
Abstract
Coccidiosis, an intestinal disease caused by Eimeria parasites, is responsible for major losses in the poultry industry by impacting chicken health. The gut microbiota is associated with health factors, such as nutrient exchange and immune system modulation, requiring understanding on the effects of Eimeria infection on the gut microbiota. This study aimed to determine the effects of Eimeria acervulina infection on the luminal and mucosal microbiota of the cecum (CeL and CeM) and ileum (IlL and IlM) at multiple time points (days 3, 5, 7, 10, and 14) post-infection. E. acervulina infection decreased evenness in CeL microbiota at day 10, increased richness in CeM microbiota at day 3 before decreasing richness at day 14, and decreased richness in IlL microbiota from day 3 to 10. CeL, CeM, and IlL microbiota differed between infected and control birds based on beta diversity at varying time points. Infection reduced relative abundance of bacterial taxa and some predicted metabolic pathways known for short-chain fatty acid production in CeL, CeM, and IlL microbiota, but further understanding of metabolic function is required. Despite E. acervulina primarily targeting the duodenum, our findings demonstrate the infection can impact bacterial diversity and abundance in the cecal and ileal microbiota.
Similar content being viewed by others

Introduction
The parasitic disease coccidiosis is a considerable health and management challenge in the poultry industry. Infection by the apicomplexan protozoa Eimeria causes damage to epithelial cells within the gastrointestinal tract (GIT), which can lead to malabsorption, inflammation, and accumulation of mucus1. Consequently, weight gain depression, inefficient feed conversion, and reduced egg production may occur, and Eimeria lesions can predispose chickens to secondary infection by Clostridium perfringens, potentially resulting in mortality by necrotic enteritis2. Together, these factors have a significant economic impact on poultry production, leading to an estimated US $14 billion in annual losses worldwide3. Eimeria acervulina, along with Eimeria maxima and Eimeria tenella, are among the most commonly found Eimeria species in poultry farms4, each targeting different sites in the GIT. For instance, E. acervulina has a predilection for the duodenum and upper jejunum of the small intestine5. In the past, antimicrobials were relied upon to treat coccidiosis, however, modern legislation has resulted in a transition away from antimicrobials and antibiotics to greater usage of vaccines and drugs. Drug resistance is a concern in control of coccidiosis, which has inspired interest in alternative solutions such as probiotics, prebiotics, antioxidants, essential oils, and feed additives6,7.
Research towards these alternative solutions has required a greater understanding of the bacterial communities in the gut microbiota and the influence of infection on these communities. Due to the growing body of 16S rRNA amplicon sequencing studies, the importance of the gut microbiota on chicken health has been shown, with factors such as nutrient exchange, immune system modulation, digestive system physiology, and pathogen exclusion being associated with the gut microbiota8,9. The gut microbiota in chickens’ small and large intestines differ both in absolute bacterial counts and bacterial taxonomic composition. Microbiota of the ileum, duodenum, and jejunum tend to be similar, having low diversity and often containing Lactobacillus, Enterococcus, Turcibacter, Clostridium sensu stricto, and bacteria from the Clostridium XI cluster (family Peptostreptococcaceae, genus Romboutsia)10. In contrast, the cecal microbiota has considerably higher absolute counts and diversity, containing hundreds of bacterial species, most of which belong to the phyla Firmicutes and Bacteroidetes, while there is minor representation from the phyla Actinobacteria and Proteobacteria10.
In analyses on the luminal and mucosal microbiota of the duodenum and jejunum, E. acervulina infection had significant effects on alpha diversity (e.g. richness and evenness) and beta diversity (e.g. differing populations in infected and control), especially in the mucosal microbiota during and after the peak of infection11. Although E. acervulina does not target the cecum and ileum directly, a study has shown an E. acervulina infection can affect cecal microbiota diversity12, and it has been suggested that E. acervulina infection could affect cecal microbiota by increasing endogenous loss of proteins and undigested dietary proteins in the GIT13,14. While there are various studies investigating mixed Eimeria species infections on the cecum and/or ileum13,15,16,17, less is known on the individual effects of E. acervulina. The aim of this study was to determine the effect of E. acervulina on the luminal and mucosal microbiota of both the cecum and ileum at multiple time points during infection.
Materials and methods
Animal care and tissue sampling
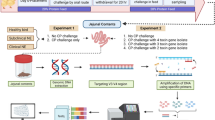
All animal care procedures were approved by the Institutional Animal Care and Use Committee (IACUC, protocol #18-025) of the Beltsville Agricultural Research Center (BARC). This study was carried out in accordance with relevant rules and regulations and was performed and reported in accordance with ARRIVE guidelines (https://arriveguidelines.org/). The following animal care procedures are as described in greater detail elsewhere11. Ross 708 male broilers (288 birds, 1 day of age) were obtained from Longnecker’s Hatchery (Elizabethtown, PA) and placed into 1.00 m2 open-top wire brooder pens (25 chicks per pen). Birds were moved at 19 days of age into 72 finisher units (Alternative Designs, Siloam Springs, AR) with 4 birds per pen. A corn-soybean-based diet (approximately 24% crude protein in crumble format) and water were provided to chicks ad libitum for the duration of the study. One-half of the birds (144 birds) were infected (IF) with 1 × 105 E. acervulina oocysts (USDA #12 isolate) in a volume of 1.0 mL per bird by oral gavage at 21 days of age, while the remaining 144 birds were sham-infected with water (control [C]). The resulting 36 pens of C birds and 36 pens of IF birds were placed in separate areas of the facility to prevent cross-transmission.
Of the 4 birds per pen, 1 bird closest to the pen’s average weight (to prevent sampling of birds with outlier weights) was euthanatized at day 0, 3, 5, 7, 10, and 14 post-infection (PI) via cervical dislocation. There were 6 pens (n = 6 replicates per treatment) for each time point, resulting in 36 C birds and 36 IF birds euthanatized. To evaluate infection, plasma carotenoid concentrations, body weight, and crypt depths were recorded at each time point. For luminal and mucosal microbiota sampling, a total of 48 of the 72 birds were randomly chosen as follows: 25 IF birds consisting of 5 replicates per day PI (3, 5, 7, 10, 14) and 23 C birds consisting of 5 replicates from day 0; 4 from each of day 3, 7, and 14; and 3 from each of day 5 and 10. The cecum and ileum were dissected, and the cecal contents (CeL), cecal epithelial scrapings (CeM), ileal contents (IlL), and ileal epithelial scrapings (IlM) were collected. The cecum was defined as paired blind sacs of the large intestine anterior to the cloaca (one sac sampled), and the ileum was defined as the part of the small intestine that is posterior to the Meckel’s diverticulum. Isolated specimens were snap frozen in liquid nitrogen and stored at − 80 °C until bacterial DNA isolation.
