
Abstract
Adults with Down syndrome have a genetic form of Alzheimer’s disease (AD) and evidence of cerebrovascular disease across the AD continuum, despite few systemic vascular risk factors. The onset and progression of AD in Down syndrome is highly age-dependent, but it is unknown at what age cerebrovascular disease emerges and what factors influence its severity. In the Alzheimer’s Biomarker Consortium-Down Syndrome study (ABC-DS; n = 242; age = 25–72), we estimated the age inflection point at which MRI-based white matter hyperintensities (WMH), enlarged perivascular spaces (PVS), microbleeds, and infarcts emerge in relation to demographic data, risk factors, amyloid and tau, and AD diagnosis. Enlarged PVS and infarcts appear to develop in the early 30s, while microbleeds, WMH, amyloid, and tau emerge in the mid to late 30s. Age-residualized WMH were higher in women, in individuals with dementia, and with lower body mass index. Participants with hypertension and APOE-ε4 had higher age-residualized PVS and microbleeds, respectively. Lifespan trajectories demonstrate a dramatic cerebrovascular profile in adults with Down syndrome that appears to evolve developmentally in parallel with AD pathophysiology approximately two decades prior to dementia symptoms.
Similar content being viewed by others

Introduction
Although typically considered a disorder that affects early life intellectual and physical development, Down syndrome is also associated with increased risk for Alzheimer’s disease (AD) in later life. The link between Down syndrome and AD was first uncovered in the 1940s1 and subsequently attributed to the triplication of the 21st chromosome2, which contains the amyloid precursor protein gene (APP). With chromosome 21 trisomy comes increased production and aggregation of beta-amyloid protein, one of the primary pathological features of AD3,4,5,6. Indeed, individuals with Down syndrome overproduce beta-amyloid protein from birth7 and by the time they are in their 40 s, most have the full pathological features of AD. Today, Down syndrome is considered a “genetic form” of AD, together with fully penetrant, autosomal dominant familial genetic mutations8,9,10,11,12,13,14.
Understanding the emergence and progression of AD-related features across the adult lifespan in Down syndrome is critical for two reasons. First, significant recent advances in medical care for individuals with Down syndrome have resulted in an average lifespan that has nearly doubled since the 1980s15. As a result, people with Down syndrome are typically living into their 60 s and almost all will suffer from AD dementia within their lifetimes15. Thus, AD represents an emerging public health crisis in this population and the time course, risk and pathogenic factors, and potential prevention or treatment targets need to be identified to mitigate its impact. Second, because the biological and clinical progression of AD among individuals with Down syndrome are very similar to late onset AD, the study of AD in this genetically at-risk population has great potential to provide insight into pathogenesis, course, and prevention or treatment strategies for the neurotypical population as well. With advances in care of medical conditions, improved social integration, and a large segment of the Down syndrome population entering older adulthood, AD threatens the health economics and quality of life of an aging society.
There is significant debate about the causes of AD. The field has embraced a single pathogenic pathway, in which accumulation of amyloid leads to tau pathology, subsequent neurodegeneration, and associated cognitive and functional decline. This “amyloid cascade hypothesis” has informed both diagnostic frameworks12,13,14 and primary treatment strategies for AD16. However, emerging evidence suggests that the clinical course and, possibly, the pathogenesis of AD are multiply determined17. Notably, the vast majority of people who die with symptomatic AD have evidence of significant cerebrovascular disease18,19. Cerebrovascular disease contributes to risk, onset, and clinical course of AD20,21 and recent studies show elevated severity of cerebrovascular disease among individuals with autosomal dominant forms of AD22,23. Supported by animal experiments that suggest that cerebrovascular disease gives rise to AD pathological features24 and genetic studies linking vascular factors to AD prevalence25, emerging evidence suggests that cerebrovascular disease is a core feature of AD.
Despite having low rates of systemic vascular risk factors, such as hypertension, we previously showed that individuals with Down syndrome have increased magnetic resonance imaging (MRI) markers of cerebrovascular disease, including white matter hyperintensities (WMH), enlarged perivascular spaces (PVS), cerebral microbleeds, and infarcts, which increase as a function of clinical AD diagnosis and its antecedent clinical conditions26 and may be mediated in part by inflammatory processes that result in neurodegeneration27,28. Notably, even in individuals without clinical symptoms of AD, we observed a significant degree of cerebrovascular changes, suggesting that they emerged earlier in life and may precede or coincide with the emergence of classical AD pathophysiology. Consistent with work in late onset and autosomal dominant AD, these findings highlight the centrality of cerebrovascular disease to the presentation of AD and possibly to its pathogenesis.
Like other genetically deterministic forms of AD, the development of AD pathological features and subsequent symptoms in Down syndrome is age-dependent and follows a somewhat prescribed pattern, highlighting that AD is a developmental component of Down syndrome. Despite our initial observations of an association between cerebrovascular disease and AD diagnosis26, it is unclear at what age cerebrovascular features typically arise in adults with Down syndrome and how the temporal evolution of cerebrovascular disease compares with other AD biomarkers, like amyloid and tau. In the current study, we examined the age “inflection point” at which MRI-based cerebrovascular disease markers emerge in adults with Down syndrome, characterizing them relative to the temporality of amyloid and tau biomarkers, measured with positron emission tomography (PET). We also examined vascular and AD-related factors that could explain a greater-than-expected cerebrovascular biomarker for a given age.
