
Abstract
Chromosomal abnormalities are the most common etiology of early spontaneous miscarriage. However, traditional karyotyping of chorionic villus samples (CVSs) is limited by cell culture and its low resolution. The objective of our study was to investigate the efficiency of molecular karyotyping technology for genetic diagnosis of early missed abortion tissues. Chromosome analysis of 1191 abortion CVSs in early pregnancy was conducted from August 2016 to June 2021; 463 cases were conducted via copy-number variations sequencing (CNV-seq)/quantitative fluorescent-polymerase chain reaction (QF-PCR) and 728 cases were conducted using SNP array. Clinically significant CNVs of CVSs were identified to clarify the cause of miscarriage and to guide the couples’ subsequent pregnancies. Among these, 31 cases with significant maternal cell contamination were removed from the study. Among the remaining 1160 samples, 751 cases (64.7%) with genetic abnormalities were identified, of which, 531 (45.8%) were single aneuploidies, 31 (2.7%) were multiple aneuploidies, 50 (4.3%) were polyploidies, 54 (4.7%) were partial aneuploidies, 77 (6.6%) had submicroscopic CNVs (including 25 with clinically significant CNVs and 52 had variants of uncertain significance), and 8 cases (0.7%) were uniparental disomies. Our study suggests that both SNP array and CNV-seq/QF-PCR are reliable, robust, and high-resolution technologies for genetic diagnosis of miscarriage.
Similar content being viewed by others

Introduction
Spontaneous miscarriage in early pregnancy occurs in ~ 10–15% of all clinically recognized pregnancies1. Embryonic chromosomal abnormalities, reproductive immune disorders, anatomic abnormalities of the uterus, thrombophilias, endocrine diseases, infectious disorders, sperm quality, and environmental issues are all related to miscarriage2. Among these, embryonic chromosomal anomalies are the most common etiology of miscarriages, representing more than half of early spontaneous abortions3.
Traditional cytogenetic analyses of chorionic villus samples (CVSs) involve cell culture and karyotyping analysis, which is hindered by the low success rates of cell culture as well as low accuracy and resolution, as selective overgrowth of maternally derived cells4. The development of molecular karyotyping techniques, such as chromosomal microarray analysis (CMA) and copy number variation (CNV) sequencing (CNV-seq) have great advantages, including no cell culture required, as well as having high throughput analysis and resolution5. Both high-resolution molecular technologies have increasingly been applied for analysis of miscarriage specimens6,7. CNV-seq is a high-resolution and low-cost sequencing technology. The combination of CNV-seq and quantitative fluorescent-polymerase chain reaction (QF-PCR) can identify chromosome aneuploidies, polyploidies, and CNVs8,9.
Embryonic chromosomal numerical abnormalities accounted for up to 90% of the causes of early miscarriage10. CNVs are genomic copy number changes with sizes ranging from above 50 bp to Mb11. It is well known that segmental deletion and/or duplication can cause miscarriage. Additionally, submicroscopic pathogenic CNVs (pCNVs) are the second-highest aberrations identified in CVSs12. However, specific data regarding the association between submicroscopic CNVs and spontaneous miscarriage is still limited. Here, we investigated the feasibility and accuracy of two approaches, microarray analysis and the combination of CNV-seq and QF-PCR, for molecular karyotyping analysis of 1191 CVSs from early missed abortions in a tertiary referral center.
Methods
Study samples and genomic DNA extraction
Among 1191 patients agreed to undergo chromosome analysis of their aborted CVSs collected via hysterosuction. All the women with missed abortions underwent a standardized diagnostic workup in our center. In short, the villi tissues were washed with 0.9% sodium chloride solution under sterile condition, and the hemagglutination and decidual tissues of the mother were separated. Then, high quality villi were chosen for genomic DNA extraction. DNA was extracted using a QIAamp DNA Mini Kit (Qiagen, Hilden, Germany) according to the manufacture’s instructions, and the purity and concentration of genomic DNA were further determined by NanoDrop 2000 Ultramicro Spectrophotometer. Peripheral blood from both the pregnant couple were also collected to rule out maternal cell contamination (MCC) and to contribute to report interpretation if necessary. The participants’ age ranged from 19 to 48 years old, with an average age of 31.2 years old. The gestational age of all the miscarriages ranged from 5 to 12 weeks, with an average gestational age of 8.5 weeks. The participants were divided into subgroups based on maternal age, the number of previous miscarriages, and gestational weeks at miscarriage (Table 1). Informed consent was obtained from all participants. The study was conducted under the guidance of the Declaration of Helsinki and approved by the Ethics Committee of the Fujian Maternal and Child Health Hospital (No. 2020KYLLD01051).
QF-PCR
After genomic DNA extraction, QF-PCR was carried out to exclude MCC as reported previously13, using 15 polymorphic short tandem repeat (STR) markers, including three markers (D21S1435, D21S11 and D21S1411) for chromosome 21, four markers (D18S1002, D18S391, D18S535 and D18S386) for chromosome 18, four markers (D13S628, D13S742, D13S634 and D13S305) for chromosome 13, and four markers (DXS981, DXS6809, X22 and AMXY) for sex chromosomes. PCR products were electrophoresed on an ABI 3500 DX genetic analyzer and results were analyzed by ABI GeneMapper software (Applied Biosystems, Foster City, CA, USA).
CNV sequencing/QF-PCR
According to its detection principle, CNV-seq alone cannot recognize SNP site to exclude MCC. Thus, for CNV-Seq technology, samples should be first investigated using QF-PCR assays or STR test to exclude significant MCC and triploidy.