Library preparation and sequencing
DNA extraction, library preparation, and sequencing were performed as described in Ref.18, utilizing a DNeasy PowerSoil kit (Qiagen, Valencia, CA), PCR primers targeting the V3-V4 region of the 16S rRNA gene, and the Illumina MiSeq platform (Illumina, Inc., San Diego, CA), respectively. The 16S rRNA gene sequences determined in this study were deposited in the NCBI Sequence Read Archive database (SRA accession no. PRJNA1066775).
Bioinformatics and data analysis
As performed in Campos et al. 11, the bioinformatics platform Quantitative Insights Into Microbial Ecology 2 (QIIME 2) version 2021.419 was used to analyze the CeL, CeM, IlL, and IlM microbiota. Demultiplexed, paired-end sequence data were denoised with DADA220 via the q2-dada2 plugin, using a quality cutoff of 30 to determine truncation settings. For taxonomic classification, the SILVA database was chosen over the commonly used Greengenes database due to previous studies demonstrating SILVA’s larger database size and more recent updates to taxonomy, potentially improving interpretation of results21,22. The SILVA reference sequences and taxonomy were obtained from the QIIME 2 Data Resources page (https://docs.qiime2.org/2022.2/data-resources/) as pre-formatted files using RESCRIPt, a process used to reduce inconsistencies and improve processing by removing duplicate sequences that are assigned different taxonomies23. Reads were extracted from reference sequences using the V3-V4 region forward and reverse primers (5ʹ-end: CCTACGGGNGGCWGCAG and 3ʹ-end: GACTACHVGGGTATCTAATCC, respectively). A feature classifier was created via q2-feature-classifier24 fit-classifier-naive-bayes using SILVA version 138 99% operational taxonomic unit reference sequences and taxonomy25. Amplicon sequence variants (ASVs) from DADA2 were assigned taxonomy via the q2-feature-classifier classify-sklearn naïve Bayes taxonomy classifier. Mitochondria and chloroplasts were filtered and excluded from the feature table. All ASVs were aligned with MAFFT26 via q2-alignment and used to construct a phylogeny with fasttree227 via q2-phylogeny. Using alpha rarefaction plots produced via q2-diversity and considering the number of samples retained in the dataset, rarefaction, or subsampling without replacement, was performed. The following sampling depths were applied for alpha and beta diversity analyses via q2-diversity: 14,132 for CeL, 19,242 for CeM, 24,469 for IlL, and 3816 for IlM.
Alpha diversity is a measure of species richness and/or evenness within an individual sample and was analyzed with four metrics: Shannon diversity index, observed features (ASVs), Faith’s phylogenetic diversity (Faith PD)28, and evenness. Observed features and Faith PD both measure richness, though Faith PD considers the phylogenetic differences in ASVs28. The non-parametric Kruskal–Wallis test was used to analyze differences in alpha diversity between treatment groups (the infection status and days post-infection). Beta diversity is a measure of distance between sample bacterial compositions and was measured using UniFrac distance metrics, which incorporate phylogenetic distances29. Unweighted UniFrac considers the presence and absence of ASVs in samples, while weighted UniFrac considers the abundance of ASVs30. To test for significance differences in groups based on UniFrac distances, the non-parametric permutational analysis of variance (PERMANOVA) test was used. To visualize UniFrac distances between groups and clustering of samples with similar microbiota, principal coordinates analysis (PCoA) was utilized. Visualizations for alpha diversity significance and beta diversity PCoA were produced in R 4.0.331 using the packages QIIME2R 0.99.3532 to import QIIME 2 PCoA results and tidyverse 1.3.033 for data wrangling with dplyr and graph production with ggplot2.
Differential bacterial abundance between IF and C birds was analyzed using the linear discriminant analysis effect size (LEfSe) algorithm34. Feature tables at the genus level were exported from QIIME 2 and analyzed at the Huttenhower Lab Galaxy web server (http://huttenhower.sph.harvard.edu/galaxy) using the default parameters. Phylogenetic Investigation of Communities by Reconstruction of Unobserved States 2 (PICRUSt2) version 2.4.2 software was used to predict functional abundances based on marker gene sequences35. The MetaCyc Metabolic Pathways Database36 was used to produce functional abundance data, and the data were analyzed and visualized using STAMP 2.1.337 to determine biological relevance of features.
Results
Infection with Eimeria acervulina
Infection was evaluated in a prior study, with plasma carotenoid concentrations being significantly lower in IF birds on days 5 and 7 post-infection, body weight being significantly lower in IF birds overall, and crypt depths being increased in IF birds, indicating an observable E. acervulina infection38.
Microbiota profiles
Sequencing summaries for the four microbiota datasets (CeL, CeM, IlL, and IlM) are presented in Table 1. The five most abundant bacterial genera in CeL microbiota were unclassified Lachnospiraceae, Lachnospiraceae NK4A136 group, Subdoligranulum, [Ruminococcus] torques group, and Bacteroides (Fig. 1A). In CeM microbiota, the five most abundant genera were unclassified Lachnospiraceae, [Ruminococcus] torques group, Bacillus, Subdoligranulum, and Lachnospiraceae NK4A136 group (Fig. 1A). IlL microbiota were primarily comprised of Lactobacillus, Romboutsia, Escherichia-Shigella, Candidatus Arthromitus, and Streptococcus (Fig. 1A). Lastly, IlM microbiota were primarily comprised of Candidatus Arthromitus, Lactobacillus, Romboutsia, unclassified Lachnospiraceae, and Subdoligranulum (Fig. 1A). Broadly, the cecum was typically dominated by the phylum Firmicutes and contained smaller percentages of Bacteroidota, Proteobacteria, and Verrucomicrobiota (Fig. 1B). The ileum was also often dominated by Firmicutes, and Proteobacteria typically composed the remaining bacteria ranging from small to large percentages (< 1% to 72%) (Fig. 1B).

Summary of bacterial composition (relative abundance, %) in combined samples for CeL, CeM, IlL, and IlM microbiota for (A) genus level and (B) phylum level. In (A), the fifteen most common genera overall are listed in the legend, and in (B), the six most common phyla overall are listed.
