
Abstract
Disulfidptosis, a novel type of programmed cell death, has attracted researchers’ attention worldwide. However, the role of disulfidptosis-related lncRNAs (DRLs) in liver hepatocellular carcinoma (LIHC) not yet been studied. We aimed to establish and validate a prognostic signature of DRLs and analyze tumor microenvironment (TME) and drug susceptibility in LIHC patients. RNA sequencing data, mutation data, and clinical data were obtained from the Cancer Genome Atlas Database (TCGA). Lasso algorithm and cox regression analysis were performed to identify a prognostic DRLs signature. Kaplan–Meier curves, principal component analysis (PCA), nomogram and calibration curve, function enrichment, TME, immune dysfunction and exclusion (TIDE), tumor mutation burden (TMB), and drug sensitivity analyses were analyzed. External datasets were used to validate the predictive value of DRLs. qRT-PCR was also used to validate the differential expression of the target lncRNAs in tissue samples and cell lines. We established a prognostic signature for the DRLs (MKLN1-AS and TMCC1-AS1) in LIHC. The signature could divide the LIHC patients into low- and high-risk groups, with the high-risk subgroup associated with a worse prognosis. We observed discrepancies in tumor-infiltrating immune cells, immune function, function enrichment, and TIDE between two risk groups. LIHC patients in the high-risk group were more sensitive to several chemotherapeutic drugs. External datasets, clinical tissue, and cell lines confirmed the expression of MKLN1-AS and TMCC1-AS1 were upregulated in LIHC and associated with a worse prognosis. The novel signature based on the two DRLs provide new insight into LIHC prognostic prediction, TME, and potential therapeutic strategies.
Similar content being viewed by others
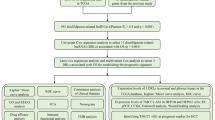

Introduction
Liver cancer is one of the most prevalent malignancies globally, with an estimated 905,677 (4.7% cancer-related new cases) new cases and 830,180 (8.3% cancer-related death) deaths worldwide in 20201. It is expected that there will be a 55.0% increase per year in new liver cancer cases between 2020 and 2040, with 1.4 million potential patients diagnosed in 20402. Liver hepatocellular carcinoma (LIHC) is the dominant type of primary liver cancer, accounting for 85–90% of all cases3. The main risk factors for developing LIHC include chronic hepatitis B/C virus infection, alcoholism, aflatoxin exposure, and metabolic risk factors4. Despite great advances in early diagnosis with molecular markers and multiple therapies, 5-year survival rates for LIHC remain unsatisfactory5. Recurrence or metastasis occurs in approximately 70% of LIHC patients who undergo surgical resection3. Worse, LIHC is a heterogeneous disorder with widespread chemotherapy and radiation resistance. Therefore, it is critical to identify and validate novel prognostic and predictive biomarkers for LIHC, which may lead to efficient detection and discovery of optimal therapies for LIHC.
Regulatory cell death (RCD) is a common type of cell death that is characterized by uncontrolled cell expansion or accumulation6. RCD plays an essential role in cancer metabolic therapy7, tissue homeostasis, inflammation, and multiple pathophysiological conditions6. Over the past two decades, several emerging RCD modalities have been highlighted, such as cuproptosis8, ferroptosis9, apoptosis, necroptosis, pyroptosis, parthanatos, autophagy-dependent cell death, immunogenic cell death, and so on6. The physiological forms of RCD are generally regarded as key therapeutic targets for the management of multiple human diseases6. Most recently, Liu et al.10 provided the first insight into a novel form of RCD known as “disulfidptosis”, which differs from previously identified cell death modalities. This study revealed that excessive expression of solute carrier family 7 member 11 (SLC7A11) in glucose-deficient cancer cells accelerates nicotinamide adenine dinucleotide phosphate (NADPH) depletion under glucose starvation conditions10. This leads to an accumulation of disulfides that cannot be reduced, altering the conformation of cytoskeletal proteins and inducing disulfide stress, which ultimately leads to disulfidptosis, an unusual form of cell death with a specific underlying mechanism7,10,11. Disulfidptosis activation may require three elements: high SLC7A11 expression, glucose deprivation conditions that impair glucose metabolism, and aberrant disulfide bonds between actin cytoskeleton proteins12. Aberrant accumulation of intracellular disulfide molecules, such as lipoic acid and cystine, triggers disulfide stress and extremely rapid cell death13. Disulfidptosis is an unusual form of cell death in morphology, genetics, and biochemistry that is involved in the regulation of tumor progression12. A link between disulfidptosis and cancer has been established via bioinformatics analysis recently11,14,15,16,17,18. Yang et al. discovered disulfidptosis-based prognostic and tumor microenvironment (TME) characteristics of clear cell renal cell carcinoma and identified AJAP1 as a possible biomarker for the disease11. Zhao et al. investigated the role of disulfidptosis-related genes (DRGs) in bladder cancer prognosis and generated a risk signature to assess patient prognosis, TME, and immunotherapy14. Wang et al. developed a risk score for DRGs and discovered that SPP1 and MYBL2 genes might predict the survival of LIHC patients15. Currently, the role of disulfidptosis in the assessment of the occurrence, progression, prognosis, and treatment of LIHC is scarce. More indicators associated with disulfidptosis with cancer susceptibility are urgent to be established.
Long non-coding RNAs (lncRNAs) are RNA molecules length greater than 200 nucleotides that cannot code for protein19. Growing evidence suggests that the role of lncRNAs in basics, transcriptomic, and clinical oncology may be comparable to, and possibly greater than that of protein-coding genes19. Over the past decade, the research on lncRNA-based clinical tools has expanded rapidly, including in cancer diagnostic and prognostic biomarkers, and therapeutic targets19. LncRNA-related signatures for prognostic prediction of LIHC have attracted more and more attention, such as cuproptosis-related lncRNAs20,21,22, ferroptosis-related lncRNAs23,24,25, necroptosis-related lncRNAs26,27, pyroptosis-related lncRNAs28,29,30, and so on. However, disulfidptosis-related lncRNAs (DRLs) as prognostic markers for LIHC have never been investigated. Therefore, we aim to identify different clustering features and construct DRLs to predict the prognosis of LIHC patients, and evaluate the correlation between the signatures and the tumor microenvironment and drug sensitivity.
Materials and methods
Dataset and data acquisition
LIHC dataset was downloaded from the TCGA database (https://portal.gdc.cancer.gov/repository). The R (version 4.1.3, https://www.r-project.org/) and relative R packages were used to analyze the TCGA cohort. After obtaining from TCGA, an FPKM gene expression matrix was transformed into TPM format31. Next, using the R package “sva”, the merged expression matrix was normalized and eliminated from batch effects32.
Identification of co-expressed DRGs and lncRNAs
Ten disulfidptosis-related genes (DRGs) were identified from the recently published studies10,11. Univariate cox regression method was applied to evaluate the hazard ratio of the DRGs to the LIHC. Then, the R package “limma” was used to acquire the expression of DRGs (see Supplementary Text files)33. According to previous documents, Pearson correlation analysis was performed to evaluate the relationship between DRGs and lncRNAs in the RNA-seq data of TCGA-LIHC samples. Next, to find adequate disulfidptosis-related lncRNAs (DRLs), we set correlation coefficient > 0.4 and P < 0.001 was the optimal cutoff value using the relevant R script (see Supplementary Text files)22. Moreover, the R packages “ggplot2” and “ggalluvial” were used to construct a Sankey diagram to show the correlation between DRGs and DRLs34.
