
Abstract
Allergens originated from Salsola kali (Russian thistle) pollen grains are one of the most important sources of aeroallergens causing pollinosis in desert and semi-desert regions. T-cell epitope-based vaccines (TEV) are more effective among different therapeutic approaches developed to alleviate allergic diseases. The physicochemical properties, and B as well as T cell epitopes of Sal k 1 (a major allergen of S. kali) were predicted using immunoinformatic tools. A TEV was constructed using the linkers EAAAK, GPGPG and the most suitable CD4+ T cell epitopes. RS04 adjuvant was added as a TLR4 agonist to the amino (N) and carboxyl (C) terminus of the TEV protein. The secondary and tertiary structures, solubility, allergenicity, toxicity, stability, physicochemical properties, docking with immune receptors, BLASTp against the human and microbiota proteomes, and in silico cloning of the designed TEV were assessed using immunoinformatic analyses. Two CD4+ T cell epitopes of Sal k1 that had high affinity with different alleles of MHC-II were selected and used in the TEV. The molecular docking of the TEV with HLADRB1, and TLR4 showed TEV strong interactions and stable binding pose to these receptors. Moreover, the codon optimized TEV sequence was cloned between NcoI and XhoI restriction sites of pET-28a(+) expression plasmid. The designed TEV can be used as a promising candidate in allergen-specific immunotherapy against S. kali. Nonetheless, effectiveness of this vaccine should be validated through immunological bioassays.
Similar content being viewed by others


Introduction
Allergic diseases are among prevalent global health problems1. More exactly, more than one-third of the world's population experiences type-I allergic reactions2. Due to the complex pathophysiology of allergic diseases, these diseases cannot affect all people3. In other words, various factors such as genetic predisposition, climate change, industrialization, and route of exposure are effective in causing these diseases4. Aeroallergens, especially pollens, are common causes of respiratory allergies such as allergic rhinitis and asthma5. Allergens originated from Salsola kali (Russian thistle) pollen grains are one of the most important sources of aeroallergens causing pollinosis in desert and semi-desert regions6,7. Seven allergens (Sal k 1 to Sal k 7) have been identified from Salsola kali and recorded in the allergen nomenclature database (http://www.allergen.org) so far. It has been reported that Sal k 1 with multiple isoforms, which is the main allergen of S. kali, causes more than 80% of sensitization in S. kali allergic patients8,9.
Allergen specific immunotherapy (AIT) with natural allergen sources is still considered as a successful therapeutic procedure for allergies to common allergen sources such as grass pollens, house dust mites, and bee venom10. There are certain challenges regarding the standardization of allergen sources that have adverse side effects11,12. Moreover, recombinant allergens are increasingly used for diagnostic and therapeutic purposes, and innovative strategies for AIT such as DNA-based vaccines or T cell or B cell targeted therapies10,13,14 have been developed. Thus, determination of the molecular factors effective in immunogenicity and allergenicity of allergens has drawn the attention of researchers. Application of T-cell epitope-based vaccines (TEV) has had successful trials as a safe method in connection with Japanese cedar pollens and grass pollens, honeybee venom, and house dust mite15,16. In this therapy, allergen-derived soluble peptides with immunodominant T-cell epitopes are utilized to treat allergic patients17. It has been reported that the use of TEV is more effective than other therapeutic approaches for the treatment of allergic diseases due to the effective reduction of adverse effects and their inability to bind to IgE- FcεRI on effector cells, mainly because of the small peptide size of T-cell epitopes (10–17 amino acids)18,19. Thus, in the present study, we intended to predict B and helper T lymphocyte (HTL) epitopes of Sal k 1 allergen by an in-silico approach. After identifying linear B and HTL epitopes by immunoinformatic tools, the best peptides containing HTL epitopes and lacking any B cell epitopes was chosen and used in designing the TEV.
Materials and methods
The protein sequence retrieval and phylogenetic analysis
The protein sequences of four isoforms or variants of Sal k 1 with accession numbers P83181, AAT99258, AAX11262, and AAX11262 were obtained from the International Union of Immunological Societies nomenclature database (IUIS) (http://www.allergen.org) and the protein database of the National Center for Biotechnology Information (NCBI) (https://www.ncbi.nlm.nih.gov/). The obtained protein sequences were aligned using CLC Genomics Workbench 3.0 software, and the consensus protein sequence was employed as the BLASTp query to find homologous sequences. Investigation of the family classification of Sal k 1 was carried out using Pfam v29.0 and InterPro v56.020. The TMHMM server 2.0 was used to predict the transmembrane helices of the Sal k 121. The phylogenetic tree was made according to the Jones–Taylor–Thornton (JTT) model using the maximum likelihood (ML) approach in MEGA-7 software22. The reliability of the phylogenetic tree of the Sal k 1 amino acid sequence was computed using the bootstrap method with 1000 replications23.
Physicochemical analysis and secondary structure prediction
The ExPASy ProtParam tool with default parameters was used to calculate the physicochemical characteristics of Sal k 1 protein24. The presence of functional motifs of the Sal k 1 was analyzed using the Prosite database25. Also, by NETPhos v 3.1 phosphorylation motifs were analyzed26. The secondary structure properties of Sal k 1 was predicted using PSIPRED and NetSurfP ver. 1.127,28.
The linear B lymphocyte epitopes prediction
The B -lymphocyte epitopes in the Sal k 1 protein were predicted using immune epitope database (IEDB) and BCepred tools. These two servers identify B lymphocyte epitopes by the physicochemical characteristics of the amino acid sequence29. Epitopes that have the features of accessibility, flexibility, antigenicity and hydrophobicity were selected as the main epitopes of B lymphocytes29. The epitopes confirmed by both IEDB and BCepred tools were chosen as the final B lymphocytes epitopes.