Alpha diversity
In CeL microbiota, Shannon diversity significantly differed based on group (infection status and time post-infection) (Kruskal–Wallis, H = 20.41, P = 0.02, Table S1). Pairwise comparisons showed there was a trend of lower Shannon diversity in IF birds compared to C birds on day 10 (H = 3.75, P = 0.053, Fig. 2A, Table S2A). Faith PD differed based on group overall (H = 18.97, P = 0.04, Table S1), however, there were no significant differences in comparisons between IF and C birds on particular days (P > 0.05, Table S2B). Evenness differed in groups overall (H = 26.58, P < 0.01, Table S1), with lower evenness in IF birds compared to C birds on day 10 (H = 5.00, P = 0.03, Fig. 2B, Table S2C). Observed features did not differ based on group (P > 0.05, Table S1).

Comparisons of alpha diversity metrics in the cecum: (A) Shannon diversity in CeL, (B) evenness in CeL, (C) observed features (ASVs) in CeM, (D) Faith’s phylogenetic diversity (Faith PD) in CeM, and (E) evenness in CeM.
In CeM microbiota, observed features differed in groups overall (H = 21.55, P = 0.02, Table S1), where observed features were higher in IF birds compared to C birds on day 5 (H = 4.46, P = 0.03, Fig. 2C, Table S3A), and there was a trend of lower observed features in IF birds on day 14 (H = 3.84, P = 0.05, Fig. 2C, Table S3A). Faith PD differed in groups overall (H = 21.19, P = 0.02, Table S1), with higher Faith PD in IF birds on day 3 (H = 6.00, P = 0.01, Fig. 2D, Table S3B). Evenness differed in groups overall (H = 21.39, P = 0.02, Table S1), and there was a trend of higher evenness on day 5 (H = 3.76, P = 0.053, Fig. 2E, Table S3C). Shannon diversity did not differ based on group (P > 0.05, Table S1).
In IlL microbiota, observed features differed in groups overall (H = 35.32, P < 0.01, Table S1), with lower observed features in IF birds compared to C birds on day 3 (H = 5.33, P = 0.03, Fig. 3A, Table S4A), day 5 (H = 4.50, P = 0.03, Fig. 3A, Table S4A), and day 7 (H = 6.05, P = 0.01, Fig. 3A, Table S4A). There was a trend of lower observed features in IF birds on day 10 (H = 3.76, P = 0.053, Fig. 3A, Table S4A). Faith PD differed in groups overall (H = 33.20, P < 0.01, Table S1), with lower Faith PD in IF birds on day 3 (H = 6.00, P = 0.01, Fig. 3B, Table S4B), day 5 (H = 4.50, P = 0.03, Fig. 3B, Table S4B), day 7 (H = 4.86, P = 0.03, Fig. 3B, Table S4B), and day 10 (H = 5.00, P = 0.03, Fig. 3B, Table S4B). There were no significant differences in groups for Shannon diversity and evenness (P > 0.05, Table S1).

Comparisons of alpha diversity metrics in the ileal luminal microbiota: (A) observed features (ASVs), (B) Faith’s phylogenetic diversity (Faith PD).
In IlM microbiota, evenness differed in groups overall (H = 19.20, P = 0.04, Table S1), however, there were no significant differences between IF and C birds on a particular day (P > 0.05, Table S5A). There were no significant differences in groups for Shannon diversity, observed features, and Faith PD (P > 0.05, Table S1).
Beta diversity
CeL microbiota of different groups were considered distinct overall under unweighted UniFrac (PERMANOVA, pseudo-F = 3.06, P < 0.01, Fig. 4A, Table S1). Microbiota were distinct between IF and C birds on day 10 (pseudo-F = 3.28, P = 0.02, Fig. 4A, Table S2D) and day 14 (pseudo-F = 2.17, P = 0.05, Fig. 4A, Table S2D). Under weighted UniFrac, CeL microbiota were distinct based on groups overall (pseudo-F = 3.75, P < 0.01, Fig. 4B, Table S2E), and there was a trend of distinct microbiota between IF and C birds on day 14 (pseudo-F = 6.18, P = 0.057, Fig. 4B, Table S2E).

Principal coordinate analysis (PCoA) comparing Eimeria acervulina-infected birds and control birds at multiple time points based on (A) unweighted UniFrac distance matrix in CeL microbiota, (B) weighted UniFrac distance matrix in CeL microbiota, (C) unweighted UniFrac distance matrix in CeM microbiota, and (D) weighted UniFrac distance matrix in CeM microbiota. Stars (*) denote significant differences (P < 0.05) between microbiota of IF and C birds at the same time point.
CeM microbiota of different groups were distinct overall under unweighted UniFrac (pseudo-F = 1.89, P < 0.01, Fig. 4C, Table S1), with distinct microbiota between IF and C birds on day 3 (pseudo-F = 2.15, P = 0.04, Fig. 4C, Table S3D) and day 14 (pseudo-F = 3.07, P = 0.01, Fig. 4C, Table S3D). Under weighted UniFrac, CeM microbiota were distinct based on groups overall (pseudo-F = 2.40, P < 0.01, Fig. 4D, Table S1), and IF and C birds had distinct microbiota on day 5 (pseudo-F = 7.60, P = 0.03, Fig. 4D, Table S3E).
IlL microbiota of different groups were distinct overall under unweighted UniFrac (pseudo-F = 3.26, P < 0.01, Fig. 5A, Table S1), with IF and C birds having distinct microbiota on day 3 (pseudo-F = 4.04, P = 0.01, Fig. 5A, Table S4C), day 7 (pseudo-F = 6.36, P = 0.01, Fig. 5A, Table S4C), and day 10 (pseudo-F = 2.39, P = 0.03, Fig. 5A, Table S4C). There was a trend of distinct microbiota on day 5 (pseudo-F = 3.01, P = 0.055, Fig. 5A, Table S4C). Under weighted UniFrac, IlL microbiota were distinct based on groups overall (pseudo-F = 1.59, P = 0.04, Fig. 5B, Table S1), and there was a trend of distinct microbiota between IF and C birds on day 3 (pseudo-F = 2.57, P = 0.056, Fig. 5B, Table S4D).

Principal coordinate analysis (PCoA) comparing Eimeria acervulina-infected birds and control birds at multiple time points based on (A) weighted UniFrac distance matrix in ileal luminal microbiota, (B) weighted UniFrac distance matrix in ileal luminal microbiota, and (C) weighted UniFrac distance matrix in ileal mucosal microbiota. Stars (*) denote significant differences (P < 0.05) between microbiota of IF and C birds at the same time point.
IlM microbiota were not distinct under unweighted UniFrac (P > 0.05, Table S1). Under weighted UniFrac, IlM microbiota were distinct based on groups overall (pseudo-F = 3.90, P < 0.01, Table S1), however, microbiota were not distinct between IF and C birds on a particular day (P > 0.05, Fig. 5C, Table S5B).