Construction of DRLs risk model
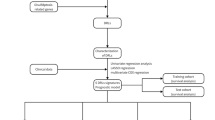
The LIHC patients were randomly assigned to the testing and training groups using the R package “caret” in a 1:1 ratio35. Univariate cox regression was utilized for obtaining prognostic DRLs (P < 0.01). Lasso cox regression analysis was used to construct the prediction model for DRLs36,37. Multivariate cox regression analysis was applied to build a two-lncRNA predictive model. The calculated formula used for disulfidptosis-related prognostic risk score was based on the published documents22,38. The model of risk score was calculated as follows: risk score = ∑i = LnCoef(i) × EXP(i). Patients were classified into low-risk and high-risk groups, depending on the median value of the risk score. The R package ‘survminer’ was used to construct Kaplan–Meier curves to examine the prognostic relevance of risk models for DRLs39. The R package “timeROC” was utilized to investigate the receiver operating characteristic (ROC) of survival39. In addition, univariate cox regression and multivariate cox regression methods were applied to evaluate the prognostic predictive potential of this risk score model. The P < 0.05 was selected by R software.
Principal component analysis (PCA) analysis and clinical features
PCA analysis was performed using the R packages “scatterplot3d”40 to demonstrate whether the DRLs prognostic risk score model can identify low-risk and high-risk groups of patients to further clarify the clinical utility of DRLs. The association between this risk score model and clinical variables, such as grade, gender, age, and stage, were evaluated by chi-square tests and Wilcoxon rank-sum test.
Nomogram and calibration curve
Using the DRLs risk model and clinicopathological factors (age, gender, grade, stage, tumor (T), metastasis (M), and positive lymph node (N)), a nomogram for predicting the 1/3/5-year survival of LIHC patients was created. We used a calibration curve to check if the nomogram predicted survival rate agreed with recognition survival rate. The R package “rms” and “regplot” were used to construct the nomogram41, and calibration curve42.
Function enrichment analysis
For the purpose of investigating majorly enriched signaling pathways and biological roles involved in the disulfidptosis-related lncRNA signature, Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) were performed43. GO analysis was performed to identify the enriched pathways in different sets by R packages “clusterProfiler”, “org.Hs.eg.db”, “enrichplot”, “ggplot2”, “circlize”, “RColorBrewer”, “dplyr”, “ggpubr”, “ComplexHeatmap” with P = 0.05 and q = 0.0544. KEGG analysis was used to reveal the pathways associated with high- and low-risk sets caring out by using R packages “DOSE”, “clusterProfiler”, “org.Hs.eg.db”, “enrichplot”, “ggplot”, “circlize, “RColorBrewer”, “dplyr”, and “ComplexHeatmap” with P = 0.05 and q = 0.0545,46,47.
Tumor microenvironment (TME) analysis
The proportion of tumor-infiltrating immune cells and immune function were displayed on boxplots for low- and high-risk sets created by R packages “limma”, “ggpubr”, and “reshape2”33. The level of immune cell infiltration in TME of LIHC was investigated using a single-sample gene set enrichment analysis (ssGSEA) algorithm48.
Immune dysfunction and exclusion (TIDE) analysis
The TIDE scoring file was acquired from the website (http://tide.dfci.harvard.edu). The R package “ggpubr” was utilized to examine the difference in TIDE between the high- and low-risk sets49.
Tumor mutation burden (TMB) analysis
The different analyses of TMB, survival analysis, the prognosis and tumor mutation between the high- and low-risk sets was performed using the R package “ggpubr” and “limma”33.
Drug susceptibility analysis
We screened potential chemotherapeutic drugs using the R packages “pRRophetic”50 to determine whether high-risk and low-risk groups have significant differential susceptibilities to the therapy (IC50 value). The filtering threshold was set at P < 0.001.
Clinical tissue samples
A total of 50 LIHC specimens and adjacent non-cancerous tissues were collected from LIHC patients who underwent surgical resection at the First Affiliated Hospital of Guangxi Medical University between June 2022 and March 2023. The following were the inclusion criteria: (I) pathologically diagnosed with LIHC the cancer for the primary time; (II) didn’t receive radiation, chemotherapy, targeted therapy, ablation, or intervention prior to the operation. Patients were excluded if they met any one of the following criteria: (I) the presence of other malignancies; (II) the occurrence of diabetes, hypertension, or other chronic conditions. All the tissues were stored at − 80 °C until the extraction of total RNA. Informed consent was obtained from all subjects involved in the study. The Medical Ethics Committee of First Affiliated Hospital of Guangxi Medical University has approved the protocol (No. 2023-E321-01).
Cell lines
Normal LO2 cells and LIHC cells (Hep G2, Huh-7, Hep-3B, MHCC-97H, QGY-7703, SMMC-7721, and HCC-LM3) were bought from the Chinese Academy of Sciences Cell Bank in Shanghai, China. All cell lines were cultured in Dulbecco’s modified Eagle’s medium (Gibco, USA) supplemented with 10% fetal bovine serum (FBS, Gibco, USA), 100 U/mL streptomycin, and 100 U/mL penicillin in a 5% CO2 environment at 37 °C.
RNA extraction and quantitative real-time polymerase chain reaction (qRT-PCR)
Total RNA from the tissues and cell lines was extracted using the TRIzol reagent. The quality and quantity of the RNA was then assessed using NanoDrop (Thermo Science). cDNA was synthesized using the Superscript II Reverse Transcriptase Kit (Takara, Japan). qRT-PCR was performed using the SYBR Green (Takara, Japan) method to detect transcript levels in tissues and cell lines by ABI 7500 Real-Time PCR System. The endogenous control gene glyceraldehyde 3-phosphate dehydrogenase (GAPDH) was used to normalize the expression of the target gene. The 2−ΔΔT approach was used to calculate the relative expression value. Primer sequences for MKLN1-AS: (f) 5′-TGACCGACACTGGGTCTGA (r) 5′-AGGCTTTCAGGAGTCCAACC; TMCC1-AS1: (f) 5′-TGCCATGCCCGTGTCAACTG (r) 5′-CTCTCTCGTTCTGCCTCCCTTATG.
Extended database validation
We used the GEPIA2: an updated and enhanced version of Gene Expression Profiling Interaction Analysis database (http://gepia2.cancer-pku.cn/#index)51 to further validate the credibility of the results. Gene expression, and overall survival (OS) survival analysis for MKLN1-AS and TMCC1-AS1 in LIHC samples were visually displayed. To divide high-expression and low-expression cohorts, the optimal cutoff was chosen automatically with log-rank P values.
Statistical analysis
We used R software (version 4.1.3) and its associated packages to analyze all statistics. RNA-seq data, mutation data, clinical information, and Ensembl IDs for lncRNAs were extracted using Strawberry-Perl (version 5.30.0.1-64bit, https://www.perl.org). The transcript levels of MKLN1-AS and TMCC1-AS1 in LIHC tissues and para-noncancerous tissues were compared using a paired t-test. The transcript levels of MKLN1-AS and TMCC1-AS1 in normal LO2 cells and LIHC cells were compared using an unpaired t-test. The proportions of the clinical features were analyzed using the chi-squared test. Statistical significance was set at P < 0.05.
Ethics approval and consent to participate
The study follows the principles of the Declaration of Helsinki. The study protocol was approved by the Ethics Committee of scientific research and clinical trial of the First Affiliated Hospital of Guangxi Medical University (Approval Number: 2023-E321-01). All patients provided written-informed consent for the collection and publication of their medical information at the first visit to our center, which was filed in their medical records.