Results
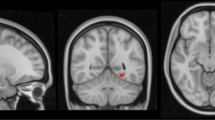
Older age (F(2,239) = 40.3, p < 0.001), greater amyloid burden (F(2,170) = 82.3, p < 0.001), and greater tau burden (Braak I/II: F(2,141) = 47.5, p < 0.001; Braak III/IV: F(2,141) = 55.9, p-value < 0.001; Braak V/VI: F(2,141) = 27.2, p < 0.001) were associated with more advanced AD-related diagnostic group from Cognitively-Stable to MCI-DS and AD dementia, highlighting the strong age-dependency of disease-related factors in this population (Table 1). The proportion of participants with hyperlipidemia was higher in those with AD dementia compared with those characterized as Cognitively-Stable; BMI was lower in those with MCI-DS compared with those characterized as cognitively-stable; and obstructive sleep apnea was common in all diagnostic groups (Table 1). Despite low frequencies of traditional vascular risk factors including hypertension (6.2%) and diabetes (5.4%), WMH volume (F(2,239) = 9.8, p < 0.001), enlarged PVS scores (F(2,167) = 5.4, p = 0.005), and the presence of infarcts (χ2(2) = 7.2, p = 0.03), but not the presence of microbleeds (χ2(2) = 4.9, p = 0.09) were associated with more advanced diagnostic group, as reported previously in a subset of the older participants26. Figure 1 displays representative images from each MRI modality.

Representative MRI scans for white matter hyperintensity volume, enlarged perivascular spaces, microbleeds, and infarcts across the lifespan of adults with Down syndrome.
Piecewise left-null regression models were fit to each cerebrovascular biomarker across the lifespan to test the hypothesis that each cerebrovascular disease biomarker emerges at a given age, reflecting disease progression in adults with DS (Fig. 2). Enlarged PVS and the presence of infarcts were the biomarkers showing the earliest age-associated increase at 31 and 32 years old, respectively. Global WMH inflected at 35 years old. Regionally, frontal and parietal WMH inflected at 35 years old, while occipital WMH inflected later at 41 years old. In relation to traditional AD biomarkers, global amyloid and tau burden in early Braak regions, measured with molecular positron emission tomography (PET) imaging, inflected at 35 years old, after enlarged PVS scores. Then, tau burden in middle and late Braak regions inflected at 39 and 37 years old, respectively, after WMH.

Piecewise left-null regressions of (A) cerebrovascular biomarkers, (B) regional white matter hyperintensity volume, and (C) traditional AD biomarkers against age across the lifespan in adults with Down syndrome. Inflection point estimates, their 95% confidence interval, and their p-value are displayed at the top.
We examined the association of age-residualized cerebrovascular biomarker levels, which reflect the extent to which the biomarker severity deviates from the value predicted by age, with demographic variables, vascular risk factors, amyloid and tau burden, and AD diagnosis (Table 2). Women had larger residuals compared with men for global WMH, driven by parietal, temporal, and occipital WMH. APOE-ε4 carriers had larger residuals compared with non-carriers for the presence of microbleeds. Individuals with hypertension had larger residuals compared with those without hypertension for enlarged PVS scores. Greater BMI was associated with lower age-residualized global WMH, driven by temporal WMH. Individuals with AD dementia had larger residuals compared with those who were cognitively-stable for global WMH, driven by parietal, temporal, and occipital WMH.
Discussion
We found that markers of cerebrovascular disease emerge in adults with Down syndrome within the same timeframe as amyloid and tau pathology and prior to the onset of AD clinical symptoms. The temporal ordering of inflection points suggests that enlarged PVS and infarcts develop in the early 30 s, while microbleeds, WMH, amyloid, and tau develop in the mid to late 30 s. Several demographic factors, vascular risk factors, and AD diagnosis were associated with a greater amount of cerebrovascular biomarkers for a given age. Therefore, vascular risk factors may exacerbate the extent of cerebrovascular disease, but they are not necessary for cerebrovascular disease to emerge across the lifespan in adults with Down syndrome.
Potential pathways between specific markers of cerebrovascular disease and AD have been studied in adults without Down syndrome29 and animal models30. Enlarged PVS generally capture impaired clearance glymphatic mechanisms due to chronic exposure to toxins like soluble beta-amyloid and may contribute to further amyloid deposition in the parenchyma31. Microbleeds among people with Down syndrome reflect amyloid deposits in the vasculature (i.e., cerebral amyloid angiopathy) that weaken the vascular endothelium and may potentiate further downstream vascular dysfunction32. White matter hyperintensities are associated with inflammation and white matter demyelination due to small vessel disease or disruption33. Infarcts reflect ischemic lesions in larger vessels; they may not be mechanistically related to amyloid and tau, but may contribute to downstream neurodegeneration and cognition34.