Differential abundance of bacterial taxa
In CeL microbiota, the genera Bacteroides (Effect size (ES) = 4.76, P < 0.01), Lactobacillus (ES = 4.17, P = 0.02), Merdibacter (ES = 2.79, P = 0.02), and [Eubacterium] nodatum group (ES = 2.17, P = 0.03) were in greater relative abundance in IF birds compared to C birds, while 8 genera, including unclassified Lachnospiraceae (ES = 4.17, P = 0.03), GCA-900066575 (ES = 3.80, P < 0.01), Clostridia UCG-014 (ES = 3.67, P < 0.01), Erysipelatoclostridium (ES = 3.65, P < 0.01), and Oscillibacter (ES = 3.60, P = 0.02), were in greater relative abundance in C birds (Fig. 6A). In CeM microbiota, 19 genera (all P < 0.05), including Bacteroides (ES = 4.51, P < 0.01), Lactobacillus (ES = 3.85, P = 0.02), Colidextribacter (ES = 3.62, P = 0.04), Lachnoclostridium (ES = 3.53, P = 0.02), and Anaerostipes (ES = 3.51, P < 0.01), were in greater relative abundance in IF birds, while Candidatus Arthromitus (ES = 4.54, P < 0.01), Anaerotruncus (ES = 3.89, P = 0.02), an unclassified bacterium (ES = 2.43, P = 0.04), and the family Butyricicoccaceae (ES = 3.65, P = 0.04) were in greater relative abundance in C birds (Fig. 6B).

Differential bacterial abundance analysis using linear discriminant analysis effect size (LEfSe) in: (A) cecal luminal (CeL) microbiota, (B) cecal mucosal (CeM) microbiota, (C) ileal luminal (IlL) microbiota, and (D) ileal mucosal (IlM) microbiota. Positive effect size (red bars) indicates higher relative abundance in Eimeria acervulina-infected birds, while negative effect size (green bars) indicates higher relative abundance in control birds.
In IlL microbiota, no taxa had increased relative abundance in IF birds, while 48 genera (all P < 0.05) and the family Aerococcaceae (ES = 3.45, P = 0.01) were in greater relative abundance in C birds, including Candidatus Arthromitus (ES = 4.77, P = 0.02), unclassified Peptostreptococcaceae (ES = 4.00, P < 0.01), uncultured Bacillaceae (ES = 3.63, P < 0.01), unclassified Bacillaceae (ES = 3.54, P < 0.01), and Brachybacterium (ES = 3.49, P = 0.01) (Fig. 6C). In IlM microbiota, Escherichia-Shigella (ES = 4.36, P < 0.01) and Lactobacillus (ES = 4.28, P < 0.01) were in greater relative abundance in IF birds, while the family Clostridiaceae (ES = 4.66, P = 0.046) and genera Serratia (ES = 4.23, P = 0.04), CHKCI001 (ES = 3.62, P = 0.03), Eisenbergiella (ES = 3.44, P = 0.01), and Colidextribacter (ES = 3.13, P < 0.01) were in greater relative abundance in C birds (Fig. 6D).
Predicted functional abundances
In CeL microbiota, genes for 18 predicted MetaCyc pathways were in greater relative abundance in IF birds compared to C birds (all P < 0.05), including “hexitol fermentation to lactate, formate, ethanol, and acetate” (P = 0.01), mycolate biosynthesis (P = 0.01), mannan degradation (P = 0.02), 8-amino-7-oxononanoate biosynthesis I (P = 0.02), and mevalonate pathway I (P = 0.02), while 11 pathways were predicted to be in greater relative abundance in C birds (all P < 0.05), including heme biosynthesis II (anaerobic) (P < 0.01), d-glucarate degradation I (P < 0.01), glutaryl-CoA degradation (P = 0.01), d-galactarate degradation I (P = 0.01), and urea cycle (P = 0.01) (top 20 significant pathways in Fig. 7A). In CeM microbiota, 24 pathways were predicted to be in greater relative abundance in IF birds (all P < 0.05), including mannan degradation (P < 0.01), palmitate biosynthesis II (bacteria and plants) (P < 0.01), 8-amino-7-oxononanoate biosynthesis I (P < 0.01), superpathway of glycol metabolism and degradation (P < 0.01), and biotin biosynthesis I (P < 0.01), while 4 pathways were predicted to be in greater relative abundance in C birds, including l-histidine degradation I (P = 0.02), and l-glutamate degradation V (via hydroxyglutarate) (P = 0.03) (Fig. 7B).

Effect of Eimeria acervulina-infection on differential mean proportion (%) of predicted MetaCyc pathways in the (A) cecal luminal (CeL), (B) cecal mucosal (CeM), (C) ileal luminal (IlL), and (D) ileal mucosal (IlM) bacterial populations.
In IlL microbiota, 115 pathways were predicted to be in greater relative abundance in IF birds (all P < 0.05), including various superpathways of menaquinol biosynthesis (all P < 0.01), superpathway of phylloquinol biosynthesis (P < 0.01), 1,4-dihydroxy-2-naphthoate biosynthesis I (P < 0.01), and enterobactin biosynthesis (P < 0.01), while 69 pathways were predicted to be in greater relative abundance in C birds (all P < 0.05), including superpathway of sulfur oxidation (Acidanus amivalens) (P < 0.01), succinate fermentation to butanoate (P < 0.01), superpathway of heme biosynthesis from glycine (P < 0.01), GDP-d-glycero-alpha-d-manno-heptose bioysynthesis (P < 0.01), and aerobic respiration I (cytochrome c) (P < 0.01) (Fig. 7C). In IlM microbiota, 92 pathways were predicted to be in greater relative abundance in IF birds (all P < 0.05), including urate biosynthesis/inosine 5ʹ-phosphate degradation (P < 0.01), superpathway of methylglyoxal degradation (P < 0.01), 3-phenylpropanoate and 3-(3-hydroxyphenyl)propanoate degradation (P < 0.01), cinnamate and 3-dhydroxycinnamate degradation (P < 0.01), and purine nucleotides degradation II (aerobic) (P < 0.01) (Fig. 7D), while 32 pathways were predicted to be in greater relative abundance in C birds (all P < 0.05), such as glutaryl-CoA degradation (P < 0.01) and l-glutamate and l-glutamine biosynthesis (P < 0.01).