The helper T lymphocyte (HTL) epitopes prediction
The prediction of 15 amino acids long HTL epitopes was performed using the MHC II prediction tool in the IEDB. Epitopes characterized by low percentile rank and SMM less than IC50 were considered as high affinity peptides for binding to the MHC-II molecule30.
Population coverage
Population coverage for the predicted HTL epitopes is determined for world population using the population coverage calculation tool in IEDB. This tool calculates the portion of individuals predicted to react to a given set of epitopes with recognized MHC limitations31.
Molecular docking of the predicted epitopes
The prediction of the 3D structure of the selected epitopes of HTL was carried out using the peptide structure prediction tool (PEP-FOLD 3)32. The protein–protein docking between the crystalized structure of the most abundant MHC-II allele reacting with Sal k 1 epitopes was performed in ClusPro 2.0 tool33. Subsequently, this molecular docking was visualized and analyzed using the PyMOL software. For this purpose, the 3D crystal structure of the most abundant MHC-II allele was retrieved from RCSPDB. Moreover, for proper docking, these 3D crystal structures were processed using UCSF Chimera software. Then, non-amino acid molecules such as ligand, ions, and solvent water were removed from it, and subsequently polar hydrogen atoms were added to it34,35.
Construction of the T-cell epitope-based vaccine
In order to construct a T-cell epitope-based vaccine (TEV), the predicted HTL epitopes were fused together and to a toll-like receptor 4 (TLR4) agonist using flexible linkers. The HTL epitopes were fused using the GPGPG flexible linker36. A synthetic TLR4 adjuvant RS04 (Sequence: GLQQVLL) was added to the N and C-terminus of the vaccine construct using an EAAAK linker37. In order to find out the epitopes' identity and similarity of the constructed vaccine to proteins in humans, they were aligned against the human proteome and microbiome using the NCBI BLASTp tool38. The PAM30 scoring matrix and an expectation value (E-value) of 0.0001 were considered as BLASTp parameters.
Prediction of allergenicity, toxicity, and antigenicity of the TEV
The antigenicity of the TEV sequence was predicted using the ANTIGENpro server39. The Algpred tool was utilized to find the allergenicity of the TEV sequence40. Furthermore, the ToxinPred tool was used to check the toxicity of the TEV41.
Physicochemical analysis of the TEV
The Expasy ProtParam tool was used to calculate the physicochemical characteristics of the designed TEV24. Molecular weight, theoretical isoelectric point (pI), aliphatic index, instability index, grand average of hydropathicity (GRAVY), and in vitro as well as in vivo half-lives were among the examined properties. The SOLPro tool was used to evaluate the vaccine's solubility upon overexpression in Escherichia coli (E. coli)42.
Prediction of the TEV's secondary structure
The TEV's secondary structure was anticipated using PSIPRED 4.0. This program was used to assess the vaccine's secondary structure for the presence of coils, -helixes, -sheets, and disordered domains43.
Codon optimization and in silico cloning preparation
The reverse translation and codon optimization of the TEV was carried out using the Java Codon Adaptation Tool (JCat). This step was evaluated using the results of the percentage of GC content and the Codon Adaptation Index (CAI), and it was carried out for expression in an E. coli O6:K15:H31 (strain 536/UPEC). CAI reveals the propensity of an organism to express a heterologous gene. Successful optimization is defined as a CAI above 0.80 and a GC content between 30 and 70%, respectively44. The NcoI and XhoI restriction sites were used to insert the enhanced nucleotide sequence's N- and C-termini, respectively. The completed sequence was added to the pET28a (+) vector and evaluated by SnapGene for viability according to predictions.
Tertiary structure prediction, refinement, and validation of the TEV
The TEV 3D model was constructed using the I-TASSER tool. The 3D model's accuracy was determined according to the confidence score (C-score). The C-score typically falls between -5 and 2, with a higher value denoting higher quality45. The constructed TEV model was refined using the Galaxy Refine tool to improve the quality of the 3D structure. This server reconstructs the side chains and repacks them using molecular dynamics modelling46.
The 3D structure of the predicted vaccine construct was verified using the PROCHECK service. The Ramachandran plot, which displays the fraction of amino acids in preferred, allowed, and banned zones based on the dihedral angles phi (φ) and psi (ψ) of each amino acid is the output of this server47. For additional validation, the ProSA online tool and ERRAT were used48,49.
Molecular docking of the TEV with TLR4 and HLA
The ClusPro 2.0 tool was used to evaluate the potential of TEV docking with MHC and TLR immune receptors. The PDB files of MHC-II (PDB Id: 1H15 for HLA-DRB1), and TLR4 (PDB ID: 3FXI) were downloaded from the PDB database50. Furthermore, before docking between TLR4 and TEV, the 3D crystal structure of TLR4 was processed using the UCSF Chimera software. Then, non-amino acid molecules such as ligand, ions, and solvent water were removed from it, and subsequently polar hydrogen atoms were added to it34,35. The docked complexes presented by ClusPro 2.0 server with the lowest energy were chosen and then checked for protein–protein interaction using PyMOL software.