CVSs samples were subjected for CNV-seq after exclusion of MCC. A total of 449 cases of chromosomal analysis was ultimately conducted by CNV sequencing (CNV-seq) due to 14 cases with significant MCC. Briefly, 10 mg of CVSs were selected under sterile conditions, DNA was extracted by Qiagen kit for later use, DNA library was constructed using dsDNA fragmentase to produce smaller fragments, end repair, A-tail and then ligated with barcoded sequencing adaptors. Single-end sequencing was performed on the the NextSeq CN500 platform to produce to produce approximately 5 million reads per sample. The number of reads after sample quality control is more than 8 M Sequence depth is ~ 1x. Each sequencing tag was matched to the corresponding chromosome by comparison software, and then standardized Z-value analysis was performed to determine chromosome copy number variation. On average, approximately 2.8 ~ 3.2 million reads were uniquely mapped for data analysis. The uniquely mapped reads were aligned to the human genomic reference sequences (hg19) using the Burrows-Wheeler algorithm and allocated to 20-kilobase (kb) bin on each chromosome14. To eliminate the influence of GC bias between different samples, GC correction was performed via LOESS regression, intrarun normalization, and linear model regression15, and CNVs were identified from 24 chromosome copy number plots. The pathogenicity of detected CNVs was assessed following American College of Medical Genetics (ACMG) guidelines. Genomic variant databases including DGV, DECIPHER , OMIM, PubMed and UCSC were used as a reference source of CNVs16.
The CNV-seq data analysis processes include (1) Quality control of raw sequencing data (fastq), removing adapters/low-quality bases, resulting in filtered fastq data. (2) Alignment of filtered fastq data to the human reference genome (hg19) using alignment software like BWA. (3) Sorting and deduplication of the alignment results. (4) Binning the sorted results using genomic intervals to obtain read-depth profiles (sample-wise median normalization is applied, where the reads count in each bin is divided by the median reads count of the bins in autosomes. This step aims to mitigate biases introduced by different sequencing depths. (5) GC correction of the read-depth profiles. (6) CNV calling based on the GC-corrected read-depth profiles to detect CNV variations. (7) Analysis and interpretation of CNVs according to ACMG/ClinGen guidelines, including classification based on their clinical significance.

SNP array
Due to the platform transition in our laboratory during the study period, totally, 711 cases of chromosomal CNVs (above 0.1 Mb) analysis were ultimately detected by SNP array due to 17 cases with significant MCC. The experimental process of SNP array was carried out using Affymetrix CytoScan 750 K array (Affymetrix Inc., Santa Clara, CA) as previously described17. The data was analyzed using Affymetrix Chromosome Analysis Suite Software (version 3.1.0.15). Parental microarray testing was carried out to determine its origin. Pathogenicity of CNVs is classified according to ACMG guideline16. Only P/LP CNVs and VOUS are reported in this study. Large CNVs (≥ 10 Mb) were classified as partial aneuploidies, and submicroscopic CNVs (< 10 Mb) were defined as microdeletions and microduplications. Large CNVs detected in three or more cases were defined as recurrent large CNVs, while submicroscopic CNVs found in two or more cases were considered as recurrent submicroscopic CNVs.
Parental conventional karyotyping analysis
Peripheral blood lymphocyte was isolated from the abortion couples when the chromosomal aberrations of CVSs suggesting parental balanced rearrangement, then were cultured and harvested after phytohemagglutinin stimulation for 72 h. Metaphase chromosomes were prepared following standard cytogenetic protocols.
Karyotypes were scanned on Leica GSL120. At least 20 metaphases were counted, and five metaphases were analyzed. The naming of abnormal karyotypes was based on ISCN 2020.
Fluorescence in situ hybridization
Fluorescence in situ hybridization (FISH) using telomere probes (Vysis, IL) was performed to identify occult reciprocal balanced translocations carriers as reported previously18. After 72 h stimulation with phytohemagglutinin, the lymphocytes were cultured and harvested after stimulation using phytohemagglutinin for 72 h, and metaphase chromosomes were fixed on slides. After degeneration for 5 min at 78 °C, the fluorescent labeled probes were hybridized with the chromosomes on the slide at 37 °C for 16 h. The slides were washed in 2 × SSC (Sigma, USA) and stained with DAPI (Vysis, IL). The signal was observed under a fluorescence microscope (Leica, Germany).
Statistical analysis
SPSS software version 19.0 (SPSS, Inc., Chicago, IL) was used for statistical analysis. Measurement data were expressed as mean ± standard deviation, statistical comparisons were performed using χ2 test (Fisher’s exact test), and p < 0.05 was considered statistically significant.
Ethics approval and consent to participate
The study complied with the principles set forth in the Declaration of Helsinki. It was approved by the Institutional Review Board of Fujian Maternal and Child Health Hospital. Written informed consent was obtained from each patient.
Results
Baseline characteristics of the participants and chromosomal abnormalities of CVSs detected by molecular karyotyping technologies
Initially, we analyzed a total of 1191 cases by CNV-seq/QF-PCR or SNP array August 2016 to June 2021. Among these, 31 cases with serious MCC were removed. The remaining 1160 cases with no MCC were available for further molecular karyotyping analysis (Table 1), of which 449 cases were tested via CNV-seq/QF-PCR, and 711 cases were tested using SNP array. All 1160 CVSs were successfully analyzed. Thus, the detection success rate was 100% (1160/1160). As detection coverage was theoretically consistent except uniparental disomy (UPD) based on SNP array and CNV-seq/QF-PCR, we counted the results except UPD detected by the two methods together. Overall, 751 cases (64.7%) with genetic abnormalities were identified, of which, 531 (45.8%) were single aneuploidies, 31 (2.7%) were multiple aneuploidies, 50 (4.3%) were polyploidies, 54 (4.7%) were partial aneuploidies, 77 (6.6%) had CNVs (including 52 (4.5%) had VOUS), and 8 cases (0.7%) were UPD (Fig. 1). In addition, a total of 213 female samples and 196 male samples were found in the normal samples, with a female-to-male ratio of 1.09.