Discussion
This study aimed to determine the effects of E. acervulina infection on bacterial diversity, abundance, and predicted metabolic function in the luminal and mucosal microbiota of the cecum and ileum during a 14-day post-infection period. We hypothesized that E. acervulina infection would have differing effects on microbiota depending on the region of microbiota, as alpha and beta diversity have been shown to differ in luminal and mucosal microbiota within the cecum, ileum, jejunum, and duodenum39. It should be noted that our feed contained a higher crude protein percentage than usual, however, another study has shown that diets with a crude protein difference of 7% did not have significantly different alpha and beta diversity results40. We observed differential effects based on microbiota region in our study, where E. acervulina infection reduced evenness on day 10 in the CeL microbiota of IF birds, while there was no effect on evenness in the CeM microbiota of IF birds. Instead, richness was increased in CeM microbiota, with observed features affected on day 5 and Faith PD affected on day 3. The difference in time points for these two metrics may be explained by observed features being a count of unique ASVs, while Faith PD considers phylogenetic branch lengths. Overall, there appeared to be a delayed effect of infection on CeL microbiota, as microbiota became distinct between IF and C birds after the typical E. acervulina peak infection period of 5–7 days PI41. Significantly reduced plasma carotenoids in IF birds of our study on days 5 and 7 PI38 supported that this period was the peak of infection. CeL microbiota were distinct on day 10 and 14 based on presence and absence of ASVs (unweighted UniFrac) and distinct on day 14 based on ASV abundances (weighted UniFrac). In contrast, CeM microbiota were affected by infection at earlier points (unweighted UniFrac: day 3, weighted UniFrac: day 5) and microbiota was then affected at a later point based on presence of ASVs (unweighted UniFrac: day 14). Despite CeL and CeM microbiota sharing some dominant genera, these results show that the luminal and mucosal microbiota in the cecum are affected differently by E. acervulina infection and at different time points.
In the ileum, dominant taxa differed between IlL and IlM microbiota, and E. acervulina infection affected bacterial diversity in IlL, but not IlM, microbiota. Lactobacillus tended to dominate IlL microbiota from day 0 to day 10, while Candidatus Arthromitus tended to dominate IlM microbiota in the same time period. Between day 10 and day 14, these dominant bacteria generally decreased in relative abundance in both IlL and IlM microbiota, while Romboutsia greatly increased and often became dominant. Lactobacillus also increased to a smaller degree in IlM microbiota during this period. E. acervulina infection reduced richness in IlL microbiota over a relatively long period, significantly affecting observed features from day 3 to day 7 and Faith PD from day 3 to day 10. Additionally, IlL microbiota were distinct between IF and C birds based on ASV presence (but not ASV abundances) on day 3, 7, and 10, further supporting that infection affected IlL microbiota over a relatively long period. Both alpha and beta diversity were not significantly affected in IlM microbiota, again demonstrating that infection can affect one region of microbiota and not the other.
To determine genera affected by E. acervulina infection, we performed differential abundance analysis comparing bacterial relative abundance in IF birds and C birds. Unclassified Lachnospiraceae were in lower relative abundance in the CeL microbiota of IF birds, a similar pattern to that observed in the CeL and CeM microbiota of chickens infected by E. tenella18. Lachnospiraceae are potentially of importance in chicken GIT health, as human gut studies have demonstrated the Lachnospiraceae group contains species associated with the production of short-chain fatty acids (SCFAs) such as butyrate42,43,44. In broiler chickens, Eimeria infection can lead to a decrease in butyrate concentration in the cecal contents45,46, which likely contributes to a decrease in body weight gain, depending on the bird breed and the Eimeria species47. Butyrate supplements, such as tributyrin or sodium butyrate, have shown promise to improve body weight gain in chickens infected with E. maxima48,49, as well as in chickens with nutrient-reduced diets50. Although supplementation may differ in terms of concentration and how the butyrate is absorbed, it has been observed that relative abundance of Lachnospiraceae may increase51,52, possibly improving natural butyrate production as well. Moreover, Lachnospiraceae abundance in the cecum has been positively correlated with body weight gain including chickens infected by E. tenella18 and those challenged with both E. maxima and Clostridium perfringens53. Fermented soybean meal supplementation has been suggested to improve Lachnospiraceae abundance in the cecum, along with Lachnoclostridium, Gastranaerophilales, and Lactobacillus, and lead to improved body weight gain and food conversion ratio in broilers54. The genus Butyricicoccus and family Butyricicoccaceae were also in lower relative abundance in the CeL and CeM microbiota, respectively, of IF birds. Although 16S data cannot pinpoint species, this taxonomic group is noteworthy because Butyricicoccus pullicaecorum, a butyrate-producing bacterium isolated from the chicken cecum55, may potentially be valuable as a probiotic that improves feed conversion and contributes to prevention of necrotic enteritis56. Lastly, another butyrate-producing bacterium Anaerotruncus57 was in lower relative abundance in CeM microbiota of IF birds, demonstrating multiple butyrate-producing taxa may be decreased in the cecum during infection.
In contrast to decreases of Butyricicoccaceae and Anaerotruncus in CeM microbiota, various taxa increased in relative abundance in infected birds. Interestingly, while infection with E. tenella tends to increase abundance of potential secondary pathogens such as Escherichia coli in the cecum18,58,59, infection with E. acervulina in our study increased relative abundance of genera that potentially contain strains with beneficial functions, though further research would be required to confirm the function of these bacteria. Some of these genera have been associated with probiotic potential, such as Bacteroides60,61, Lactobacillus62,63,64, and Anaerostipes57, or have shown to contribute beneficial health effects, such as Colidextribacter65 and Lachnoclostridium66. While E. tenella directly targets the cecum and causes physical damage, E. acervulina primarily targets the duodenum and is less pathogenic in comparison due to smaller oocysts and fewer asexual cycles in reproduction. Therefore, lesser physical damage to the cecum may explain the difference in types of bacteria in increased abundance in IF birds. For Lactobacillus in particular, high crude protein % in our feed may have affected their abundance40, however, as feed was the same for all birds in this study, the differential abundance in C and IF birds can be attributed to infection. In IlL microbiota, results generally indicated reduced abundances of various taxa in infected birds, possibly suggesting infection made it less likely for some rarer taxa to become established, consistent with the decreases in richness observed in IlL microbiota. Of these taxa, unclassified Peptostreptococcaceae, a family with butyrate-producing members in human infants67, and unclassified Lachnospiraceae were identified to follow the same pattern in duodenal luminal and jejunal luminal microbiota, respectively11. The impact on overall butyrate production in comparison to the cecum is unclear, due to the ileum, duodenum, and jejunum containing lower absolute bacterial counts and differing in microbiota composition compared to the cecum10. For IlM microbiota, IF birds were more likely to contain high relative abundances of Escherichia-Shigella (e.g. up to 48%), while C birds typically contained less than 1%.