Results
Identification and construction of DRLs risk model
RNA-seq data of 424 samples (374 tumor and 50 normal tissues) were downloaded from TCGA database. Individuals with incomplete genomes or clinical data were eliminated (n = 4), leaving 370 LIHC samples in the final group. In recent literature10, authors proposed a unique form of cell death termed disulfidptosis, which was not characterized previously. Ten DRGs were obtained from studies, and their details and correlation were presented in Fig. 1A and Supplementary Table S1. Then, a total of 1354 DRLs were obtained by Pearson analysis on the basis of R > 0.4 and P < 0.001. The significant prognostic DRLs were identified using univariate regression analysis (Fig. 1B) and Lasso cox regression analysis (see Supplementary Fig. S1A,B). The correlation between DRGs and DRLs was displayed in Fig. 1C and Supplementary Table S2. Finally, two key candidate OS-related DRLs were extracted to construct the prognostic signature, which include MKLN1-AS and TMCC1-AS1. The relation between the two DRLs and the DRGs was shown in Fig. 1D.

Construction of the differentially expressed disulfidptosis-related lncRNAs (DRLs) in TCGA-LIHC cohort. (A) The ten disulfidptosis-related genes (DRGs) and their correlation. (B) The significant prognostic DRLs were identified using univariate regression analysis. (C) The Sankey relation between DRGs and DRLs. (D) Heatmap of the correlation between DRGs and DRLs involved in model construction (*P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001).
The expression of these 10 genes between tumor and normal tissues in LIHC patients was showed in Supplementary Fig. S1C. The hazard ratio of the ten genes on the prognosis in LIHC patients was showed in Supplementary Fig. S1D. A total of 370 LIHC patients were randomly divided into the training set (n = 185) and the testing set (n = 185). Table 1 showed the clinical characteristics of the two groups of patients, demonstrating that there was no variation in clinical characteristics between the training and testing groups.
Predicting the prognosis of LIHC patients with the DRLs predictive model
The coefficients of the two DRLs were used to assess the scores for each patient. The risk score was calculated as follows: risk score = expression of MKLN1-AS × 1.010769 + expression of TMCC1-AS1 × 0.692979. Next, LIHC patients were divided into high-risk and low-risk groups based on the median risk score value (Supplementary Fig. S2A–C). Patients exhibited a higher mortality rate with risk scores increasing (Supplementary Fig. S2D–F). Survival analysis revealed that high-risk patients had significantly worse overall survival than low-risk patients (Fig. 2A–C; all P < 0.001). The expression of MKLN1-AS and TMCC1-AS1 was higher in the high-risk patients than in the low-risk patients in all sets (Fig. 2D–F), indicating that the two DRLs might be the poor indicators of prognosis.

Construction and evaluating of the prognostic value of disulfidptosis-related lncRNAs (DRLs) in all LIHC patients, test, and train sets. (A–C) Kaplan–Meier (KM) survival analysis between high-risk and low-risk LIHC patients. (D–F) Heatmaps of two DRLs between high-risk and low-risk LIHC patients (*P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001).
Univariate cox regression (see Supplementary Fig. S3A) and multivariate cox regression (Fig. 3A) analysis revealed that risk score was an independent prognostic factor for LIHC patients. The risk score’s area under the curve (AUC) was 0.745 (Fig. 3B), greater than that of the other clinical parameters, indicating that it was a more accurate predictor of LIHC patients’ overall survival. Furthermore, the survival probability of 1-, 3- and 5-year for the LIHC patient in the nomogram was 0.758, 0.551, and 0.419, respectively (Fig. 3C). The nomogram predictions were validated by the calibration curves showing there was a high degree of congruence between the clinical outcomes and predictive overall survival rates at 1, 3, and 5 years (Fig. 3D). The risk score showed its value in evaluating the 1-, 3- and 5-year survival of patients (see Supplementary Fig. S3B). In addition, ROC curves showed that the 10-year concordance-index (C-index) scored higher in the risk model than other clinical variables (see Supplementary Fig. S3C). The survival probability and clinical parameters of LIHC patients were compared according to the gender, grade, stage, and tumor between the two risk groups (see Supplementary Fig. S3D–I). The results demonstrated that all clinicopathological variables were significant differences between the high- and low-risk groups, suggesting that patients with high-risk scores had a poor prognosis (all P < 0.05).

Validation of the prognostic value of the disulfidptosis-related lncRNA (DRLs) signature model. (A) Multivariate cox regression analysis of the clinical variables and DRLs risk scores model. (B) Comparison of predictive risk score model and clinicopathological features. (C) A nomogram showing risk and clinicopathological parameters for predicting 1-, 3- and 5-OS in LIHC patients. (D) Calibration curves indicating the accuracy of risk model to predict 1-, 3- and 5-OS of LIHC patients. (E–H) PCA analysis of DRLs and clinical parameters in LIHC patients: (E) PCA of all genes. (F) PCA of disulfidptosis-associated genes. (G) PCA of disulfidptosis-associated lncRNAs. (H) PCA of the risk model (*P < 0.05, **P < 0.01, ***P < 0.001 ****P < 0.0001, ns no significance).
PCA analysis
PCA analysis was performed on the models of all genes, DRGs, DRLs, and risk lncRNAs model (Fig. 3E–H). The results indicated that our constructed model was all able to effectively classify high-risk and low-risk LIHC patients, demonstrating the accuracy of the model. All of the above findings suggest that risk scores could be a reliable prognostic factor in predicting survival in LIHC patients.
Functional and pathway analysis
GO and KEGG analyses were used to investigate the underlying different biological properties of genes in high-risk and low-risk subsets. A total of 1405 differently expressed genes were discovered between the high-risk and low-risk groups. (Supplementary Table S3). According to GO pathway analysis (Fig. 4A), biological process (BP) terminology indicated that differently expressed genes are rich in the “organelle fission”, “nuclear division”, and “positive regulation of cell adhesion”. Cell composition (CC) showed differently expressed genes were mainly enriched in “collagen-containing extracellular matrix” “apical part of cell” and “microtubule”. In term of molecular function (MF), “signaling receptor activator activity”, “receptor ligand activity” and “tubulin binding” were significantly abundant. KEGG pathway analysis (Fig. 4B) indicated that differently expressed genes were mainly concentrated in “organelle fission”, “nuclear division”, and “positive regulation of cell adhesion”. According to the results, we proposed that the DRGs primarily affect the cellular metabolic activities.

Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway enrichment analyses between high- and low-risk groups. (A) GO analysis revealed the diversity of molecular biological process (BP), cellular component (CC) and molecular function (MF). (B) KEGG pathway analysis revealed the significantly enriched pathways.
Tumor microenvironment (TME) analysis
The correlation of immune function differences between the low- and high-risk groups was depicted in Fig. 5A. “Cytolytic_activity”, “MHC_class_I”, and “Type_II_IFN_Reponse” were significantly different between the low- and high-risk groups. Furthermore, the relationship between the risk score and the content of tumor-infiltrating immune cells were surveyed. As illustrated in Fig. 5B, the “aDCs”, “B_cells”, “iDCs”, “Macrophages”, “Mast_cells”, “NK_cells”, and “Treg” were substantially different between high-risk and low-risk groups. The high-risk group had significantly higher proportion of “aDCs”, “Macrophages” and “Treg” whereas the low-risk group exhibited greater percentage of “B cells,” “Mast_cells”, and “NK_cells”. These results indicated that disulfidptosis acts a vital role in the formation of the TME.

