
Abstract
Esophageal squamous cell carcinoma (ESCC) has a dismal prognosis because of atypical early symptoms and heterogeneous therapeutic responses. 5-methylcytosine (m5C) modification plays an important role in the onset and development of many tumors and is widespread in long non-coding RNA (lncRNA) transcripts. However, the functions of m5C and lncRNAs in ESCC have not been completely elucidated. Herein, this study aimed to explore the role of m5C-related lncRNAs in ESCC. The RNA-seq transcriptome profiles and clinical information were downloaded from the TCGA-ESCC database. Pearson analysis was used to identify m5C-related lncRNAs. Then we established the m5C-related lncRNAs prognostic signature (m5C-LPS) using univariate Cox and least absolute shrinkage and selection operator (LASSO) regression analysis. Then, the prognostic value of m5C-LPS was evaluated internally and externally using the TCGA-ESCC and GSE53622 databases through multiple methods. We also detected the expression of these lncRNAs in ESCC cell lines and patient tissues. Fluorescence in situ hybridization (FISH) was used to detect the prognostic value of specific lncRNA. In addition, clinical parameters, immune status, genomic variants, oncogenic pathways, enrichment pathways, and therapeutic response features associated with m5C-LPS were explored using bioinformatics methods. We constructed and validated a prognostic signature based on 9 m5C-related lncRNAs (AC002091.2, AC009275.1, CAHM, LINC02057.1, AC0006329.1, AC037459.3, AC064807.1, ATP2B1-AS1, and UBAC2-AS1). The quantitative real-time polymerase chain reaction (qRT-PCR) revealed that most lncRNAs were upregulated in ESCC cell lines and patient tissues. And AC002091.2 was validated to have significant prognostic value in ESCC patients. A composite nomogram was generated to facilitate clinical practice by integrating this signature with the N stage. Besides, patients in the low-risk group were characterized by good clinical outcomes, favorable immune status, and low oncogenic alteration. Function enrichment analysis indicated that the risk score was associated with mRNA splicing, ncRNA processing, and DNA damage repair response. At the same time, we found significant differences in the responses to chemoradiotherapy between the two groups, proving the value of m5C-LPS in treatment decision-making in ESCC. This study established a novel prognostic signature based on 9 m5C-related lncRNAs, which is a promising biomarker for predicting clinical outcomes and therapeutic response in ESCC.
Similar content being viewed by others

Introduction
Esophageal cancer (EC) ranks eighth and sixth in terms of incidence and mortality worldwide, respectively1. Among primary esophageal cancers, approximately 88% are classified as esophageal squamous cell carcinoma (ESCC), which exhibits a relatively low 5-year survival rate ranging from 5 to 25%2,3,4,5. Currently, extensive research on molecular mechanisms has yielded promising precision cancer treatment strategies for numerous cancers6. Recently, growing research has highlighted the role of RNA post-transcriptional modifications 5-methylcytosine (m5C) on tumor development7,8. Several studies have provided evidence that m5C can influence the development of ESCC9,10. However, the precise impact of m5C on ESCC remains unclear and needs further investigation.
The reversible RNA post-transcription modification m5C, similar to N6-methyladenosine (m6A), has got enormous attention and can dynamically regulate RNA stability, translation, splicing, and exportation7,8,11. The m5C is a type of cytosine methylation that involves the addition of a methyl group to the fifth carbon position and is regulated by several enzymes including “writers” (methyltransferases: NSUN1-7, DNMT1, DNMT2 also named TRDMT1, DNMT3A, and DNMT3B), “erasers” (demethylases: TET1-3), and “readers” (YBX1 and ALYREF)12,13,14.
Mounting evidence suggests that dysregulated expression of long non-coding RNAs (lncRNAs) plays a critical role in tumor development and response to therapy15,16,17. For instance, lncRNA CASC9 has been shown to promote ESCC metastasis18. While m5C was initially found in tRNA and rRNA, emerging evidence suggests that it is also widespread presence in mRNAs and non-coding RNAs19,20,21. And the methylation density around the transcriptional start site of lncRNAs is higher than that of protein-coding genes21. Upregulated NSUN2-mediated NMR methylation in ESCC, resulting in cancer metastasis and drug resistance22, which suggested that m5C-methylated lncRNAs can regulate the biological function of cancer. However, the evidence for m5C in regulating lncRNAs in ESCC is limited and requires further research.
In this study, we aimed to investigate the function of m5C-related lncRNAs in ESCC and construed m5C-related lncRNAs prognostic signature (m5C-LPS) based on the TCGA-ESCC cohort. Additionally, we also explored the relationship between the m5C-LPS and clinical prognostic, immune status, genomic variants, enrichment pathways, as well as drug sensitivity in ESCC.
Methods
Patients cohorts
We have included patients diagnosed with ESCC in The Cancer Genome Atlas (TCGA) program (https://portal.gdc.cancer.gov/repository?facetTab=cases). Patients without complete clinical information and transcriptome profiling, or diagnosed with esophageal adenocarcinomas were excluded. Finally, the transcriptome, clinicopathologic, and somatic mutation data of 80 ESCC and 11 adjacent normal tissues were downloaded. Additionally, RNA microarray profiles and corresponding clinical information of 60 ESCC patients were downloaded from the Gene Expression Omnibus database (GSE53622, https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=gse53622). The clinicopathological parameters of TCGA-ESCC and GSE53622 cohorts were summarized in Table S1. Immunohistochemical staining images of normal esophageal tissues were obtained from the Human Protein Atlas (HPA) (https://www.proteinatlas.org/). And this study started in February 2022 and finished in July 2022.
Paraffin embedded sections of 54 ESCC patients were obtained from the First Affiliated Hospital of Xi’an JiaoTong University. Tissues were collected during surgery and were used for Fluorescence in situ hybridization (FISH) examination. And 14 ESCC and corresponding normal tissue samples were collected for the detection of lncRNA expression.
Identification of regulators of m5C and co-expression lncRNAs
We identified 16 m5C regulators from previous literature and extracted their expression from RNA-seq profiles of ESCC and adjacent normal tissues. Then, the differential expression of 16 m5C regulators was determined in the ESCC tissues versus adjacent normal tissues. The differential expression of these regulators was analyzed, and their interrelationships were visualized using the ‘corrplot’ R package. A protein–protein interaction (PPI) network of m5C regulators was constructed using the STRING database (https://cn.string-db.org/) with the gene interaction score ≥ 0.523. We selected the lncRNAs existed in both TCGA-ESCC and GSE53622 cohorts for widespread use of m5C-LPS. Pearson correlation coefficient was calculated between the expression of 16 m5C regulators and lncRNAs using the built-in function ‘cor.test’ in R. We identified 4279 m5C-related lncRNAs with |correlation coefficient| > 0.35 and the p-value < 0.01 for further analysis.