Based on previous mechanistic work, chronic exposure to soluble amyloid may impair perivascular clearance, leading to downstream amyloid deposition in the parenchyma and in the vasculature. Further, small vessel disease may be one initiator of tau phosphorylation, perhaps through an inflammatory response to damage in small vessels that upregulates kinase activity24,28,30. Our findings were consistent with this expected temporality. We observed the earliest inflection points at age 31 for enlarged PVS and, surprisingly, infarcts, demonstrating that these vascular abnormalities are among the earliest biomarker changes observed in Down syndrome. Amyloid deposition increased in the parenchyma (amyloid PET) and in the vasculature (microbleeds) at ages 35 and 36 years, respectively. Increased tau deposition in early Braak regions was also observed at 35 years, which is consistent with previous studies showing tau deposition in adults with Down syndrome who were amyloid-negative but accumulating amyloid over time35, suggesting that the emergence of amyloid and tau pathology are tightly linked together in time36,37. Tau deposition in middle and late Braak regions were later at 39 and 37 years, respectively. To investigate this somewhat unexpected finding, we ran a sensitivity analysis excluding one individual with a very low late Braak SUVR (< 0.5) at an older age, which may have biased the age trajectory after the inflection point lower and pushed the inflection point earlier; however, the estimated age inflection point in late Braak regions was very similar (37.5 [29.1, 45.9]). Longitudinal within-subject data in adults with Down syndrome demonstrated that middle Braak regions accumulate tau prior to late Braak regions according to the amyloid cascade36 White matter hyperintensities emerged at 35 years old across the brain. The temporal ordering observed is consistent with previous work in late onset AD that demonstrated that greater WMH are associated with tau burden in middle and late Braak regions, but not early Braak regions38. The estimated inflection point for microbleeds was not significant, similar to a previous study that did not show an increase across diagnostic groups in adults with Down syndrome26 and may be due to methodological limitations (e.g., GRE/SWI being more susceptible to motion artifacts than other MRI sequences, and motion artifacts being more common in older individuals with more advanced disease). Alternatively, microbleeds may start to manifest even earlier than the ages represented in these studies. Indeed, pathological studies of adults with Down syndrome suggest a particularly profound vascular amyloid profile39.
Regionally, parietal WMH, which may be more specific to AD compared with other lobar WMH, emerged at 35 years old, approximately 18 years prior to the average age of symptom onset in adults with Down syndrome15. Strikingly, in autosomal dominant AD, posteriorly distributed WMH emerged as early as 22 years before estimated onset of symptoms22,23. The majority of enlarged PVS were observed in the cortex (5.4 [5.0, 5.9]), while approximately one third of enlarged PVS were observed in the basal ganglia (2.6 [2.4, 2.8]), which have been associated with hypertension in adults without Down syndrome40. Global enlarged PVS score was strongly associated with cortical PVS (R2 = 0.89) and moderately associated with basal ganglia PVS (0.51), while cortical PVS and basal ganglia PVS scores were less correlated with each other (0.21). Cortical PVS emerged at age 31 [16.9, 45.0], while basal ganglia PVS emerged at age 42 [28.3, 54.9]. In relation to microbleeds, 1 participant had deep microbleed(s) in the absence of lobar microbleed(s) and 14 participants had lobar microbleed(s) in absence of deep microbleed(s). In relation to infarcts, 9 participants had deep infarct(s) in the absence of lobar infarct(s), 16 participants had lobar infarct(s) in absence of deep infarct(s), and 1 participant had both. Microbleeds outside of cortical lobes and infarcts in deep, subcortical structures were not common enough in adults with DS to reliably model regionally specific age trajectories.
Compared with men, women had greater global WMH than expected for participant age, particularly in parietal, temporal, and occipital lobes, which may be another contributor to age-specific sex differences in AD risk in adults with DS41. Individuals with the APOE-ε4 allele had a greater likelihood of having microbleeds than expected for age, suggesting that even in the context of amyloid precursor protein overproduction due to triplication of the 21st chromosome in Down syndrome, APOE-ε4 can still affect disease progression42. While hypertension was not common in study participants, it was associated with PVS scores that were greater than expected for participant age, suggesting a role of hemodynamics for clearance through the perivascular space. Potential mechanisms for the enlargement of PVS include atherosclerosis, arteriolosclerosis, and elastin dysfunction, which reduces the pliability and increases pulsatility31. However, autopsy studies demonstrated low prevalence of atherosclerosis and arteriosclerosis in adults with Down syndrome39, leaving the possibility that hypertension likely affects enlarged PVS through mechanisms related to vessel elasticity in this study. Hyperlipidemia was common but was not associated with cerebrovascular disease for a given age; hyperlipidemia may not operate as a vascular risk factor in the absence of metabolic disease (e.g., diabetes) in adults with DS. Lower BMI was associated with greater global WMH, highlighting a role of other disease related processes that affect diet, exercise, and weight43. While WMH may reflect Wallerian degeneration to some degree in advanced stages of late onset AD, tau burden was not associated with greater than expected WMH volume for participant age in adults with Down syndrome across the lifespan, and WMH age inflection preceded later stage tau deposition, suggesting that cerebrovascular biomarkers are upstream of advanced tau pathology. Greater age-residualized global WMH, driven by parietal, occipital, and temporal lobe WMH, was associated with a diagnosis of AD dementia. Small vessel disease in posterior brain regions is a consistent cerebrovascular process in AD pathogenesis in adults with Down syndrome26, adults with autosomal dominant AD22,23, and adults with late onset AD20,21. Therefore, these four cerebrovascular biomarkers may represent unique biological mechanisms, each with their own influence on disease pathogenesis and course29,30. Ongoing longitudinal data collection in ABC-DS will support investigations into individual-level inflection points as well as the shape and rates of these cerebrovascular disease biomarker trajectories.