Tumor microenvironment (TME), immune dysfunction and exclusion (TIDE), and tumor mutation burden (TMB) analysis of the risk score between the low and high-risk groups. (A) Immune function differences analysis. (B) Tumor-infiltrating immune cells differences analysis. (C) TIDE analysis. (D,E) The waterfall plot showed the mutated genes. (F) TMB analysis of the risk score. (G,H) TMB survival analysis and risk score (*P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001, ns no significance).
Immune dysfunction and exclusion (TIDE) analysis of the risk score
Immune escape and immunotherapy differences between high and low risk groups were examined to determine the impact of immunotherapy in LIHC patient populations. The clinical response to immune checkpoint blocking (ICB) therapy was evaluated using the TIDE score. A high TIDE score implies a low response to ICB and a poor prognosis for the cancer. TIDE score was lower in the high-risk group (Fig. 5C; P < 0.001). The lower composite TIDE score attained in the high-risk group could be explained with a higher proportion of treatment remissions.
Tumor mutation burden (TMB) analysis of the risk score
Using the “maftools” software package, we examined the somatic mutation scope of LIHC patients in high- and low-risk groups and chose 15 genes (TP53, CTNNB1, TTN, MUC16, PCLO, ALB, RYR2, APOB, CSMD3, LRP1B, XIRP2, OBSCN, ABCA13, HMCN1, FLG) with the highest mutation frequency for representation (Fig. 5D,E). The waterfall plot revealed that the top three mutated genes in high risk LIHC samples were TP53 (35%), CTNNB1 (31%), and TTN (23%), whereas in low risk LIHC samples, TTN (24%), CTNNB1 (21%), and TP53 (18%) were the most common alteration genes. In all groups, the most common variation categorization was missense mutation. Overall, there were no significant differences in TMB between the low- and high-risk groups (Fig. 5F; P = 0.42). According to a TMB survival analysis, patients in the high TMB group had a poorer prognosis (Fig. 5G; P = 0.031). When TMB and patient risk scores combined, the survival rate of high TMB + high risk score groups had the poorest prognosis (Fig. 5H; P < 0.001).
Drug susceptibility analysis
To determine whether the high-risk and low-risk groups had significantly different susceptibilities to the chemotherapy, we performed drug sensitivity analysis using “pRRophetic” package to screen prospective anti-tumor drugs (IC50 value). The sensitivity to medicines increases with decreasing IC50 values. Because too many chemotherapeutic drugs were screened based on the two DRLs, we picked nine chemotherapy agents with great differences including sorafenib, 5-fluorouracil, dasatinib, doxorubicin, imatinib, gemcitabine, sunitinib, paclitaxel, and erlotinib commonly used in clinical settings to do our further research. Doxorubicin (Fig. 6A), gemcitabine (Fig. 6B), sunitinib (Fig. 6C), paclitaxel (Fig. 6D), 5-fluorouracil (Fig. 6E), imatinib (Fig. 6F), sorafenib (Fig. 6G), and dasatinib (Fig. 6H) are eight of the nine treatments with better sensitivity/lower IC50 in the high-risk group, suggesting that these eight medications are more likely to be beneficial for high-risk LIHC patients (P < 0.0001). Erlotinib (Fig. 6I) is only one of nine treatments with better sensitivity/lower IC50 in the low-risk group (P < 0.001).

Drug sensitivity analysis and screening potential drugs for LIHC between two groups. (A) Doxorubicin; (B) gemcitabine; (C) sunitinib; (D) paclitaxel; (E) 5-fluorouracil; (F) imatinib, (G) sorafenib, (H) dasatinib; (I) erlotinib (*P < 0.05, **P < 0.01, ***P < 0.001 ****P < 0.0001).
Validation data
The relative expression levels of MKLN1-AS and TMCC1-AS1 in LIHC tissues and adjacent normal tissues, cell lines, and validation dataset were showed in Fig. 7. According to the TCGA and GEPIA2 data, the expression levels of these two lncRNAs were both considerably increased in LIHC tissues when compared to nearby normal tissues (Fig. 7A–F). MKLN1-AS and TMCC1-AS1 transcription levels were also found to be strongly linked with a poor prognosis of LIHC (Fig. 7G,H). We used qRT-PCR to examine the expression levels of MKLN1-AS and TMCC1-AS1 in 50 pairs of LIHC tissue and adjacent normal tissue (see Supplementary Original data). MKLN1-AS and TMCC1-AS1 transcription levels were significantly higher in LIHC tissues compared to adjacent normal tissues, which was consistent with the TCGA and GEPIA2 database results (Fig. 7I,K). The expression of the two lncRNAs in LIHC cells was compared to that of normal LO2 cells and LIHC cells (Hep G2, Huh-7, Hep-3B, MHCC-97H, QGY-7703, SMMC-7721, and HCC-LM3) (see Supplementary Original data). MKLN1-AS was considerably higher in Hep G2, Huh-7, MHCC-97H, QGY-7703, and HCC-LM3 cells than in normal LO2 cells (Fig. 7J), while TMCC1-AS1 was significantly higher in Hep G2, Huh-7, Hep-3B, MHCC-97H, QGY-7703,and SMMC-7721 cells (Fig. 7L). Overall, the results demonstrated that the MKLN1-AS and TMCC1-AS1 expression levels were up-regulated in LIHC and associated with the worse prognosis of LIHC.

The relative expression levels of two candidate lncRNAs in LIHC tissues compared with normal tissues, cell lines, and validation dataset. The expression of MKLN1-AS (A,B) and TMCC1-AS1 (C,D) in 374 LIHC tissues compared with 50 normal tissues in the TCGA database. The expression of MKLN1-AS (E) and TMCC1-AS1 (F) in 369 LIHC tissues compared with 160 normal tissues in the GEPAI database. Overall survival (OS) of different MKLN1-AS (G) and TMCC1-AS1 (H) expression level in LIHC samples. The expression of MKLN1-AS (I) and TMCC1-AS1 (K) in 50 pairs of LIHC and adjacent normal tissues in the validation dataset. The expression of MKLN1-AS (J) and TMCC1-AS1 (L) in HCC cell lines (*P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001).
Discussion
RCD plays a crucial role in regulating tumor proliferation and cell metabolism6. Disulfidptosis, a novel metabolic-related RCD proposed in 2023, was suggested to provide a therapeutic strategy to target disulfidptosis in cancer therapy10. Currently, there is a dearth of research on the role of disulfidptosis in the oncogenesis, progression, prognosis, immune profile, signaling pathways, and targeted therapy for LIHC. In the present study, 374 LIHC samples and 50 normal controls were obtained from the TCGA database. We focused on the expression levels of DRLs (MKLN1-AS and TMCC1-AS1) and the clinical features of LIHC to construct a comprehensive disulfidptosis-related predictive model. Based on the expression levels of the DRLs, the LIHC sample was divided into low-risk and high-risk group to investigate the effect of this disulfidptosis pattern on LIHC prognosis and immunotherapy response. In addition, 369 LIHC samples and 160 normal controls from the GEPIA2 database were used to further validate the credibility of the results. We also used qRT-PCR to examine the expression levels of MKLN1-AS and TMCC1-AS1 in 50 pairs of LIHC tissue and adjacent normal tissue, as well as in LIHC cell lines. Our data demonstrated that MKLN1-AS and TMCC1-AS1 expression levels were upregulated in LIHC and associated with a worse prognosis. In addition, the LIHC patient in high-risk group was associated with poor OS. GO and KEGG data demonstrated that DRGs primarily affect the cellular metabolic activities. Moreover, the tumor-infiltrating immune cells were significantly different between the high-risk and low-risk groups. Surprisingly, the TIDE results showed a higher immune escape potential in the low-risk group, suggesting that immunotherapy was less effective in low-risk LIHC patients. Finally, LIHC patients in the high-risk group were more sensitive to several potential chemotherapeutic drugs. Taken together, the novel signature based on the two DRLs provide new insight into LIHC prognostic prediction, immune microenvironment characteristics, and potential therapeutic strategies using a combination of disulfidptosis and drug sensitivity.