Construction and validation of m5C-related lncRNA prognosis signature
We used the ‘survival’ R package to perform univariate Cox regression analysis on the candidate m5C-related lncRNAs, filtering out those with significant prognostic value (p < 0.05). Then, we used the least absolute shrinkage and selection operator (LASSO) regression analysis with the ‘glmnet’ R package to establish a prognostic signature and calculate the coefficients for each lncRNA24,25,26. These coefficients were used to generate a risk score formula: \(\mathrm{Risk \,Score}=\sum_{i}coefficient\, of \,m5C\, related\, lncRNAi\times lncRNAi \,expression\, level\). Patients were stratified into high- and low-risk groups based on their calculated risk scores. Kaplan–Meier (K–M) analysis was performed to assess the overall survival (OS) of different groups, and time-dependent receiver operating characteristic (ROC) curve analysis was used to evaluate the predictive value of the risk score with the ‘survivalROC’ R package.
Clinical relevance investigation
A Sankey diagram was used to illustrate the one-to-one match between the m5C genes, m5C-related lncRNAs, and the corresponding risk types. Furthermore, a correlation circle graph was generated using the ‘corrplot’ and ‘circlize’ R package to visualize the co-expression status of the 9 identified lncRNAs. We also investigated the association between m5C-LPS and clinicopathological parameters. Both univariate and multivariate Cox regression analyses were conducted to investigate the independent value of the m5C-LPS and other parameters. Based on the significant prognostic variables, we constructed a nomogram to predict 1-, 2-, and 3-year survival rates using the ‘rms’ R package. And calibration curves were used to verify the agreement between nomogram-predicted survival and actual survival probabilities. Additionally, we evaluated the prognostic value of clinicopathological features by using ROC curves and calculating the area under the curve (AUC).
Evaluation of signaling pathways enrichment
We conducted functional enrichment analyses based on gene ontology (GO)27, Kyoto Encyclopedia of Genes and Genomes (KEGG)28, and Reactome29 databases to explore the biological functions and pathways associated with m5C-LPS through ‘clusterProfiler’30 and ‘ReactomePA’ R package.
Estimation of the tumor microenvironment signatures
Estimate31, single-sample gene set enrichment analysis (ssGSEA)32, Cibersort33, and xCell34 algorithms were utilized to estimate the relative abundance of immune and stromal cells in the tumor microenvironment. We also calculated the Pearson coefficients between risk scores and immune checkpoint genes and immunomodulators, such as chemokines, receptors, MHC, immunoinhibitors, and immunostimulators, which were obtained from the TISIDB database35.
Characterization of genetic alteration
The ‘maftools’ R package was utilized to identify the top 20 mutated genes based on the mutation rate across low- and high-risk groups36. Subsequently, we further investigated the fraction of affected samples and pathways based on alterations in 10 canonical oncogenic signaling pathways for different risk groups37.
Drug sensitivity analysis
We employed the ‘pRRophetic’ R package38 to predict the half-maximal inhibitory concentration (IC50) for each patient using three publicly available drug sensitivity databases (Cancer Genome Project (CGP)39, Cancer Therapeutics Response Portal (CTRP)40, and Genomics of Drug Sensitivity in Cancer (GDSC)41). Additionally, we utilized the genomic-adjusted radiation dose (GARD) model42 to predict the radiotherapy response of each patient, with higher GARD values indicating increased sensitivity to radiotherapy.
Cell lines and reagents
The human normal esophageal cell line HET-1A was purchased from American Type Culture Collection (ATCC, Virginia, USA), and the human ESCC cell lines TE-1 and KYSE150 were purchased from the Cell Bank of the Chinese Academy of Sciences Typical Culture Preservation Committee (Shanghai, China). HET-1A was cultured in Dulbecco’s Modified Eagle’s Medium (DMEM, Gibico, USA) supplemented with 10% fetal bovine serum (FBS, Gibco, USA), while TE-1, and KYSE150 were cultured in Roswell Park Memorial Institute (RPMI) 1640 medium (Gibico, USA) supplemented with 10% FBS. All cells were cultured in a 5% CO2 incubator at 37 °C.
Total RNA extraction and real-time quantitative PCR
Total RNA was extracted using the RNAfast200 kit (Fastagen, China) according to the manufacturer’s instructions. RNA concentration was quantified using NanoDrop 3000 (ThermoFisher, USA). Then, 1.0 μg of total RNA in a 20 μl reaction system was reversely transcribed into cDNAs using Evo M-MLV RT Kit with gDNA Clean for polymerase chain reaction (PCR, Accurate Biotechnology, China). Quantitative real-time PCR (qRT–PCR) was performed using 2\(\times \) RealStar Green Fast Mixture (GeneStar Technology, China). GAPDH expression was used as an internal reference. The relative expression level of lncRNAs was calculated using the 2−ΔΔCT method. Each experiment was performed in triplicate. The primer sequences used in this study are listed in Table S2.
FISH assay
The FISH probe of lncRNA AC002091.2 was synthesized by Servicebio (Wuhan, China). Paraffin embedded sections were dewaxed, rehydrated, digested, and dehydrated with dimethylbenzene, graded ethanol, protease K. Then the FISH probe was added to the hybridization mixture and incubated overnight. Next, the section was washed in the dark with washing buffer containing saline sodium citrate and PBS. Sections were stained with DAPI for 10 min and then visualized by fluorescence microscope.
Statistical analysis
All statistical analyses were performed using R software (version 4.1.1) and GraphPad Prism (version 8.0, USA). The differences between the two groups were compared using student’s t-test, while one-way analysis of variance (ANOVA) was used for multiple groups. Fisher’s exact test was used to compare categorical variables. The correlation between two continuous variables was analyzed using Pearson’s test. p-value < 0.05 was considered statistically significant.
Ethics approval and consent to participate
This study was approved by the Ethics Committee of The First Affiliated Hospital of Xi’an Jiaotong University (Approval Number: 2017-146).
Results
To facilitate the comprehension of the study, a schematic diagram is presented in Fig. S1.