The constitution of chromosome abnormalities in CVSs. Overall, 751 samples (64.7%) with abnormal chromosomes were identified among 1160 CVSs, including 531 single aneuploidies, 31multiple aneuploidies, 50 polyploidies, 54 partial aneuplodidy, 77 microdeletion/microduplication, and 8 UPD. UPD—uniparental disomy.
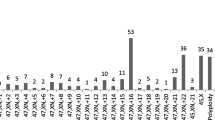
Aneuploidy was the most common abnormal result, with 531 (45.8%) single aneuploidies and 31 (2.7%) multiple aneuploidies being identified (Fig. 2). Trisomy accounted for most single aneuploidies (430/531, 81.0%). Of the single types of trisomy, trisomy 16 was the most common (115/430, 26.7%), followed by trisomy 22 (70/430, 16.3%), trisomy 21 (51/430, 11.9%), and trisomy 13, 15, and 18. Monosomy was identified in 19.0% (101/531) of the cases with single aneuploidy; of which, monosomy X accounted for 92.1% (93/101) of these cases, while the remaining autosomal monosomy comprised the remaining 8.9% (9/101), with six cases of monosomy 21, one of monosomy 14, and one of monosomy 22.

The frequency of each chromosomal aneuploidy.
Polyploidy was found in 50 (4.3%) cases: triploidy in 45 (3.9%) cases and hypotriploidy in 5 (0.4%) cases (Table 2).
Partial aneuploidy (large CNVs ≥ 10 Mb) was observed in 54 (4.7%) cases (Table 3). Both large deletions and duplications occurred most commonly in chromosome 8, terminal deletion/duplication was identified in 18 (1.6%) cases, accounting for the largest proportion of partial aneuploidies. The distributions of large CNVs and associated genes in CVSs are shown in Table 3. Large copy number losses occurred mostly in chromosome 8, followed by chromosomes X and 7. Large copy number gains also occurred most frequently in chromosome 8, followed by chromosome 9. In addition, a terminal deletion accompanied by terminal duplication involving two chromosomes was detected in 17 cases, suggesting the presence of an unbalanced translocation, which represented the second largest proportion of partial aneuploidies (17 cases; 31.5% of all partial aneuploidies and 1.5% of all analyzed cases), among these, the karyotypes of 16 couples were available, and 14 couples were reported as reciprocal translocation carriers, except for 2 that had normal karyotypes. Samples from the two couples with the normal karyotypes were further subjected to FISH analysis, and two (Case 31 and 54) of them were identified to have submicroscopic reciprocal balanced translocations (see Table 3 and Fig. 3). A terminal deletion coupled with terminal duplication involving one chromosome was detected in 8 cases, suggesting an unbalanced pericentric inversion, which accounted for 14.8% of all partial aneuploidies and 0.7% of all analyzed cases, further parental studies showed 5 of them were derived from inversion) (Table 3), in the remaining 23 cases, parental studies were not available due to their refusal.

FISH results of case 31 and 54. In case 31, SNP array on abortion villous tissue revealed a 11.8 Mb duplication on 8q24.1q24.3 and a 11.18 Mb deletion on 21q22.12q22.3, whereas parental karyotypes were normal, further FISH analysis result [ish t(8;21)(8p + ,21q + ;8q +)] revealed that copy number variation in aborted embryos is inherited from the father, chart A1 and A2 shows the results of FISH labeling 8p with green signal, 8q with red signal and 21q with red signal, indicating reciprocal translocation with father (arrow); In case 54, SNP array on abortion villous tissue revealed a 15.8 Mb deletion on 1p36.33p36.13 and a 10.8 Mb duplication on 12q24.31q24.33 , whereas parental karyotypes were normal, FISH analysis abortion villous tissue revealed ish der(1)t(1;12)(12q + ,1q +) (B1, B2), further FISH analysis result [ish t(1;12)(12q + ,1q + ;12p + ,1p +)] revealed that copy number variation in aborted embryos is inherited from the mother (C1, C2), chart c and d shows the results of FISH labeling 1p with green signal, 1q with red signal, 12q with red signal, and 12p with green signa indicating reciprocal translocation with father (arrow).
Submicroscopic CNVs (< 10 Mb) (microdeletions/ microduplications) were identified in 77 (6.6%) cases. The distributions of submicroscopic P/LP CNVs and associated genes in CVSs are shown in Table 4. Among these, 22 (1.9%) were considered pCNVs, and 3 (0.3%) and 52 (4.5%) were classified as likely pCNVs and VOUS, respectively (Table 2).
Thirty-nine and twenty-nine cases with pCNVs were found in ≤ 29-year-old and 30–34-year-old pregnant women, respectively. There were only 7 and 2 pCNVs in CVSs detected in the 35–39 and ≥ 40-year-old age groups (Fig. 4). The number of chromosomal variations in the subgroups with previous miscarriages is shown in Fig. 5. There were no significant differences in the different types of chromosomal abnormalities among the four groups (p > 0.05).

The incidence of chromosomal abnormality for subgroups of different maternal age.

The number of chromosomal abnormality in subgroups of previous miscarriage.