In this study, we identified ten genes that suppress disulfidptosis based on recent publications10,11. Six of these ten DRGs, namely GYS1, LRPPRC, NCKAPA1, RPN1, SLC3A2, and SLC7A11, were significantly associated with the risk of LIHC. It is well established that SLC7A11 is overexpressed in multiple human cancers and protects cancer cells from amino acid deprivation, oxidative stress, and metabolic stress52. Mechanistically, high levels of SLC7A11 expression, H2O2 therapy, and cystine uptake in tumor cells trigger the accumulation of harmful disulfide molecules and NADPH depletion, eventually resulting in rapid cell death—disulfidptosis12,53. Currently, few research has been conducted to investigate the involvement of DRGs in cancer11,14,15,16,17. Most recently, Feng et al. used 24 DRGs to create a classification and model that can effectively predict the prognosis and drug sensitivity of thyroid carcinoma patients17. Besides, Chen et al. reported a DRG prognostic model with ten features and found that bladder cancer patients with high DRG scores may have worse survival, inflamed TME, and an increased TMB, suggesting that the model could be used for personalized therapy16.
Increasing evidence suggests that the tumor microenvironment (TME) plays a crucial role in the progression and metastasis of tumors54. The TME of LIHC comprises extracellular matrix, endothelial cells, stromal cells, as well as various cytokines and proteins55. The infiltration of multiple immune cells within the TME promotes tumor cell growth, immune tolerance, and immune evasion, thereby facilitating cancer cell metastasis54. In this study, we observed a significant correlation between MHC_class_I responses and immune function enrichment specifically in the high-risk group. Furthermore, it has been observed that the high-risk cohort exhibited a significantly increased proportion of antigen-presenting dendritic cells (aDCs), macrophages, and regulatory T cells (Treg cells). Importantly, tumor-infiltrating macrophages have been identified as potent producers of diverse mediators within the tumor microenvironment (TME), thereby facilitating tumor proliferation, metastasis, and invasion, ultimately leading to an unfavorable prognosis56,57. Tregs play a crucial role in maintaining self-tolerance within the immune system, suppressing antitumor immunity, facilitating tumor invasion and migration, and are closely associated with cancer development and progression58. The presence of Tregs infiltrating tumors has been consistently linked to unfavorable survival outcomes in various malignancies, including LIHC58. One study revealed that the high subtypes of disulfidptosis-related gene exhibited elevated levels of immune cells and immune scores in LIHC15. Furthermore, Yang et al. demonstrated a significant increase in the infiltration level of immune cells within the high disulfidptosis-related gene score group59. Additionally, there was a positive correlation between high disulfidptosis-related gene (SLC7A11) expression in LIHC and T helper cells, macrophages, and NK CD56 bright cells60. Our study consistently demonstrated that patients with LIHC in the high-risk group exhibited higher levels of activated dendritic cells (aDCs), macrophages, and Tregs compared to those in the low-risk group. The role of B cells in LIHC proliferation and metastasis remains unclarified based on recent studies54. On one hand, research has suggested that the loss of specific TGF-β on the surface of B cells could inhibit LIHC development61. On the other hand, a study discovered that CXCR3+ B cells could interact with the LIHC microenvironment to promote polarization of M2b macrophages and enhance metastatic potential62. In this study, we observed that the low-risk group with a better prognosis had a higher percentage of B cells.
Emerging research highlights critical roles of lncRNAs to the LIHC development and progression63,64,65. Recently, Chi et al. found that the lncRNA PTOV1-AS1 is up-regulation in LIHC and associated with patients’ prognosis and sorafenib resistance64. Another study revealed that LINC01977 accelerated LIHC progression by inhibiting Notch2 ubiquitination and depletion, suggesting that LINC01977 could be a potential biomarker and therapeutic target for LIHC patients63. Similarly, Sun et al. indicated that LINC01124 played a tumor-promoting role in LIHC through regulating the miR-1247-5p-FOXO3 axis65. Interestingly, the crucial role of RCD-related abnormally expressed lncRNAs in monitoring and promoting the development and progression of LIHC is drawing growing interest. Several RCD related lncRNA signatures to forecast prognosis and tumor progression in LIHC patients have been established, such as cuproptosis-related lncRNAs20,21,22, ferroptosis-related lncRNAs23,24,25, necroptosis-related lncRNAs26,27, pyroptosis-related lncRNAs28,29,30, and so on. For instance, Guo et al. established a cuproptosis-prognostic signature for four cuproptosis-related lncRNAs (SNHG4, AC026412.3, AL590705.3, and CDKN2A-DT) that could predict prognosis and evaluate the efficacy of immunotherapy for LIHC20. Liu et al. constructed a predictive model based on eight cuproptosis-related lncRNAs (AC004112.1, AC007064.2, AC012186.2, AC026412.3, AL031985.3, AL133477.1, AL365361.1, and TMCC1-AS1) to give valuable data for predicting the prognosis of LIHC patients and developing individualized targeted therapy21. Li et al. built a five cuproptosis-related lncRNAs (FOXD2-AS1, NRAV, MED8-AS1, WARS2-AS1, and MKLN1-AS) predictive signature for LIHC and confirmed the results of bioinformatics analysis using LIHC tissue samples and cell lines22. Li et al. concluded that the predictive signature could independently predict the prognosis of LIHC patients and provide treatment references for LIHC patients22. However, the role of DRLs in the development and progression of LIHC has never been investigated. Therefore, we estimated a risk signature based on DRLs to predict the survival of LIHC patients. Ten DRGs and 1354 DRLs were co-expressed from the transcriptome data of 374 LIHC samples from the TCGA database. Among them, 160 DRLs were significantly associated with prognosis of the LIHC patients. Subsequently, two DRLs (MKLN1-AS and TMCC1-AS1) were identified by LASSO and multivariate cox regression analysis. This is the first investigation of the role of lncRNA signatures associated with disulfidptosis in the prediction of LIHC prognosis and immune landscape, as well as drug sensitivity. Previously, MKLN1-AS and TMCC1-AS1 were identified as lncRNAs related to cuproptosis22,66, pyroptosis67, autophagy68, ferroptosis and necroptosis27,69 in LIHC. For example, Zhang et al. used lncRNA expression data from TCGA to establish that five necroptosis-related lncRNAs (KDM4A-AS1, ZFPM2-AS1, AC099850.3, MKLN1-AS, and BACE1-AS) were well associated with patient prognosis, clinicopathological features, and immunotherapy effects69. More recently, a six-lncRNA prognosis model (AC012073.1, AL031985.3, LINC01060, MKLN1-AS, MSC-AS1, and TMCC1-AS1) was construed by Zhu et al.70, and this was with the potential prognosis and immunotherapy response predictive value of LIHC. On the other hand, the functional role of MKLN1AS or TMCC1‑AS1 in LIHC progression has also been reported. Gao et al. found that MKLN1AS promoted LIHC progression by acting as a molecular sponge for miR6543p to promote hepatoma‑derived growth factor (HDGF) expression71. The researchers concluded that the MKLN1AS/miR654/3p/HDGF axis might serve as a potential target for LIHC diagnosis, prognosis, and therapy71. Another study revealed that MKLN1-AS overexpression contributes to stabilizing Yes-associated transcriptional regulator 1 (YAP1) mRNA, which is essential for MKLN1-AS to be carcinogenic72. Chen et al. reported that TMCC1-AS1 were significantly overexpressed in LIHC tissues and cell lines and promoted the proliferation, migration, invasion, suggesting TMCC1-AS1 could serve as a prognostic biomarker for LIHC patients73. Collectively, both of these critical lncRNAs have a role in LIHC development and progression. According to the findings of Liu et al.10, high expression of SLC7A11 in kidney cancer cells speeds up the depletion of NADPH in the cytoplasm under glucose starvation. This leads to an accumulation of unreducible disulfides, inducing disulfide stress and eventually disulfidptosis. As one of the DRGs, SLC7A11 upregulating plays an essential role in promoting disulfidptosis and tumor development7,74, which is harmful for cancer patients. This observation implies a positive relationship between DRGs and the two target lncRNAs. This was the first study to demonstrate that these two lncRNAs were identified to be involved in the development of LIHC disulfidptosis.