Molecular dynamic simulation
The molecular dynamic simulation was carried out using the iMODS tool to examine the stability and physical movements of the TLR4-TEV and HLADRB1-TEV docked complexes51. This tool can predict the collective motions of proteins using normal mode analysis (NMA) in internal (dihedral) coordinates. The iMODS tool calculates the vaccine receptor complex’s deformability, eigenvalues, variance, covariance map, B-factor, and elastic network51,52.
Ethical approval
This article does not contain any studies with human participants or animals performed by any of the authors.
Results
The protein sequence retrieval and analysis
The Sal k 1 allergen of S. Kali consists of 399 amino acids as follows: QPIPPNPAELESWFQGAVKPVSEQKGLEPSVVQAESGGVETIEVRQDGSGKFKTISDAVKHVKVGNTKRVIITIGPGEYREKVKIERLHPYITLYGIDPKNRPTITFAGTAAEFGTVDSATLIVESDYFVGANLIVSNSAPRPDGKRKGAQASALRISGDRAAFYNCKFTGFQDTVCDDKGNHLFKDCYIEGTVDFIFGEARSLYLNTELHVVPGDPMAMITAHARKNADGVGGYSFVHCKVTGTGGTALLGRAWFEAARVVFSYCNLSDAVKPEGWSDNNKPAAQKTIFFGEYKNTGPGAAADKRVPYTKQLTEADAKTFTSLEYIEAAKWLLPPPKV.
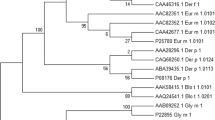
The results of Pfam v29.0 and InterPro v56.0 showed that Sal k 1 belongs to the pectin esterase family. Moreover, the results of TMHMM Server 2.0 indicated that Sal k 1 protein was located outside of the cell membrane and had no transmembrane helices. Phylogenetic analysis was performed to determine the relationships between Sal k 1 and its homologous sequences. The evolutionary tree inferred by the maximum likelihood (ML) method has been indicated in Fig. 1.

The phylogenetic relationship of the Sal k 1 allergen amino acid sequence was constructed by the ML method with 1000 bootstrap replicates.
Physicochemical and secondary structure analyses
Investigation of the physicochemical properties of Sal k 1 using the ExPASy tool indicated that its molecular weight, hypothetical pI, instability index, aliphatic index, grand average of hydropathicity (GRAVY), and the total positive and negative charges were 36.79 kDa, 7.75, 24.08, 74.54, − 0.313, 40 and 39, respectively. The total number of atoms was 5163. Moreover, Sal k 1 had a functional pectin esterase motif PS00503 in the 190–199 (IEGTVDFIFG) position. Phosphorylation sites, including five Ser (22, 49, 158, 264 and 323), four Thr (41, 54, 67 and 175), and four Tyr (128, 189, 294, and 326) residues were predicted as it has been shown in Fig. 2.

Sequence and secondary structure analyses of Sal k 1.
Furthermore, the secondary structure prediction of Sal k 1 with PSIPRED identified eight α-helices (10.32%), twenty-one β- sheets (33.92%), and thirty-six random coil (55.65%) in Sal k 1. Alternatively, NetSurfP v2 predicted eight helixes (10.32%), twenty-two β-sheets (33.62%), and thirty-one random coils (56.05%), in Sal k 1 as it has been shown in Fig. 2.
In this investigation, random coils exhibited the highest number of residues followed by β-sheets and α-helixes. The lowest and the highest numbers of residues were found in the α-helixes and the random coils of Sal k 1, respectively (Fig. 2).
Prediction of B lymphocyte epitopes
The amino acid sequence of Sal k 1 protein was subjected to Karplus & Schulz Flexibility, Emini surface accessibility, Chou & Fasman BetaTurn, Parker Hydrophilicity, Kolaskar and Tongaonkar antigenicity methods in Bcepred and IEDB tools to predict the binding to B cell (Table 1). From all the predicted B-cell epitopes, only three epitopes, i.e. GAVKPVSEQKGLEPS, VKPEGWSDNNKPAAQ, and AADKRVPYTKQ, were selected for further processing (Table 2). The predicted linear B-cell epitopes and their antigenicity, flexibility, degrees of accessibility, hydrophilicity, and the beta-turn prediction score have been summarized in Table 2.
Prediction of HTL epitopes and population coverage
The binding epitopes to specific MHC-II molecules in the protein sequence of Sal k 1 were predicted using the IEDB database. On the basis of low IC50 values, low percentile rank, and reactive with diverse MHC-II alleles only 9 potential peptides were selected for further processing. Peptides with the ability to bind to a larger number of alleles are identified as the most suitable cases due to their potential to elicit a potent protective response. The list of the most promising HTL epitopes and their correspondent binding MHC II alleles have been shown in Table 3. In order to design the TEV, population coverage was analyzed for epitopes binding to diverse alleles of MHC-II. High population coverage of the vaccine is important because many people can benefit from it with just one proper vaccine. The result of combining these methods with MHC- II limitation for the whole world, European, and Southwest Asia population with the selected MHC- II alleles has been indicated in Table 3.
Docking analysis of HTL epitopes
Using the PEP-FOLD 3 server, the 3D structures of the selected main epitopes of HTL (GTVDFIFGEARSLYL, and LGRAWFEAARVVFSY) were predicted (Fig. 3). Ten models were predicted for each epitope, and the model with the lowest energy was selected for subsequent investigation. The chosen structures were docked with the crystal structure of the human HLA.DRB1 (1H15) using ClusPro v2.0 and PyMOL tools (Fig. 3).

(A, B) docking of the predicted HTL epitopes with the HLA DRB1 (PDB: 1H15) molecule. Amino acid residues of HLADRB1 that interact with ligands have been shown in red color.