Expression patterns of m5C regulators in ESCC and normal esophageal tissue
We extracted the expression profiles of 16 m5C regulators in the TCGA-ESCC cohort and subsequently compared their expression levels between 80 ESCC tumor samples and 11 normal adjacent samples. Our analysis revealed that the expression of most genes, including DNMT3B, NOP2, DNMT1, ALYREF, NSUN2, NSUN5, TET2, TET3, DNMT3A, TET1, and YBX1 were significantly higher in ESCC tissues than in normal adjacent tissues (Fig. 1A). Moreover, the immunohistochemical staining images of normal esophageal tissues from HPA showed that 10 of 15 m5C regulators were not more than medium expression, while NSUN3 and NSUN5 were not detected (Fig. S2). To investigate the interrelationships among these 16 m5C regulators, we obtained a PPI network using the STRING database. After setting the minimum interaction score as 0.5, we identified the PPI network contains all m5C genes and 90 edges (Fig. 1B). And TRDMT1 was found to be the hub gene of the network with 11 edges (Table S3). In addition, the correlation analysis revealed significant positive correlations between TRDMT1 and other 8 m5C genes. Interestingly, all m5C regulators showed a general positive correlation, with DNMT1 exhibiting the highest correlation with ALYREF (r = 0.67) (Fig. 1C).

The expression pattern and interactive landscape of the m5C regulators in the TCGA-ESCC database. (A) Heatmap presenting the expression of 16 m5C regulators in normal esophageal (N) and ESCC (T) tissues from TCGA-ESCC database. p < 0.1, *p < 0.05, **p < 0.01, and ***p < 0.001. (B) Protein–protein interaction (PPI) network showing the interaction between m5C regulators. (C) Heatmap showing the Pearson correlation among 16 m5C regulators.
Construction of the m5C‑LPS in the TCGA database
Subsequently, we performed Pearson analysis based on the lncRNAs and m5C regulators in TCGA-ESCC profiles, and a total of 4279 lncRNAs were significantly correlated with m5C regulators (|Pearson coefficient| > 0.35 and p < 0.01). After filtering lncRNAs with the sum expression < 0.01, univariate Cox regression analysis was conducted to further explore the m5C-related lncRNAs associated with prognosis. Finally, we identified 41 lncRNAs that were significantly associated with the OS of ESCC patients (Table S4).
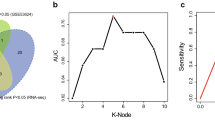
To eliminate the collinearity of variables and minimize estimation variance, LASSO regression analysis was applied to establish a prognostic signature using the 41 aforementioned lncRNAs. Subsequently, an m5C-LPS comprising 9 lncRNAs was identified based on the optimal λ value (Fig. 2A,B). Subsequently, the risk score was calculated based on the coefficients of the nine identified lncRNAs and their corresponding expression levels, yielding a concordance index (C-index) of 0.83, indicating strong discriminatory power (Fig. 2C,D). Besides, the model exhibited a sensitivity of 0.880, specificity of 0.643, positive likelihood ratio of 2.464, negative likelihood ratio of 0.187, positive predictive value of 0.524, and negative predictive value of 0.923. The m5C-LPS formula was calculated as follows: \(\mathrm{Risk\, Score}=\left(-1.45488\right)\times ATP2B1-AS1+0.78504\times LINC02057+\left(-3.09357\right)\times UBAC2-AS1+0.09339\times CAHM+\left(-1.19951\right)\times AC064807.1+\left(-2.22323\right)\times AC037459.3+0.87974\times AC002091.2+\left(-0.60497\right)\times AC006329.1+0.2869\times AC009275.1.\)

Identification and validation of the m5C-related lncRNAs prognostic signature (m5C-LPS) based on the cohort of TCGA-ESCC and GSE53622. (A,B) The minimum criterion of the LASSO regression algorithm was used to identify the most robust prognostic m5C-related lncRNAs. (C) Forest plot presenting the hazard ratio (HR) and 95% confidence interval (CI) of the 9 lncRNAs by the multivariate Cox regression. (D) The coefficients of the 9 lncRNAs contained in the m5C-LPS formula. (E) The distributions of the risk score, vital status, overall survival (OS), and expression levels of the 9 m5C-related lncRNAs in low- and high-risk groups in the cohort from TCGA-ESCC. (F) Kaplan–Meier (K–M) analysis demonstrated that patients with higher risk scores exhibited worse overall survival in the cohort from TCGA-ESCC. (G) K–M analysis demonstrated that patients with higher risk scores exhibited worse disease-free survival in the cohort from TCGA-ESCC. (H) The area under the curve (AUC) of the time-dependent ROC curves measures the predictive value of the risk score in the cohort from TCGA-ESCC. (I) The distributions of the risk score, vital status, overall survival (OS), and expression levels of the 9 m5C-related lncRNAs in low- and high-risk groups in the cohort from GSE53622. (J) K–M analysis demonstrated that patients with higher risk scores exhibited worse overall survival in the cohort from GSE53622. (K) AUC of the time-dependent ROC curves measuring the predictive value of the risk score in the cohort from GSE53622.
Subsequently, we categorized the 80 ESCC patients into low- and high-risk groups based on the median risk score. And the vital status and expression levels of the corresponding 9 lncRNAs in the cohort from TCGA-ESCC have presented in Fig. 2E. K–M analysis revealed that the patients in the high-risk group had relatively poorer OS and disease-free survival (DFS) compared with the low-risk group (OS: p < 0.0001, DFS: p = 0.064, Fig. 2F,G). Moreover, time-dependent ROC curves implied that m5C-LPS exhibited a promising ability to predict prognosis in the TCGA-ESCC cohort (1-year AUC = 0.839, 2-year AUC = 0.919, 3-year AUC = 0.898; Fig. 2H).
Validation of m5C-LPS in the cohort from the GEO database
To validate the prognostic value of m5C-LPS, we calculated risk scores for another 60 ESCC patients from the GSE53622 cohort using the same formula. ESCC patients were divided into low- and high-risk groups according to the medium value. The distribution of the risk score, survival status, and lncRNAs expression showed that patients with higher risk scores had shorter OS and higher mortality status (Fig. 2I). Consistent with the findings in the TCGA-ESCC cohort, patients in the high-risk group presented significantly poorer prognoses (p = 0.012, Fig. 2J). And the AUC of the m5C-LPS was 0.7 at 2 years, 0.715 at 3 years, and 0.79 at 4 years (Fig. 2K).