In addition, we identified 11 pCNVs (9 deletions and 2 duplications) which were detected in CVSs at least twice. These pCNVs involved deletions in 1q21.1, 8p23.1, 2q37.3, 5p15.33p14.3, 7q32.3q36.3, 17p12, 15q26.2q26.3, 4p16.3p16.1, and Xp22.3q28, as well as duplications in 5p13 and 8q23.3q24.3 with sizes ranging from 7.2 to 96.9 Mb.
UPD was detected in 8 (0.7%) cases, who all denied any known history of parental consanguinity. Among these, whole-genome UPD, suggestive of molar pregnancy, was observed in 2 cases. Single-chromosome UPD (involving chromosome 13) was identified in one case. In addition, five cases of segmental UPD (involving chromosomes 3p, 5p, 6p, 7q, 8q, 9p, 9q, 11q, 14q, 16q and 22q) were observed.
The associations between chromosomal abnormalities of CVSs and maternal age
We investigated the relationship between maternal age and chromosomal abnormalities, finding that the frequency of aneuploidy increased with maternal age. The incidence of chromosomal aneuploidy abnormalities among women aged ≤ 29, 30–34, and 35–39 years was all significantly lower than that in women ≥ 40 years old (p < 0.001), whereas the frequencies of other types of chromosomal abnormalities were not correlated with maternal age (p > 0.05). The correlation between chromosomal abnormalities and maternal age is presented in Fig. 4.
Discussion
Early pregnancy loss puts a heavy psychological burden on most couples. Embryonic numerical and structural chromosomal abnormalities are the most common cause of early pregnancy loss6,19. Chromosomal aberration analysis of CVSs is essential for assessing the causes of first-trimester miscarriages. In the current study, we aimed to evaluate the incidence and distribution of chromosomal abnormalities detected by CMA and CNV-seq/QF-PCR in CVSs samples.
Until now, karyotyping, CMA, as well as CNV-seq were the main technologies for identifying chromosomal anomalies19,20,21. Karyotyping is considered the “gold standard” for cytogenetics, its advantage is that it can detect chromosomal numerical, and visible structural abnormalities. However, this technique requires cell culture. Furthermore, its resolution is too low to detect submicroscopic CNVs. Thus, the total detection failure rate is ~ 20%22, which sometimes yields false-negative results due to the excessive growth of maternal cells relative to chorionic villus cells. CMA is a first-tier method in detecting CNVs in spontaneous miscarriages23. Although SNP array has significant advantages in detecting non-equilibrium chromosomal abnormalities, triploidy, and UPD, it has a relatively low throughput and a high detection cost. Recently, CNV-seq is gradually being used in detecting the genetic causes of CVSs24. Both molecular karyotyping technologies do not require cell culture and are reliable in detecting CNVs25. Compared with CMA, CNV-seq/QF-PCR has a higher throughput, a more accurate CNV breakpoint detection with 1 × depth, low cost, and a short reporting period, and has a lower level (≥ 10%) of mosaicism and polyploidy detection. We summarized the total detection efficiencies of SNP array and CNV-seq technologies on CVSs. In this study, all 449 and 711 CVSs were successfully examined via CNV-seq/QF-PCR and SNP array, respectively, thus, the detection success rate of both methods was 100%. More normal female samples were observed than normal male samples (213 vs. 196), which was likely caused by confusion of 69 XXX and 46 XX, which is significantly higher than the mean ratio of 0.71 for cases investigated in multiple large-scale products-of-conception studies using cytogenetic analysis26.
The overall rate of clinically significant chromosomal abnormalities was 59.7% (693/1160) (Table 2), which is similar to the reported rates in previous researches27. Chromosomal numerical anomaly is the most common genetic cause of spontaneous miscarriages28. Here, 615 cases (81.9%, 615/751) had numerical abnormalities, which is consistent with results from a recent report9, including 531 cases (45.8%) of single chromosome aneuploidy and 31 (2.7%) cases of multiple aneuploidies (Table 2). The occurrence frequency of each chromosome aneuploidy was analyzed in all 531 cases in this study (Fig. 2), which showed that non-segregation errors occurred in all chromosomes except chromosome 1, which is consistent with a previous study29.
As the longest human chromosome, trisomy 1 appears to be a very early lethal anomaly that may affect early embryonic development, causing pregnancy failure or biochemical pregnancy30. As shown in Fig. 2, trisomy 16 accounts for 81% (430/531) of autosomal trisomy cases, which is in line with the previous report31. Among the cases of single autosomal trisomy, the incidence of trisomy 16 was the highest (18.7%, 115/615), followed by trisomy 22 and trisomy 21 (Fig. 2), which is consistent with the reported research6. Non-separation of homologous chromosomes causes aneuploidies during the meiotic divisions of germ cells32. The development and survival of zygotes with trisomies are inhibited, resulting in early pregnancy loss.
Triploidy is a common chromosomal aberration that usually results in early miscarriage33. In our study, polyploidy was found in 50 (4.3%) cases: triploidy in 45 (3.9%) cases, and hypotriploidy in 5 (0.4%) cases, which are slightly lower than those reported by Wang Y.6 This difference may be due to inadequate sample size in our study.
Monosomies are lethal due to their gene dose effect; thus, embryonic monosomies often occur very early in pregnancy and lead to spontaneous abortions34. Here, monosomies were identified in 19.0% (101/531) of the cases with single aneuploidy, of which, monosomy X accounted for 91.1% (92/101). Unlike autosomal trisomy, the risk of monosomy X did not increase with maternal age, consistent with previous reports3. Hassold et al.35 found that the paternal loss of a sex chromosome was the most common reason leading to 45, X, which was likely a result of meiotic errors.