Construction of the TEV
Epitopes three (GTVDFIFGEARSLYL) and six (LGRAWFEAARVVFSY) (Tabel 3) were selected from the predicted epitopes for HTL. Subsequently, they were connected together by the GPGPG linker. A 35 amino acids sequence was produced after epitope fusion. The adjuvant sequence with a length of 7 amino acids (LPS mimic peptide RS04: GLQQVLL) was fused to the N and C terminals of the vaccine sequence by an EAAAK linker. The final designed vaccine construct was comprised of 59 amino acids (Fig. 4). Based on the result obtained from BLASTp, the vaccine construct was not identical with the human proteome and human microbiome proteins.

(A) Illustration of the 3D construct TEV of Sal k 1 in a space-filling model. (B) Sequence of the TEV of Sal k 1.
Antigenicity and safety of the constructed TEV
The probability of antigenicity predicted by ANTIGENpro was 0.108, which is indicative of its highly antigenic characteristics. Moreover, the vaccine models on both Algpred and ToxinPred tools were non-allergen and non-toxic, respectively.
Physicochemical properties of the TEV
The Expasy ProtParam tool was used to predict different physicochemical features of the designed TEV. The final composition of the TEV was composed of 59 amino acids. The theoretical pI, molecular weight, and instability index of the vaccine construct were calculated to be 6.31, 6.252 kDa, and 21.6, respectively. The half-life of the vaccine was estimated to be 30 h in mammalian reticulocytes in vitro, more than 20 h in the yeast in vivo, and more than 10 h in E. coli, in vivo. The GRAVY score of the vaccine was 0.405, and its aliphatic index was calculated to be 107.63. A solubility score of 0.916673 indicated that the TEV was soluble after the expression in E. coli.
Secondary structure properties of the TEV
The PSIPRED server predicted that the TEV contained 5.084% alpha-helix, 55.93% random coil, and 38.98% β-strand region (Fig. 5A).

(A) The secondary structure TEV of Sal k 1. (B) In silico cloning of the TEV sequence of Sal k 1 into the pET28a (+). The pink colored region shows the ORF and coding sequence of Sal k 1 in the pET28a (+) plasmid structure.
Codon optimization and in silico cloning
Reverse translation and codon optimization in the JCat tool produced a 177 bp nucleotide sequences for the constructed vaccine. The CAI values of the optimized sequences and GC contents were 1.0 and 55.36% respectively, indicating the desirable expression of the vaccines within E. coli. Subsequently, a recombinant plasmid was designed by inserting the vaccine sequence between NcoI and XhoI restriction sites in the pET28a (+) expression vector using the SnapGene software (Fig. 5B).
Modeling the 3D structure, refinement, and validation of the TEV
Five models of the 3D structure of the TEV construct were produced by the I-TASSER server using the threading templates (PDB: 1gq8A, 1gq8A, 3uw0A, 1xg2A, 1xg2A, 1gq8A, 3grhA, 3grhA, 3grhA, and 3grh). The calculated C-score values for models 1–5 were − 1.55, − 1.87, − 2.66, − 3.79, and − 4.67, respectively. The C-score is normally in the range of (− 5, 2), where high score shows high confidence in the predicted model. According to these criteria, the.
best model (C-score: − 1.55) was chosen for further investigation. Furthermore, this model had the TM score and root-mean-square deviation (RMSD) score of 0.52 ± 0.15 and 6.0 ± 3.7 Å, respectively (Fig. 6A). TM-score is a proposed scale for measuring the structural similarity between two structures53. The Galaxy Refne tool was used to improve the consistency of the vaccine model. Five models were generated from the refinement of the primary “crude” vaccine model. Model 4 was the most important model based on structure qualities, i.e. GDT-HA (0.9364), RMSD (0.491), and MolProbity (1.90), clash value 5.5), Poor rotamers (0.0) and Rama favored (87.7) (Fig. 6B). This model was chosen for further analysis. The Ramachandran plot analysis was used to validate the refined 3D structure of the vaccine construct. The results showed that 81.2% of the residues were in the best region, 12.5% were in the additional allowed region, 4.2% were in generously allowed regions, and 2.1% were in the disallowed region (Fig. 6C). The ProSA-web evaluated the Z score of the vaccine's model to be − 2.18 (Fig. 6D). Furthermore, the refinement model had an overall quality factor of 88.636% with ERRAT ((Fig. 6E).

The 3D structure, refinement and validation of the TEV. (A) The 3D structure of the TEV was predicted by I-TASSER server. (B) the 3D structure of the TEV after refinement using Galaxy Refine server. (C) ProSA-web, with a Z score of -2.18. (D) The Ramachardan plot indicated that 81.2% of the residues were in the most favored region, 12.5% were in the additional allowed region, 4.2% were in the generously allowed region, and 2.1% were in the disallowed region. (E) The ERRAT quality factor was 88.636%.
Molecular docking of the TEV with TLR4 and HLADR
Binding the TEV construct to the human immune receptors was evaluated using PyMOL and ClusPro 2.0 tools (Fig. 7A–F). The results showed that the TEV had a high affinity to bind TLR4 receptor with − 823.3 kcal/mol (Fig. 7A–C). Furthermore, it was shown that the TEV had a high affinity to bind in a grave of HLADRB1 with − 933.9 kcal/mol (Fig. 7D–F). The use of the PyMOL tool indicated that different residues of the TEV could interact with TLR4 and HLADRB1 (Table 4).