This study has some limitations, including the lack of pathological validation, using temporal ordering of cross-sectional events to infer change, and the lack of correction for multiple comparisons. Autopsy studies can identify individual plaques and tangle within specific cell layers, but amyloid and tau PET scans indicate when the amount of pathology is above the limit of detection at a spatial resolution of 2 mm. However, PET imaging is the in vivo gold standard for comparison against histopathology12. Similarly, only a proportion of microbleeds are detected on MRI with current imaging parameter44 and subtle cerebral blood flow changes likely precede the formation of WMH and infarcts45,46. Therefore, our results may be most relevant to later manifestations of pathology that can be captured with neuroimaging. Nonetheless, we used conventional radiology tools that can inform clinical evaluation by establishing normative expectations for the measured pathologies for an individual given their age. Future work will include the identification, validation, and incorporation of biofluidic measures of vascular function47,48 and comparison to autopsy data that could provide more mechanistic context for the radiological markers studied here. Conclusions about temporal ordering of the emergence of radiological abnormalities were derived with methods that are similar to those used in studies of autosomal dominant AD49,50. In both cases, there is a nearly 100% likelihood of AD incidence in the context of overproduction and/or altered metabolism of amyloid pathology with similar variability around age of dementia onset15. Multiple comparison correction was not performed as our primary interest was in characterizing the natural history of markers of cerebrovascular disease among adults with Down syndrome (i.e., inflection point models). Given the relevance of these findings to therapeutic intervention strategies in adults with Down syndrome, we wanted to minimize Type 2 statistical error (i.e., false negative) to inform future mechanistic studies of any potentially relevant pathways (i.e., age-residualized models), although we recognize the possibility of inflated Type 1 error (i.e., false positive) as a limitation. Still, our findings converge with cross-sectional literature in autosomal dominant AD, showing an early and reliable increase in WMH22,23, enlarged PVS51, microbleeds52, and infarcts53, which were later confirmed longitudinally54,55. Further, in late onset AD, APOEε-4 was associated with microbleeds56, women had faster rates of deep WMH progression57, and enlarged PVS were dependent on arterial hemodynamics58 and were associated with hypertension in a spatially-dependent manner59. Replication studies are needed in larger, longitudinal, and external datasets. Ongoing longitudinal data collection in ABC-DS will support investigations into individual-level inflection points, which may be earlier or later compared to the group estimate, as well as the shape and rates of these cerebrovascular disease biomarker trajectories.
Furthering our understanding of AD pathogenesis with respect to cerebrovascular disease is additionally important, particularly in adults with Down syndrome, because of the common incidence of edema or hemorrhage amyloid-related imaging abnormalities (ARIA) that emerge as a results of current anti-amyloid antibody therapeutics16. There is evidence of cerebrovascular disease on the group level in adults with Down syndrome along the AD continuum, but some older adults have relatively low burden. Even in the absence of existing microbleeds, which are currently the only known MRI risk factor for ARIA, adults with Down syndrome may still be at increased risk because MRI only captures a proportion of the microbleeds detected at autopsy39,52. Future research should study the extent to which other visible cerebrovascular lesions on MRI may be used to predict who is at risk for developing ARIA. Individuals with Down syndrome may be at particular risk for these side effects in anti-amyloid therapeutics. In the absence of approved pharmacological treatments for AD in adults with Down syndrome, modifiable factors like sleep60,61, diet and exercise62, and leisure activity63,64 may be potential therapeutic avenues.
MRI imaging data across the lifespan demonstrate a dramatic cerebrovascular profile in adults with Down syndrome that appears to evolve developmentally in parallel with AD pathophysiology approximately two decades prior to dementia symptoms. This work joins an emerging literature that incorporates cerebrovascular disease into our understanding of AD pathogenesis and progression and highlights new avenues towards our understanding of the cause of AD, therapeutic and preventative strategies, and safety outcomes in this unique population. Future work should emphasize the potential role of cerebrovascular pathologies in AD, beyond the way by which they impact downstream neurodegeneration and cognitive impairment as simple comorbidities as they may precede and contribute to AD pathology.
Methods
Clinical characterization
Adults with Down syndrome from the multisite Alzheimer’s Biomarker Consortium-Down Syndrome study65 (ABC-DS; n = 242; age = 25–72, 45 ± 10; 43% women) underwent MRI, amyloid PET, and tau PET under the Neurodegeneration in Aging Down Syndrome (NiAD) study and the Biomarkers of Alzheimer’s Disease in Down Syndrome (ADDS) study protocols. The studies under which data were collected were approved by the institutional review boards at participating institutions (i.e., University of Pittsburgh, Columbia University Irving Medical Center, The New York State Institute for Basic Research in Developmental Disabilities/New York State Psychiatric Institute, Harvard Medical School, University of Wisconsin-Madison, University of Cambridge, University of California, Irvine), performed in accordance with the Declaration of Helsinki, and written informed consent was obtained from participants and/or their legal guardian or legally authorized representative. Every participant gave assent prior to any study-related procedure.