The third-child policy in China led to an increasing proportion of AMA. The risk of chromosomal aneuploidies in aborted embryos increases with maternal age36. This may be due to egg aging which can cause meiosis without dissociation36. Our data also support this point.
Generally, large CNVs which contain numerous genes are expected to be lethal and are known to cause miscarriage6. In this study, 79 large CNVs (≥ 10 Mb) in 54 cases were identified. Among 54 cases of large CNVs, thirty were unbalanced rearrangements, with 21 of them confirmed to be of parental origin (16 cases of translocations and 5 cases of inversion) in the subsequent conventional karyotyping/FISH of couples. In addition, we also found that both large deletions and duplications occurred most commonly in chromosome 8, which is consistent with the recent report6,37. One likely explanation is that maternal 8p inversion, which is delimited by the olfactory receptor (OR) gene clusters, and may confer susceptibility to unequal crossovers between two OR gene clusters38. The number of previous miscarriages ranged from 0 to 4. Approximately 2.7–6.7% of chromosomal balanced rearrangement carriers have experienced recurrent spontaneous abortion (RSA), which indicates that chromosomal balanced rearrangement is one of the main causes of RSA. Our data indicate that the genetic cause of miscarriage should be conducted routinely, abnormal CVSs results can suggest balanced abnormalities in parents, and follow‐up parental testing allows for further refining recurrence risk and whether or not prenatal and/or pre-implantation genetic testing (PGT) needs to be conducted39. With exact chromosomal diagnosis, PGT was recommended to these couples40. We emphasized that an obstetrician should be aware of submicroscopic reciprocal translocation rearrangement in couples with RSA.
It is worth pointing out that in 2 couples with normal karyotypes, subsequent peripheral blood FISH was performed to confirm whether there is occult chromosomal rearrangement; both cases were proven to be hereditary from the parental submicroscopic reciprocal balanced translocations (Cases 30 and 53). Thus, submicroscopic reciprocal translocation, even in embryotic large pCNVs in couples with RSA, were sometimes ignored. Further FISH confirmation is recommended for carriers with suspected occult translocations or similar terminal chromosomal banding involving two chromosomes.
However, the role of submicroscopic CNVs in early pregnancy loss is unclear. Specific information based on large cohorts regarding the association between submicroscopic CNVs and miscarriage is limited. Nevertheless, recent studies have shown that dosage changes or mutations of genes that play an important role in early embryonic development could also cause miscarriage41. A total of 77 (6.6%) cases of submicroscopic (< 10 Mb) CNVs were identified in the study, similar to those in Levy B’s research37. Up to now, little is known about the association between a specific submicroscopic pCNV and miscarriage.
We identified submicroscopic pCNVs in 2.2% (25/1160) of our cases, which is same to 2.2% by Wang et al.6. Three recurrent pCNVs, including 22q11.2 microdeletion, 7q11.23 microdeletion, and 16p13.11 microduplication, were reported in previous studies on miscarriage21,37,42,43. The possible underlying mechanism that results in early embryonic death may be a malformed fetal cardiovascular system resulting from 22q11.2 deletion and 7q11.23 deletion6. Whereas, whether 16p13.11 microduplication contributes to miscarriage remains to be uncertain. In addition, microdeletions in 22q11.21, 2q37.3 and 9p24.3p24.2 as well as CNVs in 11p15.5 were identified as likely to be associated with miscarriage44, the potential mechanism is that aberrant methylation or duplication of imprinted genes in 11p15.5 could cause miscarriage. Likewise, Nagirnaja et al.45 identified CNV in 5p13.3, interrupting the PDZD2 and GOLPH3 genes, and showed that which was significant correlated with an increased risk of spontaneous abortion (SA). Recently, Sheng et al.46 also identified two recurrent RSA-associated CNVs (duplications at 16q24.3 and 16p13.3) compared with the SA.
In our study, we also identified microdeletion in 2q37.3 and 22q11.2 which may lead to miscarriage. In addition, eleven pCNVs, which were detected in CVSs at least twice, involved deletions at 1q21.1, 8p23.1, 2q37.3, 5p15.33p14.3, 7q32.3q36.3, 17p12, 15q26.2q26.3, 4p16.3p16.1, and Xp22.3q28, as well as duplications in 5p13 and 8q23.3q24.3. The association of miscarriage with other submicroscopic pCNVs identified in our cohort, is still unclear, and more large-scale studies are needed to confirm whether these submicroscopic pCNVs contribute to pregnancy loss. Recently, whole-exome sequencing has been applied to study RSA and a few embryonic lethal genes have been discovered47. We believe future large-scale cohort will identify more recurrent pCNVs and gene mutations concerning pregnancy loss.
Both SNP array and CNV-seq/QF-PCR were effective in identifying chromosomal aneuploidies, CNVs and polyploidies48. However, CNV-seq/QF-PCR and SNP array cannot detect all polyploidies, such as 69, XXX and tetraploidy, as well as balanced structural rearrangement. In addition, CNV-seq can detect mosaicism in at least 10% of CNVs, while microarray can detect mosaicism in at least 30% of CNVs and UPD.
Unlike embryonic chromosomal aneuploidies, the risk of pCNVs in aborted embryos is independent of maternal age49. Our data indicate that the incidence of other chromosomal abnormalities such as polyploidy, CNVs, and UPD was not directly correlated with maternal age (p > 0.05), which is partially consistent with the study reported by Larroya et al50. Based on this point, SNP array or CNV-seq/QF-PCR analysis on CVSs is strongly recommended, regardless of maternal age.