Molecular docking between the TLR4 (A–C), and HLADRB1 (D–F) with the TEV, T cell epitopes 3 and 6 interacting with HLADRB1 have been shown in cyan and yellow, respectively. Amino acid residues of HLADRB1 that interacted with TEV have been shown in red color.
Molecular dynamic (MD) simulation
We used MD simulations for the iMODS tool to examine the stability and motions of the docked complexes. Slow dynamics of the docked complexes were investigated, and their large-amplitude conformational variations were shown using normal mode analysis (NMA). Figures 8A and 9A show the NMA of the docked complexes of the TLR4-TEV and HLADRB1-TEV, respectively. The mobility features of the docked proteins are determined by the deformability and B-factor. The peaks that correlate to the regions in the proteins with deformability are shown by the deformability and B-factors of the TLR4-TEV and HLADRB1-TEV complexes, where the highest peaks reflect the regions of high deformability. The B-factor diagrams allow for a comparison of the complexes' NMA and PDB fields (Figs. 8C and 9C). The deformability and B‐factor of TLR4‐TEV and HLADRB1‐TEV complexes have been illustrated in Figs. 8 and 9, respectively. Eigenvalue, which is a crucial parameter of a stable structure, must be high to have a stable complex52. The eigenvalue rates for the simulated docking complexed TLR4-TEV and HLADRB1-TEV were 1.626365e-05 and 6.643221e-05 respectively. These rates are significantly higher for structural stability (Figs. 8D and 9D). The covariance matrix of the TLR4‐TEV and HLADRB1‐TEV complexes shows the correlations among residues in a complex. The white color in the matrix represents uncorrelated motions, while the red color shows a good correlation between the residues. Moreover, the blue color exhibits anticorrelations. The higher the correlation, the better the complex. The relationships between the atoms have been shown in the elastic maps of the docked proteins, where the stiffer regions have been exhibited by the darker grey areas (Figs. 8G and 9G).

The results of iMODS simulations of molecular dynamics (MD) for TLR4-TEV. (A) Areas of low mobility (blue) to high mobility (red) have been highlighted in the TLR4 and vaccine docking complex normal mode analysis (NMA). (B) deformability. (C) B-factor. (D) Eigenvalues (a lower value means easier deformation). (E) Variance (red color: individual variances and green color: cumulative variances). (F) Covariance map (red: correlated, white: uncorrelated, blue: anti-correlated). (G) Elastic network (darker regions mean stiffer regions).

The results of molecular dynamics simulations for HLADRB1-TEV in iMODS. (A) Areas of low mobility (blue) to high mobility (red) have been highlighted in the HLADRB1 and vaccine docking complex normal mode analysis (NMA). (B) deformability. (C) B-factor. (D) Eigenvalues (Low value indicates simpler deformation). (E) Variance (red color: individual variances and green color: cumulative variances). (F) Covariance map (red: correlated, white: uncorrelated, blue: anti-correlated). (G) Elastic network (darker regions mean stiffer regions).
Discussion
Over the past few decades, atopic illnesses in humans such as allergic rhinitis, asthma, and atopic dermatitis have become more common2,54. The increased ability to diagnose these diseases is a major concern for the global medical community55. Allergen specific immunotherapy (AIT) with an extract containing intact allergens is usually accompanied by a severe reaction in allergic patients, which can even cause anaphylactic shock56,57. Peptides prepared from allergenic substances such as tree pollens and venom insects have been used in human clinical studies and experimental animal models58. The use of T cell epitopes can cause tolerance to allergens and also prevent allergic reactions caused by IgE antibodies during conventional AIT59. Furthermore, it has been shown that when a high-dose specific allergen T cell epitope is used for treatment. It causes anergy and elimination in allergen-specific Th2 cells, and also induces Treg with IL-10 production60. Specific epitopes of CD4+ T lymphocytes are central to T cell-based epitope immunotherapy. Recognition of antigenic epitopes by T cells depend on their presentation along with the highly polymorphic molecules HLADR, HLADQ and HLADP. For this reason, epitopes in T cell-based epitope immunotherapy should be attached to different alleles of HLA molecules that can be used in different populations61. Based on this criterion, in this study, two T cell epitopes from regions 192–206 (GTVDFIFGEARSLYL) and 251–265 (LGRAWFEAARVVFSY) were selected, which were bound to various alleles of HLA molecules with high binding affinity. The predicted T cell epitopes were examined for allergenicity, toxicity, and antigenicity. Also, to prevent allergic reactions caused by IgE antibodies, these T cell epitopes were chosen so that they did not have any overlapping with the epitopes predicted to bind to B cells. In the present study, we used GPGPG linkers to link HTL epitopes, as it had already been done in previous studies62,63. The GPGPG linker promotes epitope presentation, while reducing the development of junctional epitopes64. Moreover, in this study, RS04 adjuvant was used as a TLR4 agonist in the structure of the TEV. RS04 is a synthetic peptide that mimics LPS by interacting with TLR4. It has been reported that this adjuvant, unlike alum and other adjuvants, has no side effects and can potentially stimulate immune responses through interacting with TLR437. Following performing the analysis by the PyMOL tool, it was observed that the amino acid residues of Gln55, Val57, and Leu59 related to RS04 in the structure of