis in Taiwan. PLoS ONE 8, e69919 (2013).
Association, N. G. o. N. B. o. C. M., Immunology, N. C. o. C. S. f. & Association, I. S. o. C. S. Chinese Guidelines for the Diagnosis and Management of Tumefactive Demyelinating Lesions of Central Nervous System. 中华医学杂志(英文版) 130, 1838–1850 (2017).
Velden, M. V. D., Bots, G. T. A. M. & Endtz, L. J. Cranial CT in multiple sclerosis showing a mass effect. Surg. Neurol. 12, 307–310 (1979).
Patriarca, L. et al. Is size an essential criterion to define tumefactive plaque? MR features and clinical correlation in multiple sclerosis. Neuroradiol. J. 29, 384–389 (2016).
Paty, D. W., Oger, J. J. F., Kastrukoff, L. F., Hashimoto, S. & Brandejs, V. MRI in the diagnosis of MS: A prospective study with comparison of clinical evaluation, evoked potentials, oligoclonal banding, and CT. Neurology 38, 180–185 (1988).
Mcadam, L. C., Blaser, S. I. & Banwell, B. L. Pediatric tumefactive demyelination: Case series and review of the literature. Pediatr. Neurol. 26, 18–25 (2002).
Yapici, Z. & Eraksoy, M. Bilateral demyelinating tumefactive lesions in three children with hemiparesis. J. Child Neurol. 17, 655–660 (2002).
Comi, G. Multiple sclerosis: Pseudotumoral forms. Neurol. Sci. 25, s374–s379 (2004).
Kantarci, O. Survival and predictors of disability in Turkish MS patients. Am. Acad. Neurol. 51, 765–772 (1998).
Altintas, A. et al. Clinical and radiological characteristics of tumefactive demyelinating lesions: Follow-up study. Mult. Scler. 18, 1448–1453 (2012).
Nyquist, P. A., Cascino, G. D., Mcclelland, R. L., Annegers, J. F. & Rodriguez, M. Incidence of seizures in patients with multiple sclerosis: A population-based study. Mayo Clin. Proc. Mayo Clin. 77, 910–912 (2002).
Dagher, A. P. & Smirniotopoulos, J. Tumefactive demyelinating lesions. Neuroradiology 38, 560–565 (1996).
Kalyan-Raman, U. P., Garwacki, D. J. & Elwood, P. W. Demyelinating disease of corpus callosum presenting as glioma on magnetic resonance scan: A case documented with pathological findings. Neurosurgery 21, 247–250 (1987).
Rieth, K. G. et al. Primary demyelinating disease simulating glioma of the corpus callosum. J. Neurosurg. 55, 620–624 (1981).
Xia, L. et al. Tumefactive demyelinating lesions: nine cases and a review of the literature. Neurosurg. Rev. 32, 171–179 (2009).
Javalkar, V., Manix, M., Wilson, J. & Nanda, A. Open ring enhancement in atypical brain demyelination. J. Clin. Neurosci. 19, 910–912 (2012).
Masdeu, J. C. et al. Open-ring imaging sign: Highly specific for atypical brain demyelination. Neurology 54, 1427–1433 (2000).
Masdeu, J. C., Quinto, C., Olivera, C., Leslie, D. & Visintainer, P. The open ring or white-matter-crescent pattern of enhancement differentiates demyelinating brain disease with tumor-like presentation from infection and neoplasm. Riv. Neuroradiol. 11, 15–16 (1998).
He, J., Grossman, R. I., Ge, Y. & Mannon, L. J. Enhancing patterns in multiple sclerosis: evolution and persistence. AJNR Am. J. Neuroradiol. 22, 664 (2001).
Brinar, M., Rado, M., Habek, M. & Poser, C. M. Enlargement of the spinal cord: Inflammation or neoplasm?. Clin. Neurol. Neurosurg. 108, 284–289 (2006).
Brinar, V. V. Non-MS recurrent demyelinating diseases. Clin. Neurol. Neurosurg. 106, 197–210 (2004).
Fazekas, F., Barkhof, F., Filippi, M., Grossman, R. I. & Miller, D. H. The contribution of magnetic resonance imaging to the diagnosis of multiple sclerosis. Neurology 53, 448–456 (1999).