Co-expression status and differential expression of m5C-related lncRNAs
We examined the co-expression status and differential expression of the 9 m5C-related lncRNAs. The Sankey plot showed one-to-one matches between the 7 m5C genes (5 writers: DNMT1, NSUN3, NSUN5-7; 2 erasers: TET1-2) and the 9 lncRNAs used in constructing the m5C-LPS. Additionally, the Sankey plot also depicted the risk type of each lncRNA (risk lncRNAs: AC002091.2, AC009275.1, CAHM, and LINC02057.1; protect lncRNAs: AC0006329.1, AC037459.3, AC064807.1, ATP2B1-AS1, and UBAC2-AS1, Fig. 3A). Moreover, the correlation circle plot revealed a general positive correlation among these m5C-related lncRNAs, except for CHAM and AC037459.3 had negative relationship with AC009275.1 and LINC02057, and UBAC2-AS1 showed negative correlation with AC002091.2 (Fig. 3B). Then, we compared the expression levels of these lncRNAs in normal esophageal and ESCC samples and observed that 7 lncRNAs were upregulated and 1 lncRNA was downregulated in ESCC samples (Fig. 3C).

Co-expression status and expression level of m5C-related lncRNAs in TCGA-ESCC database and ESCC cell lines. (A) Sankey plot showing one-to-one matches between m5C genes, m5C-related lncRNAs, and their risk type. (B) Circle plot presenting the co-expression status of the 9 m5C-related lncRNAs with coefficients annotated. (C) The expression level of m5C-related lncRNAs in normal esophageal and ESCC tissues based on TCGA-ESCC database. (D) Representative Fluorescence in situ hybridization (FISH) images of AC002091.2 in ESCC tissues. Scale bars represent 50 μm. (E) K–M plot for overall survival grouped by AC002091.2 expression in 54 ESCC patients. *p < 0.05, **p < 0.01, ***p < 0.001.
Subsequently, we performed qRT-PCR using normal esophageal cell line HET-1A and ESCC cell lines TE-1 and KYSE150. The boxplot revealed that the upregulation of LINC02057, UBAC2-AS1, CAHM, AC002091.2, AC006329.1, and AC009275.1 in ESCC cells, while AC037459.3 was downregulated in ESCC cells (Fig. S3A). And we also detected the expression of these lncRNAs in ESCC and adjacent normal tissues and found that most of lncRNAs were upregulated in ESCC tissues (Fig. S3B). These results indicated that the expression patterns of m5C-related lncRNAs are consistent with the findings from the TCGA database. Since AC002091.2 was upregulated in ESCC cell lines and tissues and was of great prognostic value for ESCC patients, we subsequently investigated the relationship between the expression of AC002091.2 and patients’ survival. The FISH results showed that AC002091.2 was located in the cytoplasm (Fig. 3D). And K–M plot revealed that patients with higher AC002091.2 expression had relatively poor prognosis (Fig. 3E, p = 0.0028).
Correlation of the risk score acquired from m5C-LPS and clinicopathological parameters
To evaluate the clinical significance of m5C-LPS, we assessed its association with various clinicopathological parameters of ESCC. Subgroup analysis stratified by T stage revealed a significantly higher risk score in T4 ESCC patients compared to T3 ESCC patients (p = 0.038, Fig. 4A). Stratification by M stage indicated an increased risk score in M1 patients, although the difference did not reach statistical significance (p = 0.15, Fig. 4B). No significant differences were observed between age, gender, race, tumor location, histologic grade, N stage, stage, reflux history and risk score (p > 0.05, Fig. S4A–G, Fig. 4C). Besides, ESCC patients with alcohol history exhibited a significantly elevated risk score than those without alcohol history (p = 0.022, Fig. 4D), while ESCC patients with or without smoking history had similar risk score (p = 0.71, Fig. S4H). In subgroup analysis stratified by adjuvant postoperative therapy, there was a trend towards a higher risk score in the pharmaceutical therapy and radiotherapy subgroup, although statistical significance was not achieved (p = 0.069 and 0.19, Fig. 4E,F). And ESCC patients with or without complete response after radiotherapy exhibited comparable risk scores (p = 0.47, Fig. S4I). Furthermore, ESCC patients with tumor presence, recurred/progressed, and deceased status had significantly increased risk scores (p < 0.05, Fig. 4G–I), consistent with previous results highlighting the value of m5C-LPS as a valuable prognostic marker.

The discrepancy in risk scores between different subgroups: T stage (A), M stage (B), stage (C), alcohol history (D), adjuvant postoperative pharmaceutical therapy (E), adjuvant postoperative radiotherapy (F), neoplasm status (G), disease free status (H), and overall survival status (I).
Evaluation of the prognostic value of m5C-LPS and construction of a nomogram
Univariate and multivariate Cox regression analyses were conducted to determine the independent prognostic value of m5C-LPS and other clinicopathological parameters for ESCC patients. The forest plots showed that the N stage and risk score were independent factors for the poor prognosis (p < 0.05, Fig. 5A,B). Subsequently, we used time-dependent ROC curves to evaluate the prognostic potential of the risk score, age, gender, grade, stage, and TNM stage. The AUC values of the risk score were higher than those of other clinicopathological factors for 1-, 2-, and 3-year survival (Fig. 5C). These findings highlight the significant value of the risk score in predicting patient prognosis. Meanwhile, a nomogram was constructed based on the risk score and N stage of each ESCC patient, which could be a quantitative tool to predict 1-, 2-, and 3-year survival probability (Fig. 5D). Moreover, the calibration curves showed partial agreement between the predicted and observed survival probabilities (Fig. 5E).

Verification of the independent prognostic value of m5C-LPS and construction of nomogram. Univariate (A) and multivariate (B) Cox regression analyses of the prognostic value of risk scores and other clinical parameters. (C) The ROC curves show the predictive value of the risk score and other clinical characteristics. (D) Nomogram composed of N stage and risk score was constructed to predict 1-, 2-, and 3-year survival rates. (E) Calibration plots were used to evaluate the nomogram for predicting 1-, 2-, and 3-year survival rates.