the TEV interacted well with the amino acid residues of the B chain of the TLR4 molecule (Fig. 7A–C and Table 4). This interaction had an effective role in binding the TEV and stimulating the immune system. The HTL epitopes identified in the Sal k 1 allergen have a remarkable binding affinity with DRB*0101 alleles. A large proportion of human populations have this gene, which causes the immune system to react quickly and effectively. After checking by the PyMOL tool, it was observed that several amino acid residues in the TEV structure interacted with the amino acid residues of the HLADRB1 molecule (Fig. 7D–F and Table 4), and this indicated the effective efficiency of the constructed TEV. The TEV instability index was calculated to be 21.6. If this number is less than 40, the protein is thought to be stable24. In mammalian reticulocytes, the half-life of our TEV was found to be 30 h. This finding suggests that our TEV could be exposed to the immune system for a longer period of time, the same as the vaccine created by Fanuel et al.65. The TEV’s aliphatic index was determined to be 107.63, indicating that it was thermostable65. The GRAVY value of the TEV was 0.405, and positive values for this metric denoted a hydrophobic property of the vaccine66. GRAVY was found to be 0.252 in previous studies67. However, because of the hydrophobic nature of the vaccine, it appears that micelles must be used to boost vaccine interaction inside the polar environment of the body. TLR4-TEV and HLADRB1-TEV docked complexes were also subjected to MD simulation to determine the vaccine construct’s stability. The results of the iMODS revealed that TLR4-TEV and HLADRB1-TEV complexes were stable (Figs. 8 and 9). The TEV sequence had a CAI value of 1.0 and a GC content of 55.36%. The results of this section were satisfactory for two reasons. First, CAI values more than 0.8 are thought to be excellent for expression in the host organism68. Second, it has been indicated that improved expression requires a GC content between 30 and 70%68. In this study, we designed a T-cell epitope-based vaccine. This vaccine was developed based on many in silico investigations. This approach can contribute to the rapid prediction of a vaccine's potential prior to experimental testing. The vaccine was developed based on HTL epitopes from Sal k 1, which is an allergenic protein found in S. kali pollen grains. Antigenicity and safety of the TEV were demonstrated via the evaluation of its allergenicity, solubility, and toxicity. Furthermore, the TEV designed in this study should not cause autoimmune diseases in humans due to the lack of any similarity and identity with human and microbiota proteome. Moreover, the interaction of the TEV with TLR4 and HLADRB1 demonstrated its ability to stimulate innate and adaptive immune responses. The present study is the first step in the TEV vaccine production procedure for Sal k 1 allergen. Further in vivo studies are required to investigate other aspects of this procedure.
Data availability
The datasets generated and/ or analyzed during the current study are available within the manuscript and in NCBI (https://www.ncbi.nlm.nih.gov/) with their accession numbers P83181, AAT99258, AAX11262, and AAX11262 in this manuscript.
References
Silverberg, J. I., Simpson, E. L., Durkin, H. G. & Joks, R. Prevalence of allergic disease in foreign-born American children. JAMA Pediatr. 167, 554–560 (2013).
Kim, J. S. Prevalence of allergic disease in foreign-born American children. Pediatrics 134, S137–S138 (2014).
Calzada, D., Baos, S., Cremades-Jimeno, L. & Cárdaba, B. Immunological mechanisms in allergic diseases and allergen tolerance: The role of treg cells. J. Immunol. Res. 2018 (2018).
Calzada, D., Baos, S., Cremades, L. & Cardaba, B. New treatments for allergy: Advances in peptide immunotherapy. Current Med. Chem. 25, 2215–2232 (2018).
Sierra-Heredia, C. et al. Aeroallergens in Canada: Distribution, public health impacts, and opportunities for prevention. Int. J. Environ. Res. Public Health 15, 1577 (2018).
Assarehzadegan, M. A. et al. Identification of methionine synthase (Sal k 3), as a novel allergen of Salsola kali pollen. Mol. Biol. Rep. 38, 65–73 (2011).
Tabesh, S. et al. Design and evaluation of a hypoallergenic peptide-based vaccine for Salsola kali allergy. Int. Immunopharmacol. 66, 62–68 (2019).
Barderas, R. et al. A pectin methylesterase as an allergenic marker for the sensitization to Russian thistle (Salsola kali) pollen. Clin. Exp. Allergy 37, 1111–1119 (2007).
Carnes, J. et al. Immunochemical characterization of Russian thistle (Salsola kali) pollen extracts. Purification of the allergen Sal k 1. Allergy 58, 1152–1156 (2003).
Durham, S. R. & Shamji, M. H. Allergen immunotherapy: Past, present and future. Nat. Rev. Immunol. 23, 317–328 (2023).
Jensen-Jarolim, E., Roth-Walter, F., Jordakieva, G. & Pali-Schöll, I. Allergens and adjuvants in allergen immunotherapy for immune activation, tolerance, and resilience. J. Allergy Clin. Immunol. Pract. 9, 1780–1789 (2021).
Yang, L. & Zhu, R. Allergen Preparation in AIT, Now and in the Future. Current Treat. Opt. Allergy 8, 120–132 (2021).
Zhao, J. et al. Construction of the recombinant vaccine based on T-cell epitope encoding Der p1 and evaluation on its specific immunotherapy efficacy. Int. J. Clin. Exp. Med. 8, 6436 (2015).
Marth, K., Focke-Tejkl, M., Lupinek, C., Valenta, R. & Niederberger, V. Allergen peptides, recombinant allergens and hypoallergens for allergen-specific immunotherapy. Current Treat. Opt. allergy 1, 91–106 (2014).
Worm, M. et al. Development and preliminary clinical evaluation of a peptide immunotherapy vaccine for cat allergy. J. Allergy Clin. Immunol. 127, 89-97.e14 (2011).