Lycklama, G. et al. Spinal-cord MRI in multiple sclerosis. Lancet Neurol. 2, 555–562 (2003).
Matsushita, T. et al. Extensive vasogenic edema of anti-aquaporin-4 antibody-related brain lesions. Mult. Scler. 15, 1113–1117 (2009).
Cha, S. et al. Dynamic contrast-enhanced T2*-weighted MR imaging of tumefactive demyelinating lesions. Am. J. Neuroradiol. 22, 1109–1116 (2001).
Ernst, T., Chang, L., Walot, I. & Huff, K. Physiologic MRI of a tumefactive multiple sclerosis lesion. Neurology 51, 1486–1488 (1998).
Takenaka, S. et al. Metabolic assessment of monofocal acute inflammatory demyelination using MR spectroscopy and <sup>11</sup>C-methionine-, <sup>11</sup>C-choline-, and <sup>18</sup>F-fluorodeoxyglucose-PET. Brain Tumor Pathol. 28, 229–238 (2011).
刘建国 et al. 病理证实的瘤样脱髓鞘病 60 例影像学特点. 中华神经科杂志 47 (2014).
Hardy, T. A. & Chataway, J. Tumefactive demyelination: an approach to diagnosis and management. J. Neurol. Neurosurg. Psychiatry 84, 1047–1053 (2013).
Dae, et al. Distinguishing tumefactive demyelinating lesions from glioma or central nervous system lymphoma: added value of unenhanced CT compared with conventional contrast-enhanced MR imaging. Radiology 251, 467–475 (2009).
Kiriyama, T. et al. Characteristic neuroimaging in patients with tumefactive demyelinating lesions exceeding 30 mm. J. Neuroimaging 21, e69–e77 (2011).
Wood, D. D., Bilbao, J. M., O’Connors, P. & Moscarello, M. Acute multiple sclerosis (marburg type) is associated with developmentally immature myelin basic protein. Ann. Neurol. 40, 18–24 (1996).
Beniac, D. R. et al. Marburg’s variant of multiple sclerosis correlates with a less compact structure of myelin basic protein. Mol. Cell Biol. Res. Commun. MCBRC 1, 48–51 (1999).
Maciej, J. et al. Distinct brain imaging characteristics of autoantibody-mediated CNS conditions and multiple sclerosis. Brain A J. Neurol. 140, 617–627 (2017).
Shu, Y. et al. Brain histopathological study and prognosis in MOG antibody-associated demyelinating pseudotumor. Ann. Clin. Transl. Neurol. 6, 392–396 (2019).
Ramanathan, S., Prelog, K., Barnes, E. H., Tantsis, E. M. & Dale, R. C. Radiological differentiation of optic neuritis with myelin oligodendrocyte glycoprotein antibodies, aquaporin-4 antibodies, and multiple sclerosis. Mult. Scler. 22, 470–482 (2015).
Dale, R. C. et al. Acute disseminated encephalomyelitis, multiphasic disseminated encephalomyelitis and multiple sclerosis in children. Brain 123(Pt 12), 2407–2422. https://doi.org/10.1093/brain/123.12.2407 (2000).
Jeong, I. H. et al. Tumefactive demyelinating lesions as a first clinical event: Clinical, imaging, and follow-up observations. J. Neurol. Sci. 358, 118–124. https://doi.org/10.1016/j.jns.2015.08.034 (2015).
Tremblay, M. A., Villanueva-Meyer, J. E., Cha, S., Tihan, T. & Gelfand, J. M. Clinical and imaging correlation in patients with pathologically confirmed tumefactive demyelinating lesions. J. Neurol. Sci. 381, 83–87. https://doi.org/10.1016/j.jns.2017.08.015 (2017).
Huisman, T. A. Tumor-like lesions of the brain. Cancer Imaging Off. Publ. Int. Cancer Imaging Soc. 9(A), S10-13. https://doi.org/10.1102/1470-7330.2009.9003 (2009).
Weinshenker, B. G. et al. A randomized trial of plasma exchange in acute central nervous system inflammatory demyelinating disease. Ann. Neurol. 46, 878–886. https://doi.org/10.1002/1531-8249(199912)46:6%3c878::aid-ana10%3e3.0.co;2-q (1999).