In the present study, the chromosomal structural abnormality rate (6.8%, 18/264) of CVSs samples in AMA was lower than that (12.6%, 113/896) of miscarriage samples in YMA, which is in line with a previous research51, because the frequency of embryotic chromosomal structural abnormalities did not seem to be correlated with AMA. A larger population study is needed to further clarify the noncorrelation.
UPD is a rare chromosomal abnormality. The major mechanism of UPD formation is trisomy rescue52. Specific regions on chromosomes 6, 7, 11, 14, 15, and 20 can result in genomic imprinting diseases. Chromosome-wide UPD resulted from parthenogenesis, which is one of the genetic factors for early pregnancy loss53. The incidence (0.7%) of UPD in our study might have been underestimated since UPDs were not detected in the cases via CNV-seq/QF-PCR. Single-chromosome UPD is presumably suggestive of a monosomy rescue event. Segmental UPD in these cases would likely be due to a meiotic/mitotic error, in which a meiotic non-disjunction event was followed by a mitotic cross-over between the paternal and maternal homologs, with subsequent trisomy rescue54. Pregnancy loss could be due to UPD resulting in unmasking of an underlying lethal recessive disease gene(s) or imprinted genes.
The limitation of this study is that the number of cases is not large enough; the limited data is not fully representative of the CNV characteristics of early miscarriage. Thus, the correlations among the history of previous abortions/weeks of miscarriage, modes of conception, and embryonic chromosomal abnormalities have not yet been evaluated. For this, a larger population study and enrichment analysis of genetic functions within the examined CNVs will be necessary to identify the specific genes associated with miscarriage.
In summary, our study suggests both SNP array and CNV-seq/QF-PCR are reliable, robust, and high-resolution technologies for genetic diagnosis of miscarriage, especially in detecting CNVs.
Data availability
The data that support the findings of this study are available from the corresponding author upon reasonable request.
Abbreviations
- CNV:
-
Copy number variation
- CMA:
-
Chromosomal microarray analysis
- CVSs:
-
Chorionic villus samples
- FISH:
-
Fluorescence in situ hybridization
- NGS:
-
Next generation sequencing
- SNP:
-
Single nucleotide polymorphism
- VOUS:
-
Variant of uncertain significance
- UPD:
-
Uniparental disomy
References
Rai, R. & Regan, L. Recurrent miscarriage. Lancet Lond. Eng. 368, 601–611. https://doi.org/10.1016/s0140-6736(06)69204-0 (2006).
Wang, S. et al. Th17/Treg-cell balance in the peripheral blood of pregnant females with a history of recurrent spontaneous abortion receiving progesterone or cyclosporine A. Exp. Ther. Med. 21, 37. https://doi.org/10.3892/etm.2020.9469 (2021).
Ozawa, N. et al. Maternal age, history of miscarriage, and embryonic/fetal size are associated with cytogenetic results of spontaneous early miscarriages. J. Assist. Reprod. Genet. 36, 749–757. https://doi.org/10.1007/s10815-019-01415-y (2019).
Menten, B. et al. Emerging patterns of cryptic chromosomal imbalance in patients with idiopathic mental retardation and multiple congenital anomalies: a new series of 140 patients and review of published reports. J. Med. Genet. 43, 625–633. https://doi.org/10.1136/jmg.2005.039453 (2006).
McQueen, D. B. & Lathi, R. B. Miscarriage chromosome testing: Indications, benefits and methodologies. Semin. Perinatol. 43, 101–104. https://doi.org/10.1053/j.semperi.2018.12.007 (2019).
Wang, Y. et al. Systematic analysis of copy-number variations associated with early pregnancy loss. Ultrasound Obstet. Gynecol. Off. J. Int. Soc. Ultrasound Obstet. Gynecol. 55, 96–104. https://doi.org/10.1002/uog.20412 (2020).
Pauta, M., Grande, M., Rodriguez-Revenga, L., Kolomietz, E. & Borrell, A. Added value of chromosomal microarray analysis over karyotyping in early pregnancy loss: systematic review and meta-analysis. Ultrasound Obstet. Gynecol. Off. J. Int. Soc. Ultrasound Obstet. Gynecol. 51, 453–462. https://doi.org/10.1002/uog.18929 (2018).
Liang, D. et al. Copy number variation sequencing for comprehensive diagnosis of chromosome disease syndromes. J. Mol. Diagn. JMD 16, 519–526. https://doi.org/10.1016/j.jmoldx.2014.05.002 (2014).
Li, F. X. et al. Detection of chromosomal abnormalities in spontaneous miscarriage by low-coverage next-generation sequencing. Mol. Med. Rep. 22, 1269–1276. https://doi.org/10.3892/mmr.2020.11208 (2020).
Zhang, R. et al. Prevalence of chromosomal abnormalities identified by copy number variation sequencing in high-risk pregnancies, spontaneous abortions, and suspected genetic disorders. J. Int. Med. Res. 47, 1169–1178. https://doi.org/10.1177/0300060518818020 (2019).
Lauer, S. & Gresham, D. An evolving view of copy number variants. Curr. Genet. 65, 1287–1295. https://doi.org/10.1007/s00294-019-00980-0 (2019).
Sahoo, T. et al. Comprehensive genetic analysis of pregnancy loss by chromosomal microarrays: outcomes, benefits, and challenges. Genet. Med. Off. J. Am. College Med. Genet. 19, 83–89. https://doi.org/10.1038/gim.2016.69 (2017).
Madjunkova, S. et al. QF-PCR rapid aneuploidy screen and aCGH analysis of cell free fetal (cff) DNA in supernatant of compromised amniotic fluids (AF). Prenat. Diagn. 34, 970–976. https://doi.org/10.1002/pd.4405 (2014).