Clinical diagnoses were assigned by a consensus panel that included clinicians with expertise in the assessment of adults with Down syndrome66. One of four AD consensus diagnoses was assigned to each participant based on the results from neuropsychological testing, clinical chart reviews, and interviews with knowledgeable informants, with additional consideration of health history, functional and vocational abilities, and neuropsychiatric symptoms. Results from neuroimaging or other biomarker studies were not considered in the diagnostic formulation. A diagnosis of “cognitively-stable” (CS) indicated no evidence of clinically significant cognitive decline beyond preclinical intellectual functioning and age. A diagnosis of “mild cognitive impairment-Down syndrome” (MCI-DS) indicated evidence of cognitive decline over time beyond preclinical intellectual functioning and age, but insufficient to suggest dementia. A diagnosis of “AD dementia” indicated clear evidence of substantial cognitive and functional decline of breadth and severity greater than MCI-DS, with a high degree of confidence. Eleven participants in the neuroimaging sample (4.2%) were excluded based on complications or concerns unrelated to neurodegenerative disorders (e.g., severe sensory loss, new psychiatric diagnosis).
Neuroimaging acquisition and analysis
Participants were scanned on 3 T MRI and PET scanners, following protocols put forth by the Alzheimer’s Disease Neuroimaging Initiative (ADNI). Participants underwent a high-resolution T1-weighted anatomical scan (repetition time [TR]/echo time [TE]/inversion time [TI] = 2,300/2.96/900 ms, voxel size = 1 × 1 × 1 mm3), a T2-weighted fluid-attenuated inversion recovery (FLAIR) scan (TR/TE/TI = 5,000/386/1,800 ms, voxel size = 0.4 × 0.4 × 0.9mm3), and a T2*-weighted gradient echo (GRE) scan (TR/TE = 650/20 ms, voxel size = 0.8 × 0.8 × 4 mm3) or susceptibility-weighted image (SWI; TR/TE = 27/20 ms, voxel size = 0.9 × 0.9 × 1.5 mm3). Participants underwent amyloid PET with [11C]PiB (15 mCi, 50–70 min post-injection scan, 5 min frames) or [18F]Florbetapir (AV45; 10 mCi, 80–100 min post-injection scan, 5 min frames); participants also underwent tau PET with [18F]Flortaucipir (AV1451; 10 mCi, 75–105 min post-injection, 5 min frames). All PET data were corrected for attenuation, detection dead time, scanner normalization, scatter, and radioactive decay.
Magnetic resonance imaging scans were analyzed for cerebrovascular disease with analytic pipelines developed in-house. For white matter hyperintensities, total and lobar (frontal, temporal, parietal, occipital) WMH volumes were semi-automatically segmented from T2-weighted FLAIR scans. Images were interpolated to a standard matrix in MNI152 space (256 × 256 × 256; 1 mm3), skull stripped, bias field corrected, and intensity normalized (0–255). Percentile thresholds initiated a Gaussian mixture model, separating dark/bright and bright/brightest intensities; estimated percentile thresholds were then relaxed by an intensity of 10 to account for variations in FLAIR quality. Roberts edge detection removed any hyperintense labels from non-white matter before visual inspection. Enlarged perivascular spaces (PVS) were visually inspected on T1-weighted scans and classified as hypointensities across 13 brain regions, rated from 0 to 2 based on FLAIR characteristics (hyperintense ring), and combined into a global score ranging from 0 (no enlarged PVS in any region) to 26 (most severe enlarged PVS in each region)67,68. We developed an algorithm58 based on anatomical location, appearance on FLAIR, and size to determine the most likely underlying pathology of a given lesion. A hyperintense FLAIR rim around a T1 void is by far the single most important determining factor to distinguish enlarged PVS from infarcts, although the need for a FLAIR rim is lower in areas in which enlarged PVS rarely exist (such as the brain stem or the upper basal ganglia). Microbleeds were visually rated as hypointense round or ovoid lesions on GRE or SWI, surrounded at least halfway by parenchyma with a “blooming” effect and no hyperintensity on accompanying T1-weighted or FLAIR scans to distinguish them from iron or calcium deposits, bone, or vessel flow voids. Due to a skewed distribution (i.e., if present, most scans had 1 microbleed), microbleeds were scored across the whole brain as present or not present. Infarcts were visually rated on T2-weighted FLAIR scans as discrete hypointense lesions greater than 5 mm with a partial or complete hyperintense ring, confirmed on T1 scans as hypointense areas, and scored across the whole brain as present or not present due to a skewed distribution. Amyloid PET with [11C]PiB or [18F]Florbetapir were harmonized into the centiloid scale using reported formulas available on Global Alzheimer’s Association Information Network (GAAIN; http://www.gaain.org)69,70. Tau PET with [18F]Flortaucipir was quantified as standard uptake value ratio (SUVR, 80–100 min post-injection, FreeSurfer-defined cerebellar gray matter reference region) in Braak I/II, Braak III/IV, and Braak V/VI for early, middle, and late tau burden. Biomarkers were available in 242 participants for WMH, 182 participants for enlarged PVS score, 140 participants for microbleeds, and 237 participants for infarcts; 215 participants for amyloid PET, and 175 participants for tau PET.
Genotyping
Participants were genotyped for APOE (rs429358 and rs7412) with the Kompetitive allele-specific polymerase chain reaction genotyping system (LGC Genomics; Berlin, Germany). For these analyses, individuals with at least one copy of the APOE-ε4 allele were classified as APOE-ε4 carriers.