Exploration of immune microenvironment affected by m5C-LPS
We further investigated the relationship between the immune microenvironment and the risk score obtained from m5C-LPS. The relative abundance of immune and stromal cells of each sample was estimated using Estimate, Cibersort, ssGSEA, and xCELL algorithms. The heatmap revealed the different distribution patterns of various cell types between the low- and high-risk groups (Fig. 6A). Comparison of the Cibersort results revealed significant enrichments of CD8+ T cells, memory activated CD4+ T cells, and T follicular helper cells in the low-risk group, while M2 macrophages were found to be enriched in the high-risk group (Fig. 6B). The ssGSEA results showed that central memory CD8+ T cell, gamma delta T cell, macrophage, NK cell, plasmacytoid dendritic cell, Tregs, and T follicular helper cell were significantly enriched in the high-risk group (p < 0.05, Fig. S5A). However, there were no significant differences in stromal cells between the low- and high-risk groups (Fig. S5B). The correlation heatmap identified three main clustering modules: function immune cells, resting immune cells, and stromal cells (Fig. S6A). Furthermore, the correlation coefficient indicated a negative association between the risk scores and multiple well-known immune checkpoint molecules, except for IDO1 (Fig. 6C). The histogram and heatmap revealed inverse relationships between the risk score and most immunomodulators, including chemokines, receptors, MHC, immunoinhibitors, and immunostimulators (Fig. 6D, Fig. S6B). These findings indicated that the activation of immune components in the tumor microenvironment may contribute to better outcomes for patients in the low-risk group.

Investigation of immune status in different risk groups. (A) Heatmap revealing the immune and stromal cells infiltration in ESCC immune microenvironment. (B) Box plots showing the infiltration of the immune cells based on the Cibersort algorithm in different risk groups; *p < 0.05 and **p < 0.01. (C) Estimation of the coefficients for risk score with immune checkpoint genes. (D) Histogram showing the relationships between risk score and chemokines, receptors, MHC, immunoinhibitors, and immunostimulators.
Comprehensive analysis of enriched pathways between different risk groups
To elucidate the biological functions of the differentially expressed genes associated with m5C-LPS, we performed GO, KEGG, and Reactome enrichment analyses. The prominent GO terms in molecular function (MF), cellular component (CC), and biological process (BP) were catalytic activity acting on RNA, nuclear speck, and ncRNA metabolic process, respectively (Fig. S7A). Furthermore, the top five enriched KEGG terms included spliceosome, cell cycle, ribosome biogenesis in eukaryotes, RNA degradation, and Homologous recombination (Fig. S7B). The three main key modules identified in the Reactome analysis were rRNA processing, mRNA splicing and processing, and DNA damage repair response (Fig. S7C).
The genomic alteration difference between two m5C-LPS groups
By analyzing the MuTect2 mutation annotation files, we identified the top 20 most frequently mutated genes in the low- and high-risk groups, as illustrated in Fig. S8A,B, respectively. The waterfall plots revealed that TP53, TTN, and KMT2D were most frequently mutated in both groups. However, the ranking of mutated genes showed slight changes between the two groups. For example, the mutation frequency of MUC16 was ranked third in the high-risk group (20%), but it dropped out of the top 20 mutated genes in the low-risk group. Furthermore, the mutation rates of seven oncogenic pathways (NOTCH, WNT, PI3K, MYC, TP53, TGF-beta, Cell-Cycle) were higher in the high-risk group compared to the low-risk group (Fig. S8C,D). These findings suggested that ESCC patients in the low- and high-risk groups may have different mutation driver genes and pathways.
m5C-LPS predict therapeutic response in ESCC patients
Given that chemotherapy and radiotherapy are crucial in ESCC treatments, and DNA damage repair response plays a pivotal role in regulating chemoradiotherapy response, we attempted to evaluate the therapeutic response of the low- and high-risk groups. We estimated the IC50 levels of several commonly used chemotherapeutic drugs in each patient using CGP, CTRP, and GDSC-derived drug response data. The heatmap showed that the estimated IC50 levels of these drugs were reduced in the low-risk group, indicating that patients in low-risk group were more sensitive to chemotherapy (Fig. 7A). Boxplots further demonstrated that patients in the low-risk group exhibited greater sensitivity to five CGP-derived compounds (5-fluorouracil, cisplatin, docetaxel, vinorelbine, and etoposide), two CTRP-derived compounds (docetaxel and gemcitabine), and four GDSC-derived compounds (docetaxel, paclitaxel, oxaliplatin, and vinorelbine). And significant differences in the IC50 level of docetaxel were observed among three database-derived results (Fig. 7B–D). Besides, the radiation-sensitivity index (RSI) increased in the high-risk group, suggesting that patients in the high-risk group might require a higher radiotherapy dose, although there was no statistical significance (p = 0.23, Fig. 7E).

Identification of therapeutic response features of different risk groups. (A) Heatmap revealing IC50 for chemotherapeutic agents and radiation-sensitivity index (RSI) for radiotherapy. Box plots showing the sensitivity of selected chemotherapeutic agents for patients in low- and high-risk groups based on CGP (B), CTRP (C), and GDSC (D) databases. (E) Boxplot showing the radiotherapy sensitivity for patients in low- and high-risk groups.
Discussion
ESCC accounts for about 90% of the incidence of EC annually with a dismal 5-year survival rate of 5–25% worldwide1,3,43. To date, molecular-related target therapy had emerged as new therapeutic strategies for prolonging patients’ prognosis. In recent years, RNA post-transcriptional methylation modification, including m6A, m5C, and m1A, has arrested substantial attention among researchers worldwide44,45. Over the past decade, numerous m5C regulators have been found to play pivotal roles in regulating gene expression and disease progression, including cancer46,47. For instance, NSUN2, which plays crucial roles in tissue homeostasis, spindle stability, and early embryogenesis as a nucleolar protein, is overexpressed and possesses prognostic survival value in various tumors48,49. While the function of m5C modification in other cancers has been extensively studied12,50,51, its effect on ESCC has not been fully explored. In the present study, we observed the upregulation of 11 m5C regulators in ESCC tissues compared to normal adjacent tissues (Fig. 1A). Thus, we aimed to investigate the role of m5C in ESCC further.
Existing evidences have testified that m5C methylated lncRNA can regulate the occurrence and development of cancer20. The “writer” NSUN2 modifies the lncRNA H19 and recruits the oncoprotein G3BP1 in hepatocellular carcinoma, suggesting that m5C modifications are involved in malignant tumor progression52. Furthermore, as dysregulation of lncRNAs plays a crucial role in tumor development, and they can be detected in easily accessible bodily fluids like urine, saliva, and serum, they have great potential as prognostic biomarkers and therapeutic targets for tumors53. We believe that investigating the interplay between m5C regulators and lncRNAs will become a promising area for identifying prognostic markers and therapeutic targets for cancers. Nonetheless, the role of lncRNAs involved in m5C regulation in ESCC remains unclear. To our knowledge, this is the first comprehensive analysis of the function of m5C-related lncRNAs in ESCC.