To search for potential links between differential abundance of taxa and functional profiles of the microbiota related to SCFA production, we utilized predictive functional analysis. Although accuracy of functional prediction can be limited outside of human studies68, patterns observed may assist in designing future research studies using methods such as shotgun metagenome sequencing, transcriptomics, and metabolomics. In a previous study on luminal microbiota of the duodenum and jejunum, genes predicted to influence pathways related to acetyl-CoA production were in lower relative abundance in IF birds11. Two major pathways utilize acetyl-CoA in butyrate production57, presenting the possibility that reduced butyrate production in microbiota may contribute to decreased body weight gain during infection. We observed a similar pattern in CeL microbiota, where genes predicted to affect pathways for glutaryl-CoA degradation, l-glutamate degradation V (via hydroxyglutarate), and pyruvate fermentation to butanoate (butyrate) were in lower relative abundance in IF birds. Furthermore, l-glutamate degradation V was predicted to be lower in the CeM microbiota of IF birds, and the pathways succinate fermentation to butanoate and acetyl-CoA fermentation to butanoate II were predicted to be lower in the IlL microbiota of IF birds.
Conclusions
Eimeria acervulina infection affected alpha and beta diversity in the CeL, CeM, and IlL microbiota at varying time points, with CeL microbiota affected after the peak of infection, CeM microbiota affected both early and late in infection, and IlL microbiota for a more prolonged period (day 3–day 10). This study provided further evidence that Eimeria infections may reduce the relative abundance of potential SCFA-producing bacteria, with CeL, CeM, and IlL microbiota demonstrating this pattern. Predicted functional abundances suggest some pathways related to butyrate production could be affected as relative abundance of these bacteria are reduced during infection. Continued research to confirm the function of gut bacteria in SCFA production and determine the effectiveness of probiotics or SCFA supplementation on performance and health during Eimeria infections will be of importance in development of alternative solutions to antimicrobials.
Data availability
The 16S rRNA gene sequences determined in this study were deposited in the NCBI Sequence Read Archive (SRA) database (https://www.ncbi.nlm.nih.gov/bioproject; SRA accession no. PRJNA1066775).
References
Blake, D. P. & Tomley, F. M. Securing poultry production from the ever-present Eimeria challenge. Trends Parasitol. 30, 12–19. https://doi.org/10.1016/j.pt.2013.10.003 (2014).
Moore, R. J. Necrotic enteritis predisposing factors in broiler chickens. Avian Pathol. 45, 275–281. https://doi.org/10.1080/03079457.2016.1150587 (2016).
Blake, D. P. et al. Re-calculating the cost of coccidiosis in chickens. Vet. Res. 51, 1–14. https://doi.org/10.1186/s13567-020-00837-2 (2020).
Györke, A., Pop, L. & Cozma, V. Prevalence and distribution of Eimeria species in broiler chicken farms of different capacities. Parasite. https://doi.org/10.1051/parasite/2013052 (2013).
Lillehoj, H. S. & Trout, J. M. Avian gut-associated lymphoid tissues and intestinal immune responses to Eimeria parasites. Clin. Microbiol. Rev. 9, 349–360. https://doi.org/10.1128/cmr.9.3.349 (1996).
Bozkurt, M. et al. Efficacy of in-feed preparations of an anticoccidial, multienzyme, prebiotic, probiotic, and herbal essential oil mixture in healthy and Eimeria spp.-infected broilers. Poultry Sci. 93, 389–399. https://doi.org/10.3382/ps.2013-03368 (2014).
Khater, H. F. et al. Avian coccidiosis: Recent advances in alternative control strategies and vaccine development. Agrobiol. Rec. 1, 11–25. https://doi.org/10.47278/journal.abr/2020.003 (2020).
Stanley, D., Hughes, R. J. & Moore, R. J. Microbiota of the chicken gastrointestinal tract: Influence on health, productivity and disease. Appl. Microbiol. Biotechnol. 98, 4301–4310. https://doi.org/10.1007/s00253-014-5646-2 (2014).
Clavijo, V. & Flórez, M. J. V. The gastrointestinal microbiome and its association with the control of pathogens in broiler chicken production: A review. Poultry Sci. 97, 1006–1021. https://doi.org/10.3382/ps/pex359 (2018).
Rychlik, I. Composition and function of chicken gut microbiota. Animals 10, 103. https://doi.org/10.3390/ani10010103 (2020).
Campos, P. M., Miska, K. B., Jenkins, M. C., Yan, X. & Proszkowiec-Weglarz, M. Effects of Eimeria acervulina infection on the luminal and mucosal microbiota of the duodenum and jejunum in broiler chickens. Front. Microbiol. 14, 750. https://doi.org/10.3389/fmicb.2023.1147579 (2023).
Perez, V. et al. Effect of corn distillers dried grains with solubles and Eimeria acervulina infection on growth performance and the intestinal microbiota of young chicks. Poultry Sci. 90, 958–964. https://doi.org/10.3382/ps.2010-01066 (2011).
Choi, J. et al. Effects of different Eimeria inoculation doses on growth performance, daily feed intake, gut health, gut microbiota, foot pad dermatitis, and Eimeria gene expression in broilers raised in floor pens for 35 days. Animals 13, 2237. https://doi.org/10.3390/ani13132237 (2023).
Diether, N. E. & Willing, B. P. Microbial fermentation of dietary protein: An important factor in diet–microbe–host interaction. Microorganisms 7, 19. https://doi.org/10.3390/microorganisms7010019 (2019).
Bortoluzzi, C. et al. Hops β-acids and zinc bacitracin affect the performance and intestinal microbiota of broilers challenged with Eimeria acervulina and Eimeria tenella. Anim. Feed Sci. Technol. 207, 181–189. https://doi.org/10.1016/j.anifeedsci.2015.06.006 (2015).
Martynova-Van Kley, M. A., Oviedo-Rondón, E. O., Dowd, S. E., Hume, M. & Nalian, A. Effect of Eimeria infection on cecal microbiome of broilers fed essential oils. Int. J. Poultry Sci. 11, 747–755 (2012).
Villar-Patiño, G. et al. Effect of an Alliaceae encapsulated extract on growth performance, gut health, and intestinal microbiota in broiler chickens challenged with Eimeria spp. Animals 13, 3884. https://doi.org/10.3390/ani13243884 (2023).
Campos, P. M. et al. Effects of Eimeria tenella on cecal luminal and mucosal microbiota in broiler chickens. Avian Diseases 66, 39–52. https://doi.org/10.1637/21-00068 (2022).
Bolyen, E. et al. Reproducible, interactive, scalable and extensible microbiome data science using QIIME 2. Nat. Biotechnol. 37, 852–857. https://doi.org/10.1038/s41587-019-0209-9 (2019).