Statistical analysis
Age, amyloid burden, and tau burden were compared across clinical diagnostic groups to support the use of chronological age as disease progression in adults with Down syndrome, while demographic characteristics (i.e., sex, premorbid intellectual developmental disability, APOE status) and vascular risk factors (i.e., hypertension, diabetes, hyperlipidemia, body mass index (BMI), and obstructive sleep apnea (OSA)) were compared across clinical diagnostic groups to assess potential covariates. Vascular risk factors were reported by participants or their informants as part of their health history (“Do you currently have or have you ever had a diagnosis of [disease]”) or obtained from medical/health records. Information was aggregated across sources and binarized as “yes” or “no”. Body mass index was objectively measured and used continuously. Cerebrovascular biomarkers were fit with piece-wise, left null regression models against age to estimate the age inflection point at which these cerebrovascular markers emerge, adjusting for study protocol (i.e., NiAD vs ADDS). The Davies test71 was used to determine if the slope after the estimated inflection point was different from the slope before the estimated inflection point (i.e., zero). General linear models were used for continuous variables (i.e., WMH, enlarged PVS) and logistic regression models were used for dichotomous variables (i.e., the presence of microbleeds, the presence of infarcts). Log transformations for skewed variables (i.e., WMH volume) and multisite harmonization methods (e.g., ComBat72) were explored, and led to minimal improvements in model fits with similar results; therefore, untransformed WMH volumes were used and study was included as a simple covariate in reported models. Further, the interpretation of the inflection point as the age at which cerebrovascular biomarkers visibly emerge on MRI is preserved in comparison to the age at which a transformed variable deviates from zero.
As amyloid precursor protein is overexpressed from birth in adults with Down syndrome, age can reasonably represent disease duration and cross-sectional models can provide pseudo-longitudinal trajectories of cerebrovascular biomarker development11,15. Amyloid chronicity73 has also been shown to represent disease progression beyond chronological age, but would limit our sample size to those with amyloid PET. To determine the emergence of cerebrovascular disease relative to classical AD biomarkers, including amyloid and tau, we also fit left null regression models with age for amyloid PET Centiloids and tau PET SUVRs.
Traditional vascular risk factors, including hypertension and diabetes type 2, are lower in adults with Down syndrome compared with adults without Down syndrome; however, some vascular risk factors are present at similar or higher rates, including hyperlipidemia, high BMI, and OSA. The residuals from the piecewise left-null regression against age (i.e., higher or lower biomarker level than expected for a given age) were fit against demographic data, vascular risk factors, amyloid and tau burden, and AD diagnosis (Cognitively-Stable (reference group), MCI-DS, AD dementia) to determine their influence on the development of each cerebrovascular disease biomarker. All statistics were run in R v4.2.2.
Data availability
ABC-DS is committed to providing rapid public access to all clinical, cognitive and biomarker (fluid and imaging) data, without embargo, and access to the biological samples by qualified scientific investigators. ABC-DS data are transferred to the Laboratory of Neuro Imaging (LONI), for harmonization, documentation and de-identification; biospecimen samples are transferred and managed by the National Centralized Repository for Alzheimer’s Disease and Related Dementias (NCRAD) and an ABC-DS biospecimen bank. As of May 2021, data from the first and second waves of longitudinal data are available for requests. Qualified investigators can submit requests for access to data and samples (https://pitt.co1.qualtrics.com/jfe/form/SV_cu0pNCZZlrdSxUN), and all requests will be reviewed by ABC-DS investigators and NIH staff. Approved data requests will be managed by the ABC-DS Biostatistics and Data Management Core for access to the clinical, cognitive, and neuroimaging data listed below. Upon approval and availability of biospecimen samples, NCRAD will distribute DNA, plasma and serum, and the ABC-DS biospecimen bank will distribute CSF. LONI will store the associated data for access by approved investigators (https://ida.loni.usc.edu/collaboration/access/appLicense.jsp;jsessionid=AC572158DA02C57FD870AE42D137FFF0).
References
Jervis, G. A. Early senile dementia in mongoloid idiocy. Am. J. Psychiatry 105, 102–106 (1948).
Glenner, G. G., Wong, C. W., J. B. & communications. Alzheimer’s disease and Down’s syndrome: Sharing of a unique cerebrovascular amyloid fibril protein. Biochem. Biophys. Res. Commun. 122, 1131–1135 (1984).
Beyreuther, K. & Masters, C. L. Amyloid precursor protein (APP) and ΒZA4 amyloid in the etiology of Alzheimer’s disease: Precursor-product relationships in the derangement of neuronal function. Brain Pathol. 1, 241–251 (1991).
Hardy, J. & Allsop, D. Amyloid deposition as the central event in the aetiology of Alzheimer’s disease. Trends Pharmacol. Sci. 12, 383–388 (1991).
Selkoe, D. J. The molecular pathology of Alzheimer’s disease. Neuron 6, 487–498 (1991).
Hardy, J. A. & Higgins, G. A. J. S. Alzheimer’s disease: The amyloid cascade hypothesis. Science 256, 184–185 (1992).
Oyama, F. et al. Down’s syndrome: Up-regulation of β-amyloid protein precursor and τ mRNAs and their defective coordination. J. Neurochem. 62(1062), 1066 (1994).
Rafii, M. S. et al. The AT (N) framework for Alzheimer’s disease in adults with Down syndrome. Alzheimer’s Dement.: Diagn. Assess. Dis. Monit. 12, e12062 (2020).
Hartley, S. L. et al. AT (N) biomarker profiles and Alzheimer’s disease symptomology in Down syndrome. Alzheimer’s Dement. 20(1), 366–375 (2023).
Fortea, J. et al. Alzheimer’s disease associated with Down syndrome: A genetic form of dementia. Lancet Neurol. 20(930), 942 (2021).