Li, H. & Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinform. Oxf. Eng. 25, 1754–1760. https://doi.org/10.1093/bioinformatics/btp324 (2009).
Liao, C. et al. Noninvasive prenatal diagnosis of common aneuploidies by semiconductor sequencing. Proc. Natl. Acad. Sci. U. S. A. 111, 7415–7420. https://doi.org/10.1073/pnas.1321997111 (2014).
Riggs, E. R. et al. Technical standards for the interpretation and reporting of constitutional copy-number variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics (ACMG) and the Clinical Genome Resource (ClinGen). Genet. Med. Off. J. Am. College Med. Genet. 22, 245–257. https://doi.org/10.1038/s41436-019-0686-8 (2020).
Xue, H. et al. Efficiency of expanded noninvasive prenatal testing in the detection of fetal subchromosomal microdeletion and microduplication in a cohort of 31,256 single pregnancies. Sci. Rep. 12, 19750. https://doi.org/10.1038/s41598-022-24337-9 (2022).
Xue, H. et al. Molecular cytogenetic identification of small supernumerary marker chromosomes using chromosome microarray analysis. Mol. Cytogenet. 12, 13. https://doi.org/10.1186/s13039-019-0425-5 (2019).
Zhang, T., Sun, Y., Chen, Z. & Li, T. Traditional and molecular chromosomal abnormality analysis of products of conception in spontaneous and recurrent miscarriage. BJOG Int. J. Obstet. Gynaecol. 125, 414–420. https://doi.org/10.1111/1471-0528.15052 (2018).
Finley, J. et al. The genomic basis of sporadic and recurrent pregnancy loss: A comprehensive in-depth analysis of 24,900 miscarriages. Reprod. Biomed. Online 45, 125–134. https://doi.org/10.1016/j.rbmo.2022.03.014 (2022).
Peng, G. et al. Estimation on risk of spontaneous abortions by genomic disorders from a meta-analysis of microarray results on large case series of pregnancy losses. Mol. Genet. Genomic Med. 11, e2181. https://doi.org/10.1002/mgg3.2181 (2023).
Shearer, B. M., Thorland, E. C., Carlson, A. W., Jalal, S. M. & Ketterling, R. P. Reflex fluorescent in situ hybridization testing for unsuccessful product of conception cultures: A retrospective analysis of 5555 samples attempted by conventional cytogenetics and fluorescent in situ hybridization. Genet. Med. Off. J. Am. College Med. Genet. 13, 545–552. https://doi.org/10.1097/GIM.0b013e31820c685b (2011).
Gregg, A. R. et al. Noninvasive prenatal screening for fetal aneuploidy, 2016 update: A position statement of the American college of medical genetics and genomics. Genet. Med. Off. J. Am. College Med. Genet. 18, 1056–1065. https://doi.org/10.1038/gim.2016.97 (2016).
Tan, S., Pan, P., Yang, Z., Su, J. & Wei, H. Study on the correlation between the ultrasound phenotype and copy number variation of abnormal embryo in spontaneous abortion. J. Obstet. Gynaecol. Res. 47, 3779–3788. https://doi.org/10.1111/jog.14987 (2021).
Mazzonetto, P. C. et al. Low-pass whole genome sequencing is a reliable and cost-effective approach for copy number variant analysis in the clinical setting. Ann. Hum. Genet. https://doi.org/10.1111/ahg.12532 (2023).
Menasha, J., Levy, B., Hirschhorn, K. & Kardon, N. B. Incidence and spectrum of chromosome abnormalities in spontaneous abortions: new insights from a 12-year study. Genet. Med. Off. J. Am. College Med. Genet. 7, 251–263. https://doi.org/10.1097/01.gim.0000160075.96707.04 (2005).
Shen, J. et al. Chromosomal copy number analysis on chorionic villus samples from early spontaneous miscarriages by high throughput genetic technology. Mol. Cytogen. 9, 7. https://doi.org/10.1186/s13039-015-0210-z (2016).
Park, S. J. et al. Chromosomal abnormalities of 19,000 couples with recurrent spontaneous abortions: A multicenter study. Fertil. Steril. 117, 1015–1025. https://doi.org/10.1016/j.fertnstert.2022.01.011 (2022).
Wang, B. T. et al. Abnormalities in spontaneous abortions detected by G-banding and chromosomal microarray analysis (CMA) at a national reference laboratory. Mol. Cytogenet. 7, 33. https://doi.org/10.1186/1755-8166-7-33 (2014).
Vicić, A., Roje, D., Strinić, T. & Stipoljev, F. Trisomy 1 in an early pregnancy failure. Am. J. Med. Genet. Part A 146, 2439–2441. https://doi.org/10.1002/ajmg.a.32481 (2008).
Tamura, Y. et al. Chromosomal copy number analysis of products of conception by conventional karyotyping and next-generation sequencing. Reprod. Med. Biol. 20, 71–75. https://doi.org/10.1002/rmb2.12351 (2021).
Mikwar, M., MacFarlane, A. J. & Marchetti, F. Mechanisms of oocyte aneuploidy associated with advanced maternal age. Mutat. Res. Rev. Mutation Res. 785, 108320. https://doi.org/10.1016/j.mrrev.2020.108320 (2020).
Walsh, R. & Sharma, A. Extended survival of a premature infant with a postnatal diagnosis of complete triploidy. BMJ Case Rep. 15, 244551. https://doi.org/10.1136/bcr-2021-244551 (2022).
Peng, J. P. & Yuan, H. M. Application of chromosomal microarray analysis for a cohort of 2600 Chinese patients with miscarriage. Yichuan Hereditas 40, 779–788. https://doi.org/10.16288/j.yczz.18-120 (2018).