In this study, we evaluated the prognostic value of m5C-related lncRNAs in ESCC patients. A prognostic model based on 9 m5C-related lncRNAs was constructed using univariate and LASSO Cox regression analyses, and a formula for the calculation of risk score was established. The prognostic value of m5C-LPS was then tested in both training (TCGA-ESCC) and validation (GSE53622) datasets (Fig. 2). These results suggest that m5C-LPS could serve as a powerful tool for predicting the prognosis of ESCC patients.
Limited information is currently available on the lncRNAs identified in our study. However, the functions that have been reported for CAHM, ATP2B1-AS1, and UBAC2-AS1 provide important insights into the potential roles of these 9 novel m5C-related lncRNAs. The well-established functions of CAHM, which is also known as colorectal adenocarcinoma hypermethylated, as a prognostic biomarker in colorectal and thyroid carcinoma54,55, and its regulation by DNMT1 in glioma cells, suggest its involvement in glioma grade, subtype, malignant behavior, and prognosis56. Similarly, the involvement of ATP2B1-AS1 in the NF-kappa-B signaling pathway, which plays a crucial role in tumorigenesis, particularly in gastrointestinal cancers57,58, suggests its potential as a target for therapeutic intervention. Furthermore, the close association between UBAC2-AS1 and autophagy genes highlights its potential involvement in cancer-related processes and its possible therapeutic implications59. Our study has revealed the overexpression of these lncRNAs in ESCC. However, further research is needed to elucidate the precise functions and mechanisms of these lncRNAs. Nonetheless, our study provides a foundation for exploring these lncRNAs as potential therapeutic targets in cancer treatment.
Due to the association between m5C-LPS and immune status being weak, we further investigated the signaling pathways and biological functions related to m5C-LPS. Our analysis revealed a significant enrichment of functions associated with mRNA and ncRNA processing, as well as DNA damage repair response. These findings align with the established functions of m5C and lncRNAs previously reported in the literature14,60,61,62. For instance, the mRNA and translation levels enhanced when NSUN6-targeted mRNAs were methylated63. TRDMT1–FMRP–TET1-mediated m5C regulation can promote transcription-coupled homologous recombination64. And TRDMT1 can mediate m5C mRNA methylation at DNA damage sites and regulate homologous recombination60. Besides, DNA damage repair response can regulate the response effectiveness of chemoradiotherapy65,66, which is the mainstay for ESCC treatment43,67, we evaluated the therapeutic response of ESCC patients in the TCGA-ESCC cohort. Our analysis demonstrated that patients in the low-risk group exhibited a higher sensitivity to chemoradiotherapy. Additionally, studies in leukemia have shown that NSUN3 and DNMT2 can regulate the chromatin structures by directly binding hnRNPK and further modulating 5-Azacitidine response68. These observations provide valuable insights into the potential role of m5C-LPS as a predictive marker and highlight the need for further exploration of m5C function in cancer treatment.
This study has several limitations that should be acknowledged. Firstly, the small sample size, retrospective nature, and non-uniform patient source and race of the TCGA-ESCC and GSE53622 cohorts may have influenced the results. And the absence of an independent clinical cohort limits the validation of the prognostic signature. Thus, more high-quality cohort data are needed in the future to validate the prognostic value and chemoradiotherapy response of m5C-LPS. Secondly, although we detected the expression of the 9 identified lncRNAs in m5C-LPS in ESCC and normal esophageal cell lines, further in vitro and in vivo experiments are required to support our in silico results.
In this study, we constructed and validated a prognostic signature based on 9 m5C-related lncRNAs for ESCC patients. And we found that stratification of ESCC patients based on m5C-LPS is associated with different clinical features, immune status, genomic variants, oncogenic pathways, enrichment pathways, and therapeutic responses. In summary, our study provides a valuable tool for understanding the potential role of m5C-related lncRNAs and guiding personalized management of ESCC.
Data availability
The data presented in this study can be found in TCGA (https://portal.gdc.cancer.gov/repository?facetTab=cases) and GSE53622 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=gse53622) databases.
Abbreviations
- AUC:
-
Area under the curve
- ESCC:
-
Esophageal squamous cell carcinoma
- CGP:
-
Cancer Genome Project
- CTRP:
-
Cancer therapeutics response portal
- GARD:
-
Genomic-adjusted radiation dose
- GDSC:
-
Genomics of drug sensitivity in cancer
- GO:
-
Gene ontology
- HPA:
-
Human Protein Atlas
- IC50:
-
Half-maximal inhibitory concentration
- KEGG:
-
Kyoto Encyclopedia of Genes and Genomes
- K–M:
-
Kaplan–Meier
- LASSO:
-
Least absolute shrinkage and selection operator
- lncRNAs:
-
Long non-coding RNA
- m5C:
-
5-Methylcytosine
- m5C-LPS:
-
m5C-Related lncRNAs prognostic signature
- OS:
-
Overall survival
- PPI:
-
Protein–protein interaction
- qRT-PCR:
-
Quantitative real-time polymerase chain reaction
- ROC:
-
Receiver operating characteristic
- ssGSEA:
-
Single-sample gene set enrichment analysis
- TCGA:
-
The Cancer Genome Atlas
References
Sung, H. et al. Global cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 71(3), 209–249 (2021).
Arnold, M., Ferlay, J., van Berge Henegouwen, M. I. & Soerjomataram, I. Global burden of oesophageal and gastric cancer by histology and subsite in 2018. Gut 69(9), 1564–1571 (2020).
Abnet, C. C., Arnold, M. & Wei, W. Q. Epidemiology of esophageal squamous cell carcinoma. Gastroenterology 154(2), 360–373 (2018).
Thrift, A. P. Global burden and epidemiology of Barrett oesophagus and oesophageal cancer. Nat. Rev. Gastroenterol. Hepatol. 18(6), 432–443 (2021).
Waters, J. K. & Reznik, S. I. Update on management of squamous cell esophageal cancer. Curr. Oncol. Rep. 24(3), 375–385 (2022).