Boerwinkle, A. H. et al. Comparison of amyloid burden in individuals with Down syndrome versus autosomal dominant Alzheimer’s disease: A cross-sectional study. Lancet Neurol. 22(55), 65 (2023).
Jack, C. R. Jr. et al. NIA-AA research framework: Toward a biological definition of Alzheimer’s disease. Alzheimers Dement. 14, 535–562. https://doi.org/10.1016/j.jalz.2018.02.018 (2018).
Hampel, H. et al. Developing the ATX (N) classification for use across the Alzheimer disease continuum. Nat. Rev. Neurol. 17, 580–589 (2021).
Workgroup, A. s. A. Revised criteria for diagnosis and staging of Alzheimer's disease. (2023).
Iulita, M. F. et al. Association of Alzheimer disease with life expectancy in people with Down syndrome. JAMA Netw. Open 5, e2212910–e2212910 (2022).
Cummings, J. J. D. Anti-amyloid monoclonal antibodies are transformative treatments that redefine Alzheimer’s disease therapeutics. Drugs 83(7), 569–576 (2023).
Livingston, G. et al. Dementia prevention, intervention, and care: 2020 report of the Lancet Commission. Lancet 396, 413–446 (2020).
Kapasi, A., DeCarli, C. & Schneider, J. A. Impact of multiple pathologies on the threshold for clinically overt dementia. Acta Neuropathol. 134, 171–186. https://doi.org/10.1007/s00401-017-1717-7 (2017).
Barnes, L. L. et al. Mixed pathology is more likely in black than white decedents with Alzheimer dementia. Neurology 85, 528–534. https://doi.org/10.1212/wnl.0000000000001834 (2015).
Brickman, A. M. et al. Reconsidering harbingers of dementia: Progression of parietal lobe white matter hyperintensities predicts Alzheimer’s disease incidence. Neurobiol. Aging 36, 27–32 (2015).
Lao, P. J. et al. Amyloid, cerebrovascular disease, and neurodegeneration biomarkers are associated with cognitive trajectories in a racially and ethnically diverse, community-based sample. Neurobiol. Aging 117, 83–96 (2022).
Lee, S. et al. White matter hyperintensities are a core feature of Alzheimer’s disease: Evidence from the dominantly inherited Alzheimer network. Ann. Neurol. 79, 929–939. https://doi.org/10.1002/ana.24647 (2016).
Schoemaker, D. et al. White matter hyperintensities are a prominent feature of autosomal dominant Alzheimer’s disease that emerge prior to dementia. Alzheimer’s Res. Ther. 14, 89 (2022).
Laing, K. K. et al. Cerebrovascular disease promotes tau pathology in Alzheimer’s disease. Brain Commun. 2(2), fcaa132 (2020).
Yang, A. C. et al. A human brain vascular atlas reveals diverse mediators of Alzheimer’s risk. Nature 603(885), 892 (2022).
Lao, P. J. et al. Alzheimer-related cerebrovascular disease in Down syndrome. Ann. Neurol. 88, 1165–1177 (2020).
Moni, F. et al. Probing the proteome to explore potential correlates of increased Alzheimer’s-related cerebrovascular disease in adults with Down syndrome. Alzheimer’s Dement. 18(1744), 1753 (2022).
Edwards, N. C. et al. Cerebrovascular disease drives Alzheimer plasma biomarker concentrations in adults with Down syndrome. Medrxiv. https://doi.org/10.1101/2023.11.28.23298693 (2023).
Zlokovic, B. V. J. N. R. N. Neurovascular pathways to neurodegeneration in Alzheimer’s disease and other disorders. Nat. Rev. Neurosci. 12(723), 738 (2011).
Scheffer, S. et al. Vascular hypothesis of Alzheimer disease: Topical review of mouse models. Arterioscler. Thromb. Vasc. Biol. 41(1265), 1283 (2021).
Gouveia-Freitas, K. & Bastos-Leite, A. J. J. N. Perivascular spaces and brain waste clearance systems: Relevance for neurodegenerative and cerebrovascular pathology. Neuroradiology 63(1581), 1597 (2021).
van Dalen, J. W. et al. Cortical microinfarcts detected in vivo on 3 Tesla MRI: Clinical and radiological correlates. Stroke 46, 255–257. https://doi.org/10.1161/strokeaha.114.007568 (2015).
Prins, N. D. & Scheltens, P. White matter hyperintensities, cognitive impairment and dementia: An update. Nat. Rev. Neurol. 11, 157–165 (2015).
Vermeer, S. E. et al. Silent brain infarcts and the risk of dementia and cognitive decline. N. Engl. J. Med. 348, 1215–1222 (2003).
Zammit, M. D. et al. Neurofibrillary tau depositions emerge with subthreshold cerebral beta-amyloidosis in Down syndrome. NeuroImage Clin. 31, 102740 (2021).
Zammit, M. D. et al. Characterizing the emergence of amyloid and tau burden in Down syndrome. Alzheimer’s Dement. 20(1), 388–398 (2023).
Tudorascu, D. et al. Relationship of amyloid beta and neurofibrillary tau deposition in Neurodegeneration in Aging Down Syndrome (NiAD) study at baseline. Transl. Res. Clin. Interv. 6, e12096 (2020).
Royse, S. K. et al. Unhealthy white matter connectivity, cognition, and racialization in older adults. Alzheimer’s Dement. 20(3), 1483–1496 (2023).