There are some limitations in this study. First, our results were obtained from the TCGA database, and we were unable to extract external validation from the GEO database due to the lack of relevant clinical data for the two lncRNAs. Second, the findings of our research depended mostly on integrative bioinformatics and preliminary qRT-PCR results. More basic research is needed to confirm the biological function of our identified lncRNAs in disulfidptosis.
In conclusion, we succeeded in firstly constructing a signature of disulfidptosis-related lncRNAs that predicts prognosis of LIHC patients. Besides, this signature can be a valuable tool for a deeper understanding of tumor immune microenvironment and the immunotherapy response of LIHC, providing us new insights into the therapeutic strategies of LIHC patients.
Data availability
The datasets used and/or analyzed during the current study are available from the corresponding author on reasonable request.
References
Sung, H. et al. Global cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 71(3), 209–249. https://doi.org/10.3322/caac.21660 (2021).
Rumgay, H. et al. Global burden of primary liver cancer in 2020 and predictions to 2040. J. Hepatol. 77(6), 1598–1606. https://doi.org/10.1016/j.jhep.2022.08.021 (2022).
Llovet, J. M. et al. Hepatocellular carcinoma. Nat. Rev. Dis. Primers 2, 16018. https://doi.org/10.1038/nrdp.2016.18 (2016).
McGlynn, K. A., Petrick, J. L. & El-Serag, H. B. Epidemiology of hepatocellular carcinoma. Hepatology 73(Suppl 1), 4–13. https://doi.org/10.1002/hep.31288 (2021).
Grandhi, M. S. et al. Hepatocellular carcinoma: From diagnosis to treatment. Surg. Oncol. 25(2), 74–85. https://doi.org/10.1016/j.suronc.2016.03.002 (2016).
Galluzzi, L. et al. Molecular mechanisms of cell death: Recommendations of the Nomenclature Committee on cell death 2018. Cell Death Differ. 25(3), 486–541. https://doi.org/10.1038/s41418-017-0012-4 (2018).
Zheng, P., Zhou, C., Ding, Y. & Duan, S. Disulfidptosis: A new target for metabolic cancer therapy. J. Exp. Clin. Cancer Res. 42(1), 103. https://doi.org/10.1186/s13046-023-02675-4 (2023).
Tsvetkov, P. et al. Copper induces cell death by targeting lipoylated TCA cycle proteins. Science 375(6586), 1254–1261. https://doi.org/10.1126/science.abf0529 (2022).
Stockwell, B. R. Ferroptosis turns 10: Emerging mechanisms, physiological functions, and therapeutic applications. Cell 185(14), 2401–2421. https://doi.org/10.1016/j.cell.2022.06.003 (2022).
Liu, X. et al. Actin cytoskeleton vulnerability to disulfide stress mediates disulfidptosis. Nat. Cell Biol. 25(3), 404–414. https://doi.org/10.1038/s41556-023-01091-2 (2023).
Yang, L. et al. Based on disulfidptosis, revealing the prognostic and immunological characteristics of renal cell carcinoma with tumor thrombus of vena cava and identifying potential therapeutic target AJAP1. J. Cancer Res. Clin. Oncol. https://doi.org/10.1007/s00432-023-04877-x (2023).
Zheng, T., Liu, Q., Xing, F., Zeng, C. & Wang, W. Disulfidptosis: A new form of programmed cell death. J. Exp. Clin. Cancer Res. 42(1), 137. https://doi.org/10.1186/s13046-023-02712-2 (2023).
Liu, X. et al. Cystine transporter regulation of pentose phosphate pathway dependency and disulfide stress exposes a targetable metabolic vulnerability in cancer. Nat. Cell Biol. 22(4), 476–486. https://doi.org/10.1038/s41556-020-0496-x (2020).
Zhao, S. et al. Crosstalk of disulfidptosis-related subtypes, establishment of a prognostic signature and immune infiltration characteristics in bladder cancer based on a machine learning survival framework. Front. Endocrinol. (Lausanne) 14, 1180404. https://doi.org/10.3389/fendo.2023.1180404 (2023).
Wang, T. et al. Disulfidptosis classification of hepatocellular carcinoma reveals correlation with clinical prognosis and immune profile. Int. Immunopharmacol. 120, 110368. https://doi.org/10.1016/j.intimp.2023.110368 (2023).
Chen, H., Yang, W., Li, Y., Ma, L. & Ji, Z. Leveraging a disulfidptosis-based signature to improve the survival and drug sensitivity of bladder cancer patients. Front. Immunol. 14, 1198878. https://doi.org/10.3389/fimmu.2023.1198878 (2023).
Feng, Z. et al. Identification a unique disulfidptosis classification regarding prognosis and immune landscapes in thyroid carcinoma and providing therapeutic strategies. J. Cancer Res. Clin. Oncol. https://doi.org/10.1007/s00432-023-05006-4 (2023).
Xue, W. et al. Disulfidptosis-associated long non-coding RNA signature predicts the prognosis, tumor microenvironment, and immunotherapy and chemotherapy options in colon adenocarcinoma. Cancer Cell Int. 23(1), 218. https://doi.org/10.1186/s12935-023-03065-8 (2023).
Evans, J. R., Feng, F. Y. & Chinnaiyan, A. M. The bright side of dark matter: lncRNAs in cancer. J. Clin. Investig. 126(8), 2775–2782. https://doi.org/10.1172/jci84421 (2016).
Guo, D. F., Fan, L. W., Zeng, H. H., Huang, C. B. & Wu, X. H. Establishment and validation of a cuproptosis-related lncRNA signature that predicts prognosis and potential targeted therapy in hepatocellular carcinoma. Biotechnol. Genet. Eng. Rev. 1, 1–26. https://doi.org/10.1080/02648725.2023.2190640 (2023).
Liu, Y. & Jiang, J. A novel cuproptosis-related lncRNA signature predicts the prognosis and immunotherapy for hepatocellular carcinoma. Cancer Biomark. 37(1), 13–26. https://doi.org/10.3233/cbm-220259 (2023).