Ramchandani, R., Hossenbaccus, L. & Ellis, A. K. Immunoregulatory T cell epitope peptides for the treatment of allergic disease. Immunotherapy 13, 1283–1291 (2021).
Prickett, S. et al. Ara h 1 CD4+ T cell epitope-based peptides: Candidates for a peanut allergy therapeutic. Clin. Exp. Allergy 43, 684–697 (2013).
Calzada, D., Cremades-Jimeno, L., López-Ramos, M. & Cárdaba, B. Peptide allergen immunotherapy: A new perspective in olive-pollen allergy. Pharmaceutics https://doi.org/10.3390/pharmaceutics13071007 (2021).
Verhoef, A., Alexander, C., Kay, A. B. & Larché, M. T cell epitope immunotherapy induces a CD4+ T cell population with regulatory activity. PLoS Med. 2, e78 (2005).
Mitchell, A. et al. The InterPro protein families database: The classification resource after 15 years. Nucleic Acids Res. 43, D213–D221 (2015).
Krogh, A., Larsson, B., Von Heijne, G. & Sonnhammer, E. L. Predicting transmembrane protein topology with a hidden Markov model: Application to complete genomes. J. Mol. Biol. 305, 567–580 (2001).
Chen, H., Yang, H. W., Wei, J. F. & Tao, A. L. In silico prediction of the T-cell and IgE-binding epitopes of Per a 6 and Bla g 6 allergens in cockroaches. Mol. Med. Rep. 10, 2130–2136 (2014).
Ren, A., Ishida, T. & Akiyama, Y. Assessing statistical reliability of phylogenetic trees via a speedy double bootstrap method. Mol. Phylogenet. Evol. 67, 429–435 (2013).
Gasteiger, E. et al. Protein identification and analysis tools on the ExPASy server. (Springer, 2005).
Sigrist, C. J. et al. New and continuing developments at PROSITE. Nucleic Acids Res. Nucleic Acids Res. 41, D344–D347 (2012).
Blom, N., Gammeltoft, S. & Brunak, S. Sequence and structure-based prediction of eukaryotic protein phosphorylation sites. J. Mol. Biol. 294, 1351–1362 (1999).
Jones, D. T. Protein secondary structure prediction based on position-specific scoring matrices. J. Mol. Biol. 292, 195–202 (1999).
Petersen, B., Petersen, T. N., Andersen, P., Nielsen, M. & Lundegaard, C. A generic method for assignment of reliability scores applied to solvent accessibility predictions. BMC Struct. Biol. 9, 51 (2009).
Jespersen, M. C., Peters, B., Nielsen, M. & Marcatili, P. BepiPred-2.0: improving sequence-based B-cell epitope prediction using conformational epitopes. Nucleic Acids Res. 45, W24–W29 (2017).
Fleri, W. et al. The immune epitope database and analysis resource in epitope discovery and synthetic vaccine design. Front. Immunol. 8, 278. https://doi.org/10.3389/fimmu.2017.00278 (2017).
Bui, H. H. et al. Predicting population coverage of T-cell epitope-based diagnostics and vaccines. BMC Bioinform. 7, 153. https://doi.org/10.1186/1471-2105-7-153 (2006).
Shen, Y., Maupetit, J., Derreumaux, P. & Tufféry, P. Improved PEP-FOLD approach for peptide and miniprotein structure prediction. J. Chem. Theory Comput. 10, 282–284 (2014).
Desta, I. T., Porter, K. A., Xia, B., Kozakov, D. & Vajda, S. Performance and its limits in rigid body protein-protein docking. Structure 28, 1071-1081.e1073 (2020).
Kausar, M. A. et al. In silico comparative exploration of allergens of Periplaneta americana, Blattella germanica and Phoenix dactylifera for the diagnosis of patients suffering from IgE-mediated allergic respiratory diseases. Molecules 27, 8740 (2022).
Mitra, D., Pandey, J., Jain, A. & Swaroop, S. In silico design of multi-epitope-based peptide vaccine against SARS-CoV-2 using its spike protein. J. Biomol. Struct. Dynam. 40, 5189–5202 (2022).
Martinelli, D. D. In silico vaccine design: A tutorial in immunoinformatics. Healthcare Anal. 2, p100044 (2022).
Shanmugam, A. et al. Synthetic Toll like receptor-4 (TLR-4) agonist peptides as a novel class of adjuvants. PloS one 7, e30839 (2012).
Alzarea, S. I. Identification and construction of a multi-epitopes vaccine design against Klebsiella aerogenes: Molecular modeling study. Sci. Rep. 12, 14402 (2022).
Magnan, C. N. et al. High-throughput prediction of protein antigenicity using protein microarray data. Bioinform. 26, 2936–2943. https://doi.org/10.1093/bioinformatics/btq551 (2010).
Sharma, N. et al. AlgPred 2.0: An improved method for predicting allergenic proteins and mapping of IgE epitopes. Brief. Bioinform. https://doi.org/10.1093/bib/bbaa294 (2021).
Gupta, S. et al. In silico approach for predicting toxicity of peptides and proteins. PloS One 8, e73957. https://doi.org/10.1371/journal.pone.0073957 (2013).
Mahapatra, S. R. et al. Immunoinformatics and molecular docking studies reveal a novel multi-epitope peptide vaccine against pneumonia infection. Vaccine 39, 6221–6237 (2021).
Drozdetskiy, A., Cole, C., Procter, J. & Barton, G. J. JPred4: A protein secondary structure prediction server. Nucleic Acids Res. 43, W389–W394 (2015).