Zugazagoitia, J. et al. Current challenges in cancer treatment. Clin. Ther. 38(7), 1551–1566 (2016).
Zhang, Q. et al. The role of RNA m(5)C modification in cancer metastasis. Int. J. Biol. Sci. 17(13), 3369–3380 (2021).
Haruehanroengra, P., Zheng, Y. Y., Zhou, Y., Huang, Y. & Sheng, J. RNA modifications and cancer. RNA Biol. 17(11), 1560–1575 (2020).
Su, J. et al. NSUN2-mediated RNA 5-methylcytosine promotes esophageal squamous cell carcinoma progression via LIN28B-dependent GRB2 mRNA stabilization. Oncogene 40(39), 5814–5828 (2021).
Murata, A. et al. TET family proteins and 5-hydroxymethylcytosine in esophageal squamous cell carcinoma. Oncotarget 6(27), 23372–23382 (2015).
Zhou, M., Liu, W., Zhang, J. & Sun, N. RNA m(6)A modification in immunocytes and DNA repair: The biological functions and prospects in clinical application. Front. Cell. Dev. Biol. 9, 794754 (2021).
Guo, G. et al. Advances in mRNA 5-methylcytosine modifications: Detection, effectors, biological functions, and clinical relevance. Mol. Ther. Nucleic Acids 26, 575–593 (2021).
García-Vílchez, R., Sevilla, A. & Blanco, S. Post-transcriptional regulation by cytosine-5 methylation of RNA. Biochim. Biophys. Acta Gene Regul. Mech. 1862(3), 240–252 (2019).
Bohnsack, K. E., Höbartner, C. & Bohnsack, M. T. Eukaryotic 5-methylcytosine (m5C) RNA methyltransferases: Mechanisms, cellular functions, and links to disease. Genes 10(2), 102 (2019).
Ransohoff, J. D., Wei, Y. & Khavari, P. A. The functions and unique features of long intergenic non-coding RNA. Nat. Rev. Mol. Cell Biol. 19(3), 143–157 (2018).
Fang, Y. & Fullwood, M. J. Roles, functions, and mechanisms of long non-coding RNAs in cancer. Genom. Proteom. Bioinform. 14(1), 42–54 (2016).
Zhang, X. et al. Role of non-coding RNAs and RNA modifiers in cancer therapy resistance. Mol. Cancer 19(1), 47 (2020).
Liang, Y. et al. LncRNA CASC9 promotes esophageal squamous cell carcinoma metastasis through upregulating LAMC2 expression by interacting with the CREB-binding protein. Cell Death Differ. 25(11), 1980–1995 (2018).
Torsin, L. I. et al. Editing and chemical modifications on non-coding RNAs in cancer: A new tale with clinical significance. Int. J. Mol. Sci. 22(2), 581 (2021).
He, Y., Shi, Q., Zhang, Y., Yuan, X. & Yu, Z. Transcriptome-wide 5-methylcytosine functional profiling of long non-coding RNA in hepatocellular carcinoma. Cancer Manag. Res. 12, 6877–6885 (2020).
Squires, J. E. et al. Widespread occurrence of 5-methylcytosine in human coding and non-coding RNA. Nucleic Acids Res. 40(11), 5023–5033 (2012).
Li, Y. et al. Novel long noncoding RNA NMR promotes tumor progression via NSUN2 and BPTF in esophageal squamous cell carcinoma. Cancer Lett. 430, 57–66 (2018).
Szklarczyk, D. et al. The STRING database in 2021: Customizable protein–protein networks, and functional characterization of user-uploaded gene/measurement sets. Nucleic Acids Res. 49(D1), D605–D612 (2021).
Tibshirani, R. The lasso method for variable selection in the Cox model. Stat. Med. 16(4), 385–395 (1997).
Ternès, N., Rotolo, F. & Michiels, S. Empirical extensions of the lasso penalty to reduce the false discovery rate in high-dimensional Cox regression models. Stat. Med. 35(15), 2561–2573 (2016).
Pak, K. et al. A user-friendly, web-based integrative tool (ESurv) for survival analysis: Development and validation study. J. Med. Internet Res. 22(5), e16084 (2020).
Consortium GO. The Gene Ontology resource: Enriching a GOld mine. Nucleic Acids Res. 49(D1), D325–D334 (2021).
Kanehisa, M., Furumichi, M., Tanabe, M., Sato, Y. & Morishima, K. KEGG: New perspectives on genomes, pathways, diseases and drugs. Nucleic Acids Res. 45(D1), D353–D361 (2017).
Jassal, B. et al. The reactome pathway knowledgebase. Nucleic Acids Res. 48(D1), D498–D503 (2020).
Yu, G., Wang, L. G., Han, Y. & He, Q. Y. clusterProfiler: An R package for comparing biological themes among gene clusters. OMICS 16(5), 284–287 (2012).
Yoshihara, K. et al. Inferring tumour purity and stromal and immune cell admixture from expression data. Nat. Commun. 4, 2612 (2013).
Hänzelmann, S., Castelo, R. & Guinney, J. GSVA: Gene set variation analysis for microarray and RNA-seq data. BMC Bioinform. 14, 7 (2013).
Newman, A. M. et al. Robust enumeration of cell subsets from tissue expression profiles. Nat. Methods 12(5), 453–457 (2015).
Aran, D., Hu, Z. & Butte, A. J. xCell: Digitally portraying the tissue cellular heterogeneity landscape. Genome Biol. 18(1), 220 (2017).
Ru, B. et al. TISIDB: An integrated repository portal for tumor-immune system interactions. Bioinformatics 35(20), 4200–4202 (2019).
Mayakonda, A., Lin, D. C., Assenov, Y., Plass, C. & Koeffler, H. P. Maftools: Efficient and comprehensive analysis of somatic variants in cancer. Genome Res. 28(11), 1747–1756 (2018).
Sanchez-Vega, F. et al. Oncogenic signaling pathways in the cancer genome atlas. Cell 173(2), 321–337 (2018).
Geeleher, P., Cox, N. & Huang, R. S. pRRophetic: An R package for prediction of clinical chemotherapeutic response from tumor gene expression levels. PLoS ONE 9(9), e107468 (2014).
Garnett, M. J. et al. Systematic identification of genomic markers of drug sensitivity in cancer cells. Nature 483(7391), 570–575 (2012).