Li, D. et al. Comprehensive analysis of cuproptosis-related lncRNAs for prognostic significance and immune microenvironment characterization in hepatocellular carcinoma. Front. Immunol. 13, 991604. https://doi.org/10.3389/fimmu.2022.991604 (2022).
Li, G. et al. A novel ferroptosis-related long non-coding RNA prognostic signature correlates with genomic heterogeneity, immunosuppressive phenotype, and drug sensitivity in hepatocellular carcinoma. Front. Immunol. 13, 929089. https://doi.org/10.3389/fimmu.2022.929089 (2022).
Yang, X. et al. Ferroptosis-related long non-coding RNA signature predicts the prognosis of hepatocellular carcinoma. Aging 14(9), 4069–4084. https://doi.org/10.18632/aging.204073 (2022).
Zhang, Z. et al. Construction and validation of a ferroptosis-related lncRNA signature as a novel biomarker for prognosis, immunotherapy and targeted therapy in hepatocellular carcinoma. Front. Cell Dev. Biol. 10, 792676. https://doi.org/10.3389/fcell.2022.792676 (2022).
Wang, J., Shen, B., Liu, X. & Jiang, J. A novel necroptosis-related lncRNA signature predicts the prognosis and immune microenvironment of hepatocellular carcinoma. Front Genet. 13, 985191. https://doi.org/10.3389/fgene.2022.985191 (2022).
Wang, W. et al. Construction of a necroptosis-associated long non-coding RNA signature to predict prognosis and immune response in hepatocellular carcinoma. Front. Mol. Biosci. 9, 937979. https://doi.org/10.3389/fmolb.2022.937979 (2022).
Wu, T. et al. Screening prognostic markers for hepatocellular carcinoma based on pyroptosis-related lncRNA pairs. BMC Bioinform. 24(1), 176. https://doi.org/10.1186/s12859-023-05299-9 (2023).
Wu, Z. H., Li, Z. W., Yang, D. L. & Liu, J. Development and validation of a pyroptosis-related long non-coding RNA signature for hepatocellular carcinoma. Front. Cell Dev. Biol. 9, 713925. https://doi.org/10.3389/fcell.2021.713925 (2021).
Zhang, Z. et al. Prognosis and tumour immune microenvironment of patients with hepatocellular carcinoma by a novel pyroptosis-related lncRNA signature. Front. Immunol. 13, 836576. https://doi.org/10.3389/fimmu.2022.836576 (2022).
Zhao, S., Ye, Z. & Stanton, R. Misuse of RPKM or TPM normalization when comparing across samples and sequencing protocols. RNA 26(8), 903–909. https://doi.org/10.1261/rna.074922.120 (2020).
Parker, H. S. et al. Preserving biological heterogeneity with a permuted surrogate variable analysis for genomics batch correction. Bioinformatics 30(19), 2757–2763. https://doi.org/10.1093/bioinformatics/btu375 (2014).
Ritchie, M. E. et al. Limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 43(7), e47. https://doi.org/10.1093/nar/gkv007 (2015).
Liu, S., Xie, X., Lei, H., Zou, B. & Xie, L. Identification of key circRNAs/lncRNAs/miRNAs/mRNAs and pathways in preeclampsia using bioinformatics analysis. Med. Sci. Monit. 25, 1679–1693. https://doi.org/10.12659/MSM.912801 (2019).
Nukui, T. & Onogi, A. An R package for ensemble learning stacking. Bioinform. Adv. 3(1), 139. https://doi.org/10.1093/bioadv/vbad139 (2023).
Tibshirani, R. The Lasso method for variable selection in the Cox model. Stat. Med. 16(4), 385–395 (1997).
Simon, N., Friedman, J., Hastie, T. & Tibshirani, R. Regularization paths for Cox’s proportional hazards model via coordinate descent. J. Stat. Softw. 39(5), 1–13. https://doi.org/10.18637/jss.v039.i05 (2011).
Pang, Y. et al. Cuproptosis-related LncRNA-based prediction of the prognosis and immunotherapy response in papillary renal cell carcinoma. Int. J. Mol. Sci. 24(2), 1464. https://doi.org/10.3390/ijms24021464 (2023).
Blanche, P., Dartigues, J. F. & Jacqmin-Gadda, H. Estimating and comparing time-dependent areas under receiver operating characteristic curves for censored event times with competing risks. Stat. Med. 32(30), 5381–5397. https://doi.org/10.1002/sim.5958 (2013).
Ligges, U. & Maechler, M. Scatterplot3d—An R package for visualizing multivariate data. J. Stat. Softw. 8(11), 1–20. https://doi.org/10.18637/jss.v008.i11 (2003).
Zhang, Z. & Kattan, M. W. Drawing nomograms with R: Applications to categorical outcome and survival data. Ann. Transl. Med. 5(10), 211. https://doi.org/10.21037/atm.2017.04.01 (2017).
Kalasekar, S. M., Garrido-Laguna, I. & Evason, K. J. Immune checkpoint inhibitors in combinations for hepatocellular carcinoma. Hepatology 73(6), 2591–2593. https://doi.org/10.1002/hep.31706 (2021).
Subramanian, A. et al. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. U.S.A. 102(43), 15545–15550. https://doi.org/10.1073/pnas.0506580102 (2005).
Gu, Z., Gu, L., Eils, R., Schlesner, M. & Brors, B. Circlize implements and enhances circular visualization in R. Bioinformatics 30(19), 2811–2812. https://doi.org/10.1093/bioinformatics/btu393 (2014).
Gu, Z., Eils, R. & Schlesner, M. Complex heatmaps reveal patterns and correlations in multidimensional genomic data. Bioinformatics 32(18), 2847–2849. https://doi.org/10.1093/bioinformatics/btw313 (2016).
Wu, T. et al. clusterProfiler 4.0: A universal enrichment tool for interpreting omics data. Innovation (Camb.) 2(3), 100141. https://doi.org/10.1016/j.xinn.2021.100141 (2021).
Yu, G., Wang, L. G., Yan, G. R. & He, Q. Y. DOSE: An R/bioconductor package for disease ontology semantic and enrichment analysis. Bioinformatics 31(4), 608–609. https://doi.org/10.1093/bioinformatics/btu684 (2015).
Hänzelmann, S., Castelo, R. & Guinney, J. GSVA: Gene set variation analysis for microarray and RNA-seq data. BMC Bioinform. 14, 7. https://doi.org/10.1186/1471-2105-14-7 (2013).
Jiang, P. et al. Signatures of T cell dysfunction and exclusion predict cancer immunotherapy response. Nat. Med. 24(10), 1550–1558. https://doi.org/10.1038/s41591-018-0136-1 (2018).
Geeleher, P., Cox, N. & Huang, R. S. pRRophetic: An R package for prediction of clinical chemotherapeutic response from tumor gene expression levels. PLoS ONE 9(9), e107468. https://doi.org/10.1371/journal.pone.0107468 (2014).
Tang, Z., Kang, B., Li, C., Chen, T. & Zhang, Z. GEPIA2: An enhanced web server for large-scale expression profiling and interactive analysis. Nucleic Acids Res. 47(W1), W556–W560. https://doi.org/10.1093/nar/gkz430 (2019).
Koppula, P., Zhuang, L. & Gan, B. Cystine transporter SLC7A11/xCT in cancer: Ferroptosis, nutrient dependency, and cancer therapy. Protein Cell 12(8), 599–620. https://doi.org/10.1007/s13238-020-00789-5 (2021).