Callahan, B. J. et al. DADA2: High-resolution sample inference from Illumina amplicon data. Nat. Methods 13, 581–583. https://doi.org/10.1038/nmeth.3869 (2016).
Balvočiūtė, M. & Huson, D. H. SILVA, RDP, Greengenes, NCBI and OTT—How do these taxonomies compare?. BMC Genom. 18, 114. https://doi.org/10.1186/s12864-017-3501-4 (2017).
Campos, P. M., Darwish, N., Shao, J. & Proszkowiec-Weglarz, M. Research Note: Choice of microbiota database affects data analysis and interpretation in chicken cecal microbiota. Poultry Sci. 101, 971. https://doi.org/10.1016/j.psj.2022.101971 (2022).
Robeson, M. S. II. et al. RESCRIPt: Reproducible sequence taxonomy reference database management. PLOS Comput. Biol. 17, e1009581. https://doi.org/10.1371/journal.pcbi.1009581 (2021).
Bokulich, N. A. et al. Optimizing taxonomic classification of marker-gene amplicon sequences with QIIME 2’s q2-feature-classifier plugin. Microbiome 6, 1–17. https://doi.org/10.1186/s40168-018-0470-z (2018).
Quast, C. et al. The SILVA ribosomal RNA gene database project: Improved data processing and web-based tools. Nucleic Acids Res. 41, D590–D596. https://doi.org/10.1093/nar/gks1219 (2012).
Katoh, K., Misawa, K., Kuma, K. I. & Miyata, T. MAFFT: A novel method for rapid multiple sequence alignment based on fast Fourier transform. Nucleic Acids Res. 30, 3059–3066. https://doi.org/10.1093/nar/gkf436 (2002).
Price, M. N., Dehal, P. S. & Arkin, A. P. FastTree 2–approximately maximum-likelihood trees for large alignments. PloS One 5, e9490. https://doi.org/10.1371/journal.pone.0009490 (2010).
Faith, D. P. Conservation evaluation and phylogenetic diversity. Biol. Conserv. 61, 1–10. https://doi.org/10.1016/0006-3207(92)91201-3 (1992).
Lozupone, C. & Knight, R. UniFrac: A new phylogenetic method for comparing microbial communities. Appl. Environ. Microbiol. 71, 8228–8235. https://doi.org/10.1128/AEM.71.12.8228-8235.2005 (2005).
Lozupone, C. A., Hamady, M., Kelley, S. T. & Knight, R. Quantitative and qualitative β diversity measures lead to different insights into factors that structure microbial communities. Appl. Environ. Microbiol. 73, 1576–1585. https://doi.org/10.1128/AEM.01996-06 (2007).
R: A language and environment for statistical computing (R Foundation for Statistical Computing, Vienna, 2020).
Bisanz, J. qiime2R: Importing QIIME2 artifacts and associated data into R sessions. Version 0.99 13 (2018).
Wickham, H. et al. Welcome to the Tidyverse. J. Open Source Softw. 4, 1686. https://doi.org/10.21105/joss.01686 (2019).
Segata, N. et al. Metagenomic biomarker discovery and explanation. Genome Biol. 12, 1–18. https://doi.org/10.1186/gb-2011-12-6-r60 (2011).
Douglas, G. M. et al. PICRUSt2 for prediction of metagenome functions. Nat. Biotechnol. 38, 685–688. https://doi.org/10.1038/s41587-020-0548-6 (2020).
Caspi, R. et al. The MetaCyc database of metabolic pathways and enzymes—A 2019 update. Nucleic Acids Res. 48, D445–D453. https://doi.org/10.1093/nar/gkz862 (2020).
Parks, D. H., Tyson, G. W., Hugenholtz, P. & Beiko, R. G. STAMP: Statistical analysis of taxonomic and functional profiles. Bioinformatics 30, 3123–3124. https://doi.org/10.1093/bioinformatics/btu494 (2014).
Cloft, S. E. et al. Temporal changes of genes associated with intestinal homeostasis in broiler chickens following a single infection with Eimeria acervulina. Poultry Sci. 102, 537. https://doi.org/10.1016/j.psj.2023.102537 (2023).
Proszkowiec-Weglarz, M. in Sturkie's Avian Physiology 485–527 (Elsevier, 2022).
De Cesare, A. et al. Effect of a low protein diet on chicken ceca microbiome and productive performances. Poultry Sci. 98, 3963–3976. https://doi.org/10.3382/ps/pez132 (2019).
Allen, P. C. & Fetterer, R. H. Recent advances in biology and immunobiology of Eimeria species and in diagnosis and control of infection with these coccidian parasites of poultry. Clin. Microbiol. Rev. 15, 58–65. https://doi.org/10.1128/CMR.15.1.58-65.2002 (2002).
Barcenilla, A. et al. Phylogenetic relationships of butyrate-producing bacteria from the human gut. Appl. Environ. Microbiol. 66, 1654–1661. https://doi.org/10.1128/AEM.66.4.1654-1661.2000 (2000).
Duncan, S. H., Barcenilla, A., Stewart, C. S., Pryde, S. E. & Flint, H. J. Acetate utilization and butyryl coenzyme A (CoA):acetate-CoA transferase in butyrate-producing bacteria from the human large intestine. Appl. Environ. Microbiol. 68, 5186–5190. https://doi.org/10.1128/AEM.68.10.5186-5190.2002 (2002).
Biddle, A., Stewart, L., Blanchard, J. & Leschine, S. Untangling the genetic basis of fibrolytic specialization by lachnospiraceae and ruminococcaceae in diverse gut communities. Diversity 5, 627–640. https://doi.org/10.3390/d5030627 (2013).
Lin, Y. & Olukosi, O. A. Exogenous enzymes influenced Eimeria-induced changes in cecal fermentation profile and gene expression of nutrient transporters in broiler chickens. Animals 11, 2698. https://doi.org/10.3390/ani11092698 (2021).
Liu, G. et al. The effects of arginine and branched-chain amino acid supplementation to reduced-protein diet on intestinal health, cecal short-chain fatty acid profiles, and immune response in broiler chickens challenged with Eimeria spp. Poultry Sci. 102, 102773. https://doi.org/10.1016/j.psj.2023.102773 (2023).
Bumstead, N. & Millard, B. Genetics of resistance to coccidiosis: Response of inbred chicken lines to infection by eimeria tenella and eimeria maxima. Br. Poultry Sci. 28, 705–715. https://doi.org/10.1080/00071668708417006 (1987).
Hansen, V. L. et al. The effects of tributyrin supplementation on weight gain and intestinal gene expression in broiler chickens during Eimeria maxima-induced coccidiosis. Poultry Sci. 100, 100984. https://doi.org/10.1016/j.psj.2021.01.007 (2021).