Hassold, T., Arnovitz, K., Jacobs, P. A., May, K. & Robinson, D. The parental origin of the missing or additional chromosome in 45, X and 47, XXX females. Birth Defects Orig. Artic. Ser. 26, 297–304 (1990).
Gomez, R. et al. Genetic findings in miscarriages and their relation to the number of previous miscarriages. Arch. Gynecol. Obstet. 303, 1425–1432. https://doi.org/10.1007/s00404-020-05859-x (2021).
Levy, B. et al. Genomic imbalance in products of conception: single-nucleotide polymorphism chromosomal microarray analysis. Obstet. Gynecol. 124, 202–209. https://doi.org/10.1097/aog.0000000000000325 (2014).
Sugawara, H. et al. Complex low-copy repeats associated with a common polymorphic inversion at human chromosome 8p23. Genomics 82, 238–244. https://doi.org/10.1016/s0888-7543(03)00108-3 (2003).
Definitions of infertility and recurrent pregnancy loss. A committee opinion. Fertil. Steril. 99, 63. https://doi.org/10.1016/j.fertnstert.2012.09.023 (2013).
Fiorentino, F. et al. PGD for reciprocal and Robertsonian translocations using array comparative genomic hybridization. Hum. Reprod. Oxf. Eng. 26, 1925–1935. https://doi.org/10.1093/humrep/der082 (2011).
Colley, E. et al. Potential genetic causes of miscarriage in euploid pregnancies: A systematic review. Hum. Reprod. Updat. 25, 452–472. https://doi.org/10.1093/humupd/dmz015 (2019).
Rosenfeld, J. A. et al. Diagnostic utility of microarray testing in pregnancy loss. Ultrasound Obstet. Gynecol. Off. J. Int. Soc. Ultrasound Obstet. Gynecol. 46, 478–486. https://doi.org/10.1002/uog.14866 (2015).
Maisenbacher, M. K. et al. Incidence of the 22q11.2 deletion in a large cohort of miscarriage samples. Mol. Cytogen. 10, 6. https://doi.org/10.1186/s13039-017-0308-6 (2017).
Zhang, H. et al. Implementation of a high-resolution single-nucleotide polymorphism array in analyzing the products of conception. Genet. Test. Mol. Biomark. 20, 352–358. https://doi.org/10.1089/gtmb.2016.0035 (2016).
Nagirnaja, L. et al. Structural genomic variation as risk factor for idiopathic recurrent miscarriage. Hum. Mutat. 35, 972–982. https://doi.org/10.1002/humu.22589 (2014).
Sheng, Y. R. et al. Characterization of copy-number variations and possible candidate genes in recurrent pregnancy losses. Genes 12, 141. https://doi.org/10.3390/genes12020141 (2021).
Qiao, Y. et al. Whole exome sequencing in recurrent early pregnancy loss. Mol. Hum. Reprod. 22, 364–372. https://doi.org/10.1093/molehr/gaw008 (2016).
Zheng, Y. et al. Experience of low-pass whole-genome sequencing-based copy number variant analysis: a survey of chinese tertiary hospitals. Diagn. Basel Switz. 12, 125055. https://doi.org/10.3390/diagnostics12051098 (2022).
Wapner, R. J. et al. Expanding the scope of noninvasive prenatal testing: Detection of fetal microdeletion syndromes. Am. J. Obstet. Gynecol. 212(332), e331-339. https://doi.org/10.1016/j.ajog.2014.11.041 (2015).
Larroya, M. et al. Have maternal or paternal ages any impact on the prenatal incidence of genomic copy number variants associated with fetal structural anomalies?. PloS ONE 16, e0253866. https://doi.org/10.1371/journal.pone.0253866 (2021).
Xanthopoulou, L. et al. Chromosome breakage in human preimplantation embryos from carriers of structural chromosomal abnormalities in relation to fragile sites, maternal age, and poor sperm factors. Cytogenet. Genome Res. 136, 21–29. https://doi.org/10.1159/000334836 (2012).
Eggermann, T. Prenatal detection of uniparental disomies (UPD): Intended and incidental finding in the era of next generation genomics. Genes 11, 112475. https://doi.org/10.3390/genes11121454 (2020).
Gogiel, M. et al. Genome-wide paternal uniparental disomy mosaicism in a woman with Beckwith-Wiedemann syndrome and ovarian steroid cell tumour. Eur. J. Hum. Genet. EJHG 21, 788–791. https://doi.org/10.1038/ejhg.2012.259 (2013).
Kotzot, D. Complex and segmental uniparental disomy updated. J. Med. Genet. 45, 545–556. https://doi.org/10.1136/jmg.2008.058016 (2008).
Acknowledgements
We thank the family members who participated in our study. We also appreciate the obstetricians, radiographers, sonographer and pediatricians who offered assistance to our study.
Funding
This study was sponsored by the Joint Funds for the Innovation of Science and Technology, Fujian Province (no.2020Y9149), 2021 Fujian provincial health technology project (no.2021GGA051), and Natural Science Foundation of Fujian Province (no.2022J01421).
Author information
Authors and Affiliations
Contributions
H.X. and A.Y. prepared the main manuscript; X.C., L.X., M.L., and Q.G. prepared the experiment. All authors have read and approved the final article. Informed consent was obtained from all patients for being included in the study.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Xue, H., Guo, Q., Yu, A. et al. Genetic analysis of chorionic villus tissues in early missed abortions. Sci Rep 13, 21719 (2023). https://doi.org/10.1038/s41598-023-48358-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-023-48358-0
- Springer Nature Limited