Grote, A. et al. JCat: A novel tool to adapt codon usage of a target gene to its potential expression host. Nucleic Acids Res. 33, W526–W531 (2005).
Zheng, W. et al. Folding non-homologous proteins by coupling deep-learning contact maps with I-TASSER assembly simulations. Cell Rep. Methods 1, 100014 (2021).
Heo, L., Park, H. & Seok, C. GalaxyRefine: Protein structure refinement driven by side-chain repacking. Nucleic Acids Res. 41, W384–W388 (2013).
Laskowski, R. A., MacArthur, M. W., Moss, D. S. & Thornton, J. M. PROCHECK: A program to check the stereochemical quality of protein structures. J. Appl. Crystallogr. 26, 283–291 (1993).
Sippl, M. J. Recognition of errors in three-dimensional structures of proteins. Proteins Struct. Funct. Bioinform. 17, 355–362 (1993).
Colovos, C. & Yeates, T. O. Verification of protein structures: patterns of nonbonded atomic interactions. Prorein Sci. 2, 1511–1519 (1993).
Berman, H. M. et al. The protein data bank. Nucleic Acids Res. 28, 235–242 (2000).
López-Blanco, J. R., Aliaga, J. I., Quintana-Ortí, E. S. & Chacón, P. iMODS: Internal coordinates normal mode analysis server. Nucleic Acids Res. 42, W271–W276 (2014).
Lopéz-Blanco, J. R., Garzón, J. I. & Chacón, P. iMod: Multipurpose normal mode analysis in internal coordinates. Bioinformatics 27, 2843–2850 (2011).
Zhang, Y. & Skolnick, J. Scoring function for automated assessment of protein structure template quality. Proteins Struct. Funct. Bioinform. 57, 702–710 (2004).
Edwards-Salmon, S. E., Padmanabhan, S. L., Kuruvilla, M. & Levy, J. M. Increasing prevalence of allergic disease and its impact on current practice. Current Otorhinolaryngol. Rep. 10, 278–284 (2022).
Marth, K., Focke-Tejkl, M., Lupinek, C., Valenta, R. & Niederberger, V. Allergen peptides, recombinant allergens and hypoallergens for allergen-specific immunotherapy. Current Treat Opt. Allergy 1, 91–106 (2014).
Larché, M. J. V. A. A. T cell epitope-based allergy vaccines. 107–119 (2011).
Makatsori, M., Pfaar, O., Lleonart, R. & Calderon, M. A. Recombinant allergen immunotherapy: Clinical evidence of efficacy—a review. Current Allergy Asthma Rep. 13, 371–380 (2013).
Gupta, K., Kumar, S., Das, M. & Dwivedi, P. D. Peptide based immunotherapy: A pivotal tool for allergy treatment. Int. Immunopharmacol. 19, 391–398 (2014).
van de Veen, W. et al. IgG4 production is confined to human IL-10–producing regulatory B cells that suppress antigen-specific immune responses. J. Allergy Clin. Immunol. 131, 1204–1212 (2013).
Prickett, S., Rolland, J. & O’Hehir, R. Immunoregulatory T cell epitope peptides: The new frontier in allergy therapy. Clin. Exp. Allergy 45, 1015–1026 (2015).
Mutschlechner, S. et al. Naturally processed T cell–activating peptides of the major birch pollen allergen. J. Allergy Clin. Immunol. 125, 711–718712 (2010).
Khatoon, N., Pandey, R. K. & Prajapati, V. K. Exploring Leishmania secretory proteins to design B and T cell multi-epitope subunit vaccine using immunoinformatics approach. Sci. Rep. 7, 1–12 (2017).
Sanami, S. et al. Design of a multi-epitope vaccine against cervical cancer using immunoinformatics approaches. Sci. Rep. 11, 12397 (2021).
Livingston, B. et al. A rational strategy to design multiepitope immunogens based on multiple Th lymphocyte epitopes. J. Immunol. 168, 5499–5506 (2002).
Fanuel, S. et al. Construction of a recombinant B-cell epitope vaccine based on a Der p1-derived hypoallergen: A bioinformatics approach. Immunotherapy 10, 537–553 (2018).
Wang, H., Zhong, H., Gao, C., Zang, J. & Yang, D. The distinct properties of the consecutive disordered regions inside or outside protein domains and their functional significance. Int. J. Mol. Sci. 22, 10677 (2021).
Negahdaripour, M. et al. Structural vaccinology considerations for in silico designing of a multi-epitope vaccine. Infection, Genet. Evol. 58, 96–109 (2018).
Morla, S., Makhija, A. & Kumar, S. Synonymous codon usage pattern in glycoprotein gene of rabies virus. Gene 584, 1–6 (2016).
Acknowledgements
The authors would like to express their sincere gratitude to the officials of Hepatitis Research Center of Lorestan University of Medical Sciences, Khorramabad, Iran, for their support (Grant No: IR.LUMS.REC.1402.150).
Author information
Authors and Affiliations
Contributions
Designed and performed all the experiments: M.H.S., M.A.A. & S.M.S.; Analyzed the data and wrote the manuscript: A.H. and A.S.; Edited the draft: P.A.B.; Finalized the manuscript: R.J. and H.M.; Conceived, designed, and supervised the study: A.F.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Shams, M.H., Sohrabi, S.M., Jafari, R. et al. Designing a T-cell epitope-based vaccine using in silico approaches against the Sal k 1 allergen of Salsola kali plant. Sci Rep 14, 5040 (2024). https://doi.org/10.1038/s41598-024-55788-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-024-55788-x
- Springer Nature Limited