Proszkowiec-Weglarz, M. et al. Research Note: Effect of butyric acid glycerol esters on ileal and cecal mucosal and luminal microbiota in chickens challenged with Eimeria maxima. Poultry Sci. 99, 5143–5148. https://doi.org/10.1016/j.psj.2020.06.022 (2020).
Bortoluzzi, C. et al. Sodium butyrate improved performance while modulating the cecal microbiota and regulating the expression of intestinal immune-related genes of broiler chickens. Poultry Sci. 96, 3981–3993. https://doi.org/10.3382/ps/pex218 (2017).
Wu, W., Xiao, Z., An, W., Dong, Y. & Zhang, B. Dietary sodium butyrate improves intestinal development and function by modulating the microbial community in broilers. PLOS ONE 13, e0197762. https://doi.org/10.1371/journal.pone.0197762 (2018).
Xiao, C. et al. Dietary sodium butyrate improves female broiler breeder performance and offspring immune function by enhancing maternal intestinal barrier and microbiota. Poultry Sci. 102, 102658. https://doi.org/10.1016/j.psj.2023.102658 (2023).
Lu, M. et al. Effects of Eimeria maxima and Clostridium perfringens infections on cecal microbial composition and the possible correlation with body weight gain in broiler chickens. Res. Vet. Sci. 132, 142–149. https://doi.org/10.1016/j.rvsc.2020.05.013 (2020).
Li, Y. et al. Effects of fermented soybean meal supplementation on the growth performance and Cecal microbiota community of broiler chickens. Animals 10, 1098. https://doi.org/10.3390/ani10061098 (2020).
Eeckhaut, V. et al. Butyricicoccus pullicaecorum gen. nov., sp. nov., an anaerobic, butyrate-producing bacterium isolated from the caecal content of a broiler chicken. Int. J. Syst. Evolut. Microbiol. 58, 2799–2802. https://doi.org/10.1099/ijs.0.65730-0 (2008).
Eeckhaut, V. et al. The probiotic Butyricicoccus pullicaecorum reduces feed conversion and protects from potentially harmful intestinal microorganisms and necrotic enteritis in broilers. Front. Microbiol. https://doi.org/10.3389/fmicb.2016.01416 (2016).
Polansky, O. et al. Important metabolic pathways and biological processes expressed by chicken cecal microbiota. Appl. Environ. Microbiol. 82, 1569–1576. https://doi.org/10.1128/AEM.03473-15 (2016).
Macdonald, S. E. et al. Effects of Eimeria tenella infection on chicken caecal microbiome diversity, exploring variation associated with severity of pathology. PLoS One 12, e0184890. https://doi.org/10.1371/journal.pone.0184890 (2017).
Huang, G. et al. Eimeria tenella infection perturbs the chicken gut microbiota from the onset of oocyst shedding. Vet. Parasitol. 258, 30–37 (2018).
Kollarcikova, M. et al. Different bacteroides species colonise human and chicken intestinal tract. Microorganisms. https://doi.org/10.3390/microorganisms8101483 (2020).
Papouskova, A., Rychlik, I., Harustiakova, D. & Cizek, A. (2023) Research Note: A mixture of Bacteroides spp. and other probiotic intestinal anaerobes reduces colonization by pathogenic E. coli strain O78:H4-ST117 in newly hatched chickens. Poultry Sci. 102, 102529. https://doi.org/10.1016/j.psj.2023.102529
Zhang, G., Ma, L. & Doyle, M. P. Salmonellae reduction in poultry by competitive exclusion Bacteria Lactobacillus salivarius and Streptococcus cristatus. J. Food Protect. 70, 874–878. https://doi.org/10.4315/0362-028X-70.4.874 (2007).
Mappley, L. J. et al. Oral treatment of chickens with Lactobacillus reuteri LM1 reduces Brachyspira pilosicoli-induced pathology. J. Med. Microbiol. 62, 287–296. https://doi.org/10.1099/jmm.0.051862-0 (2013).
De Cesare, A. et al. Effect of Lactobacillus acidophilus D2/CSL (CECT 4529) supplementation in drinking water on chicken crop and caeca microbiome. PLOS ONE 15, e0228338. https://doi.org/10.1371/journal.pone.0228338 (2020).
Liu, Y. et al. Effects of dietary Bopu powder supplementation on intestinal development and microbiota in broiler chickens. Front. Microbiol. https://doi.org/10.3389/fmicb.2022.1019130 (2022).
Lei, J. et al. Intestinal microbiota regulate certain meat quality parameters in chicken. Front. Nutr. https://doi.org/10.3389/fnut.2022.747705 (2022).
Appert, O. et al. Initial butyrate producers during infant gut microbiota development are endospore formers. Environ. Microbiol. 22, 3909–3921. https://doi.org/10.1111/1462-2920.15167 (2020).
Sun, S., Jones, R. B. & Fodor, A. A. Inference-based accuracy of metagenome prediction tools varies across sample types and functional categories. Microbiome 8, 46. https://doi.org/10.1186/s40168-020-00815-y (2020).
Acknowledgements
The authors wish to thank Lori Schreier for laboratory support in this study. Mention of trade name, proprietary product, or specific equipment does not constitute guarantee or warranty by USDA and does not imply its approval to the exclusion of other suitable products. This research was supported in part by an appointment to the Agricultural Research Service (ARS) Research Participation Program administered by the Oak Ridge Institute for Science and Education (ORISE) through an interagency agreement between the U.S. Department of Energy (DOE) and the U.S. Department of Agriculture (USDA). ORISE is managed by ORAU under DOE contract number DE-SC0014664. All opinions expressed in this paper are the author’s and do not necessarily reflect the policies and views of USDA, DOE, or ORAU/ORISE.
Funding
This research was funded by the in-house USDA-ARS CRIS project number 8042-31000-108-00D.
Author information
Authors and Affiliations
Contributions
K.B.M., M.C.J., and M.P.-W. contributed to the conception, design, and investigation of the study. X.Y. contributed to the initial statistical analysis and bioinformatics analysis. P.M.C. completed the final statistical analysis, bioinformatics analysis, visualization, and wrote the first draft of the manuscript. P.M.C., K.B.M., M.C.J., and M.P.-W. contributed to manuscript revision. All authors have read and approved the submitted version of the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Campos, P.M., Miska, K.B., Jenkins, M.C. et al. Effects of Eimeria acervulina infection on the luminal and mucosal microbiota of the cecum and ileum in broiler chickens. Sci Rep 14, 10702 (2024). https://doi.org/10.1038/s41598-024-61299-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-024-61299-6
- Springer Nature Limited