Munarriz, P. M. et al. Tumefactive multiple sclerosis requiring emergency craniotomy: Case report and literature review. Neurocirugia (Asturias, Spain) 24, 220–224. https://doi.org/10.1016/j.neucir.2013.02.008 (2013).
Malhotra, H. S. et al. Characterization of tumefactive demyelinating lesions using MR imaging and in-vivo proton MR spectroscopy. Mult. Scler. 15, 193–203. https://doi.org/10.1177/1352458508097922 (2009).
Puri, V., Chaudhry, N., Gulati, P., Tatke, M. & Singh, D. Recurrent tumefactive demyelination in a child. J. Clin. Neurosci. Off. J. Neurosurg. Soc. Australas. 12, 495–500. https://doi.org/10.1016/j.jocn.2004.07.001 (2005).
Sinha, M. K., Garg, R. K., Bhatt, M. L. & Chandra, A. Tumefactive demyelinating lesion: Experience with two unusual patients. J. Postgrad. Med. 56, 146–149. https://doi.org/10.4103/0022-3859.65292 (2010).
Totaro, R. et al. Occurrence and long-term outcome of tumefactive demyelinating lesions in multiple sclerosis. Neurol. Sci. Off. J. Ital. Neurol. Soc. Ital. Soc. Clin. Neurophysiol. 37, 1113–1117. https://doi.org/10.1007/s10072-016-2558-1 (2016).
Braverman, D. L., Lachmann, E. A., Tunkel, R. & Nagler, W. Multiple sclerosis presenting as a spinal cord tumor. Arch. Phys. Med. Rehabil. 78, 1274–1276. https://doi.org/10.1016/s0003-9993(97)90344-0 (1997).
Checrallah, A., Geha, S., Rizk, T. & Koussa, S. Multiple sclerosis mimicking brain tumor: An unusual presentation. Leban. Med. J. 53, 45–49 (2005).
Di Patre, P. L. et al. “Tumor-mimicking” multiple sclerosis. Clin. Neuropathol. 22, 235–239 (2003).
Iwamoto, K., Oka, H., Utsuki, S., Ozawa, T. & Fujii, K. Late-onset multiple sclerosis mimicking brain tumor: A case report. Brain Tumor Pathol. 21, 83–86. https://doi.org/10.1007/bf02484515 (2004).
Lolekha, P. & Kulkantrakorn, K. Tumor-like manifestation, uncommon form of multiple sclerosis: Report of a patient. Chotmaihet Thangphaet 89, 721–726 (2006).
Poser, S. et al. Acute demyelinating disease. Classification and non-invasive diagnosis. Acta Neurol. Scand. 86, 579–585. https://doi.org/10.1111/j.1600-0404.1992.tb05490.x (1992).
Erome, J. Neuromyelitis optica and concentric rings of Balo in the brainstem. Arch. Neurol. 66, 274 (2009).
Hayashi, T. et al. Inflammatory demyelinating disease mimicking malignant glioma. J. Nucl. Med. Off. Publ. Soc. Nucl. Med. 44, 565 (2003).
Kumar, K., Toth, C. & Jay, V. Focal plaque of demyelination mimicking cerebral tumor in a pediatric patient. Pediatr. Neurosurg. 29, 60–63 (1998).
Siri, A. et al. Isolated tumefactive demyelinating lesions: diagnosis and long-term evolution of 16 patients in a multicentric study. J. Neurol. 262, 1637–1645 (2015).
Funding
This article was funded by Tianjin Key Medical Discipline (Specialty) Construction Project (No. TJYXZDXK-052B).
Author information
Authors and Affiliations
Contributions
Y.Z.: Conceptualization, Methodology, statistics, Writing- Original draft preparation. X.Z. and T.Z.: Data curation, Writing- Original draft preparation. C.Z.: Visualization, Investigation. R.L. /X.Y./J.L.: Supervision. Y.Y. and J.Z./Y.Z.: statistics, Validation. W.Y.: Writing- Reviewing and Editing.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Zhang, Y., Zhang, T., Zhang, X. et al. Clinical spectrum and prognosis of pathologically confirmed atypical tumefactive demyelinating lesions. Sci Rep 13, 7773 (2023). https://doi.org/10.1038/s41598-023-34420-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-023-34420-4
- Springer Nature Limited