Rees, M. G. et al. Correlating chemical sensitivity and basal gene expression reveals mechanism of action. Nat. Chem. Biol. 12(2), 109–116 (2016).
Yang, W. et al. Genomics of drug sensitivity in cancer (GDSC): A resource for therapeutic biomarker discovery in cancer cells. Nucleic Acids Res. 41, D955–D961 (2013).
Scott, J. G. et al. A genome-based model for adjusting radiotherapy dose (GARD): A retrospective, cohort-based study. Lancet Oncol. 18(2), 202–211 (2017).
Leng, X. F., Daiko, H., Han, Y. T. & Mao, Y. S. Optimal preoperative neoadjuvant therapy for resectable locally advanced esophageal squamous cell carcinoma. Ann. N. Y. Acad. Sci. 1482(1), 213–224 (2020).
Zhao, B. S., Roundtree, I. A. & He, C. Post-transcriptional gene regulation by mRNA modifications. Nat. Rev. Mol. Cell Biol. 18(1), 31–42 (2017).
Gilbert, W. V., Bell, T. A. & Schaening, C. Messenger RNA modifications: Form, distribution, and function. Science 352(6292), 1408–1412 (2016).
Chen, Y. S., Yang, W. L., Zhao, Y. L. & Yang, Y. G. Dynamic transcriptomic m(5) C and its regulatory role in RNA processing. Wiley Interdiscip. Rev. RNA 12(4), e1639 (2021).
Wood, S., Willbanks, A. & Cheng, J. X. The role of RNA modifications and RNA-modifying proteins in cancer therapy and drug resistance. Curr. Cancer Drug Targets 21(4), 326–352 (2021).
Blanco, S. & Frye, M. Role of RNA methyltransferases in tissue renewal and pathology. Curr. Opin. Cell Biol. 31, 1–7 (2014).
Chellamuthu, A. & Gray, S. G. The RNA methyltransferase NSUN2 and its potential roles in cancer. Cells 9(8), 1758 (2020).
Yin, H. et al. 5-Methylcytosine (m(5)C) modification in peripheral blood immune cells is a novel non-invasive biomarker for colorectal cancer diagnosis. Front. Immunol. 13, 967921 (2022).
Pan, J., Huang, Z. & Xu, Y. m5C-related lncRNAs predict overall survival of patients and regulate the tumor immune microenvironment in lung adenocarcinoma. Front. Cell. Dev. Biol. 9, 671821 (2021).
Sun, Z. et al. Aberrant NSUN2-mediated m(5)C modification of H19 lncRNA is associated with poor differentiation of hepatocellular carcinoma. Oncogene 39(45), 6906–6919 (2020).
Sarfi, M., Abbastabar, M. & Khalili, E. Long noncoding RNAs biomarker-based cancer assessment. J. Cell Physiol. 234(10), 16971–16986 (2019).
Pedersen, S. K. et al. CAHM, a long non-coding RNA gene hypermethylated in colorectal neoplasia. Epigenetics 9(8), 1071–1082 (2014).
Xiao, Y., Tu, Y. & Li, Y. Expression level of long non-coding RNA colon adenocarcinoma hypermethylated serves as a novel prognostic biomarker in patients with thyroid carcinoma. Biosci. Rep. 41(4), 284 (2021).
Xu, Y. et al. DNMT1 mediated CAHM repression promotes glioma invasion via SPAK/JNK pathway. Cell. Mol. Neurobiol. 1, 1–11 (2021).
Hoesel, B. & Schmid, J. A. The complexity of NF-κB signaling in inflammation and cancer. Mol. Cancer 12, 86 (2013).
Peng, C., Ouyang, Y., Lu, N. & Li, N. The NF-κB signaling pathway, the microbiota, and gastrointestinal tumorigenesis: Recent advances. Front. Immunol. 11, 1387 (2020).
Jiang, Q., Xue, D., Shi, F. & Qiu, J. Prognostic significance of an autophagy-related long non-coding RNA signature in patients with oral and oropharyngeal squamous cell carcinoma. Oncol. Lett. 21(1), 29 (2021).
Chen, H. et al. m(5)C modification of mRNA serves a DNA damage code to promote homologous recombination. Nat. Commun. 11(1), 2834 (2020).
Su, M. et al. LncRNAs in DNA damage response and repair in cancer cells. Acta Biochim. Biophys. Sin. (Shanghai) 50(5), 433–439 (2018).
Li, M. et al. 5-methylcytosine RNA methyltransferases and their potential roles in cancer. J. Transl. Med. 20(1), 214 (2022).
Selmi, T. et al. Sequence- and structure-specific cytosine-5 mRNA methylation by NSUN6. Nucleic Acids Res. 49(2), 1006–1022 (2021).
Yang, H. et al. FMRP promotes transcription-coupled homologous recombination via facilitating TET1-mediated m5C RNA modification demethylation. Proc. Natl. Acad. Sci. U.S.A. 119(12), e2116251119 (2022).
O’Connor, M. J. Targeting the DNA damage response in cancer. Mol. Cell 60(4), 547–560 (2015).
Huang, R. X. & Zhou, P. K. DNA damage response signaling pathways and targets for radiotherapy sensitization in cancer. Signal Transduct Target Ther. 5(1), 60 (2020).
Sasaki, Y. & Kato, K. Chemoradiotherapy for esophageal squamous cell cancer. Jpn. J. Clin. Oncol. 46(9), 805–810 (2016).
Cheng, J. X. et al. RNA cytosine methylation and methyltransferases mediate chromatin organization and 5-azacytidine response and resistance in leukaemia. Nat. Commun. 9(1), 1163 (2018).
Acknowledgements
The authors sincerely thank the data provided by TCGA and GEO databases.
Funding
This research was funded by the National Natural Science Foundation of China (Grant Number 81773239).
Author information
Authors and Affiliations
Contributions
T.G. and X.Z. conceived, designed, and supervised the study. Y.M., Y.S., and X.F. performed formal analysis and data interpretation; Y.M. wrote the original draft; X.Z., J.L., and X.Z. provided critical revisions and contributed to the editing of the paper. All authors read and approved the final manuscript. All authors have agreed to the publication of this research.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Ma, Y., Sun, Y., Zhao, X. et al. Identification of m5C-related lncRNAs signature to predict prognosis and therapeutic responses in esophageal squamous cell carcinoma patients. Sci Rep 13, 14499 (2023). https://doi.org/10.1038/s41598-023-41495-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-023-41495-6
- Springer Nature Limited