Yan, Y. et al. SLC7A11 expression level dictates differential responses to oxidative stress in cancer cells. Nat. Commun. 14(1), 3673. https://doi.org/10.1038/s41467-023-39401-9 (2023).
Chen, Y. et al. Effect of infiltrating immune cells in tumor microenvironment on metastasis of hepatocellular carcinoma. Cell Oncol. (Dordr.). https://doi.org/10.1007/s13402-023-00841-6 (2023).
Ribeiro Franco, P. I., Rodrigues, A. P., de Menezes, L. B. & Pacheco Miguel, M. Tumor microenvironment components: Allies of cancer progression. Pathol. Res. Pract. 216(1), 152729. https://doi.org/10.1016/j.prp.2019.152729 (2020).
Sugihara, H. et al. Identification of miR-30e* regulation of Bmi1 expression mediated by tumor-associated macrophages in gastrointestinal cancer. PLoS ONE 8(11), e81839. https://doi.org/10.1371/journal.pone.0081839 (2013).
Liu, Y. & Cao, X. The origin and function of tumor-associated macrophages. Cell Mol. Immunol. 12(1), 1–4. https://doi.org/10.1038/cmi.2014.83 (2015).
Shan, F., Somasundaram, A., Bruno, T. C., Workman, C. J. & Vignali, D. A. A. Therapeutic targeting of regulatory T cells in cancer. Trends Cancer 8(11), 944–961. https://doi.org/10.1016/j.trecan.2022.06.008 (2022).
Yang, L., Zhang, W. & Yan, Y. Identification and characterization of a novel molecular classification based on disulfidptosis-related genes to predict prognosis and immunotherapy efficacy in hepatocellular carcinoma. Aging 15, 204809. https://doi.org/10.18632/aging.204809 (2023).
Li, X. M. et al. Identification of disulfidptosis-related genes with immune infiltration in hepatocellular carcinoma. Heliyon 9(8), e18436. https://doi.org/10.1016/j.heliyon.2023.e18436 (2023).
Shalapour, S. et al. Inflammation-induced IgA+ cells dismantle anti-liver cancer immunity. Nature 551(7680), 340–345. https://doi.org/10.1038/nature24302 (2017).
Liu, R. X. et al. Chemokine (C-X-C motif) receptor 3-positive B cells link interleukin-17 inflammation to protumorigenic macrophage polarization in human hepatocellular carcinoma. Hepatology 62(6), 1779–1790. https://doi.org/10.1002/hep.28020 (2015).
Xia, A. et al. The cancer-testis lncRNA LINC01977 promotes HCC progression by interacting with RBM39 to prevent Notch2 ubiquitination. Cell Death Discov. 9(1), 169. https://doi.org/10.1038/s41420-023-01459-1 (2023).
Chi, X. et al. Upregulation of lncRNA PTOV1-AS1 in hepatocellular carcinoma contributes to disease progression and sorafenib resistance through regulating miR-505. J. Biochem. Mol. Toxicol. 37, e23437. https://doi.org/10.1002/jbt.23437 (2023).
Sun, L. et al. Long noncoding RNA LINC01124 activates hepatocellular carcinoma cell proliferation, migration, and invasion by absorbing microRNA-1247-5p and overexpressing FOXO3. Oncol. Res. 29(3), 175–187. https://doi.org/10.32604/or.2022.03550 (2021).
Yuan, W. et al. Identification of a cuproptosis and copper metabolism gene-related lncRNAs prognostic signature associated with clinical and immunological characteristics of hepatocellular carcinoma. Front. Oncol. 13, 1153353. https://doi.org/10.3389/fonc.2023.1153353 (2023).
Wang, T. et al. The pyroptosis-related long noncoding RNA signature predicts prognosis and indicates immunotherapeutic efficiency in hepatocellular carcinoma. Front. Cell Dev. Biol. 10, 779269. https://doi.org/10.3389/fcell.2022.779269 (2022).
Deng, X. et al. Identification of a five-autophagy-related-lncRNA signature as a novel prognostic biomarker for hepatocellular carcinoma. Front. Mol. Biosci. 7, 611626. https://doi.org/10.3389/fmolb.2020.611626 (2020).
Zhao, L., You, Z., Bai, Z. & Xie, J. Machine learning-based construction of a ferroptosis and necroptosis associated lncRNA signature for predicting prognosis and immunotherapy response in hepatocellular cancer. Front. Oncol. 13, 1171878. https://doi.org/10.3389/fonc.2023.1171878 (2023).
Zhu, L. et al. Identification of a CD4+ conventional T cells-related lncRNAs signature associated with hepatocellular carcinoma prognosis, therapy, and tumor microenvironment. Front. Immunol. 13, 1111246. https://doi.org/10.3389/fimmu.2022.1111246 (2022).
Gao, W., Chen, X., Chi, W. & Xue, M. Long non-coding RNA MKLN1-AS aggravates hepatocellular carcinoma progression by functioning as a molecular sponge for miR-654-3p, thereby promoting hepatoma-derived growth factor expression. Int. J. Mol. Med. 46(5), 1743–1754. https://doi.org/10.3892/ijmm.2020.4722 (2020).
Guo, C. et al. Long non-coding RNA muskelin 1 antisense RNA (MKLN1-AS) is a potential diagnostic and prognostic biomarker and therapeutic target for hepatocellular carcinoma. Exp. Mol. Pathol. 120, 104638. https://doi.org/10.1016/j.yexmp.2021.104638 (2021).
Chen, C., Su, N., Li, G., Shen, Y. & Duan, X. Long non-coding RNA TMCC1-AS1 predicts poor prognosis and accelerates epithelial–mesenchymal transition in liver cancer. Oncol. Lett. 22(5), 13034. https://doi.org/10.3892/ol.2021.13034 (2021).
Kobayashi, H., Yoshimoto, C., Matsubara, S., Shigetomi, H. & Imanaka, S. A comprehensive overview of recent developments on the mechanisms and pathways of ferroptosis in cancer: The potential implications for therapeutic strategies in ovarian cancer. Cancer Drug Resist. 6(3), 547–566. https://doi.org/10.20517/cdr.2023.49 (2023).
Acknowledgements
Thanks to TCGA databases and GEPIA2 for making the data available.
Funding
This research was supported by National Natural Science Foundation of China (81960390) and Key Laboratory of Early Prevention and Treatment for Regional High-Incidence-Tumor, Guangxi Medical University, Ministry of Education (GKE2019-05, GKE-ZZ202132).
Author information
Authors and Affiliations
Contributions
Y.L. wrote, drafted, reviewed and edited interpretation of results and wrote the paper; Y.L. and J.M. performed the experiment and analyzed the data; X.R., F.W., and J.M. collected the tissue sample and performed the experiment. F.Z. for laboratory assistance. X.Q. designed the research study and provided clinical advice. All authors contributed to this article and read and approved the final manuscript. All authors had full access to all of the data in the study and had final responsibility for the decision to submit for publication. All participants gave both their informed consent for study participation and for the publication of their images/data in an online open-access publication.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Liu, Y., Meng, J., Ruan, X. et al. A disulfidptosis-related lncRNAs signature in hepatocellular carcinoma: prognostic prediction, tumor immune microenvironment and drug susceptibility. Sci Rep 14, 746 (2024). https://doi.org/10.1038/s41598-024-51459-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-024-51459-z
- Springer Nature Limited