Head, E. et al. Cerebrovascular pathology in Down syndrome and Alzheimer disease. Acta Neuropathol. Commun. 5(1), 9 (2017).
Charidimou, A. et al. White matter perivascular spaces: An MRI marker in pathology-proven cerebral amyloid angiopathy?. Neurology 82, 57–62 (2014).
Mhatre, P. G. et al. The association between sex and risk of Alzheimer’s disease in adults with Down syndrome. J. Clin. Med. 10, 2966 (2021).
Schupf, N. et al. Onset of dementia is associated with apolipoprotein E ε4 in Down’s syndrome. Ann. Neurol.: Off. J. Am. Neurol. Assoc. Child Neurol. Soc. 40, 799–801 (1996).
Fleming, V. et al. Weight loss and Alzheimer’s disease in Down Syndrome. J. Alzheimer’s Dis. 91(3), 1215–1227 (2023).
Jolink, W. M. et al. Histopathology of cerebral microinfarcts and microbleeds in spontaneous intracerebral hemorrhage. Transl. Stroke Res. 14(174), 184 (2023).
Wardlaw, J. M., Valdes Hernandez, M. C. & Munoz-Maniega, S. What are white matter hyperintensities made of? Relevance to vascular cognitive impairment. J. Am. Heart Assoc. https://doi.org/10.1161/jaha.114.001140 (2015).
Lao, P. et al. White matter regions with low microstructure in young adults are associated with white matter hyperintensities in late life. bioRxiv, 517763, doi:https://doi.org/10.1101/517763 (2019).
Wilcock, D. et al. MarkVCID cerebral small vessel consortium: I Enrollment, clinical, fluid protocols. Alzheimer’s Dement. 17(704), 715 (2021).
Foley, K. E. & Wilcock, D. M. Soluble biomarkers of cerebrovascular pathologie. Stroke 55, 801–811 (2024).
Bateman, R. J. et al. Clinical and biomarker changes in dominantly inherited Alzheimer’s disease. N. Engl. J. Med. 367, 795–804. https://doi.org/10.1056/NEJMoa1202753 (2012).
Johnson, E. C. et al. 2023 Cerebrospinal fluid proteomics define the natural history of autosomal dominant Alzheimer’s disease. Nat. Med. 29, 1979 (1988).
Littau, J. L. et al. Evidence of beta amyloid independent small vessel disease in familial Alzheimer’s disease. Brain Pathol. 32, e13097 (2022).
Carmona-Iragui, M. et al. Cerebral amyloid angiopathy in Down syndrome and sporadic and autosomal-dominant Alzheimer’s disease. Alzheimer’s Dement. 13, 1251–1260 (2017).
Tang, M. et al. Neurological manifestations of autosomal dominant familial Alzheimer’s disease: A comparison of the published literature with the dominantly inherited Alzheimer Network observational study (DIAN-OBS). Lancet Neurol. 15, 1317–1325 (2016).
Joseph-Mathurin, N. et al. Longitudinal accumulation of cerebral microhemorrhages in dominantly inherited Alzheimer disease. Neurology 96, e1632–e1645 (2021).
Shirzadi, Z. et al. Etiology of white matter hyperintensities in autosomal dominant and sporadic Alzheimer disease. JAMA Neurol. 80, 1353–1363 (2023).
Ingala, S. et al. The relation between APOE genotype and cerebral microbleeds in cognitively unimpaired middle-and old-aged individuals. Neurobiol. Aging 95(104), 114 (2020).
Van Den Heuvel, D. et al. Different progression rates for deep white matter hyperintensities in elderly men and women. Neurology 63(1699), 1701 (2004).
Gutierrez, J. et al. Pulsatile and steady components of blood pressure and subclinical cerebrovascular disease: The Northern Manhattan Study. J. Hypertens. 33, 2115 (2015).
Charidimou, A. et al. MRI-visible perivascular spaces in cerebral amyloid angiopathy and hypertensive arteriopathy. Neurology 88, 1157–1164 (2017).
Lao, P. et al. Obstructive sleep apnea, cerebrovascular disease, and amyloid in older adults with Down syndrome across the Alzheimer’s continuum. Sleep Adv. 3(1), 130 (2022).
Cody, K. A. et al. Association of sleep with cognition and beta amyloid accumulation in adults with Down syndrome. Neurobiol. Aging 93, 44–51 (2020).
Kenshole, A. V., Gallichan, D., Pahl, S. & Clibbens, J. Lifestyle factors and Alzheimer’s disease in people with Down syndrome. J. Appl. Res. Intell. Disabil. 30, 58–66 (2017).
Mihaila, I. et al. Leisure activity and caregiver involvement in middle-aged and older adults with Down syndrome. Intell. Dev. Disabil. 55, 97–109 (2017).
Mihaila, I. et al. Leisure activity brain β-amyloid, and episodic memory in adults with Down Syndrome. Dev. Neurobiol. 79, 738–749 (2019).
Handen, B. L. et al. The Alzheimer’s biomarker consortium-Down syndrome: Rationale and methodology. Alzheimer’s Dement.: Diagn. Assess. Dis. Monit. 12, e12065 (2020).
Krinsky-McHale, S. J. et al. Promising outcome measures of early Alzheimer’s dementia in adults with Down syndrome. Alzheimer’s Dement.: Diagn. Assess. Dis. Monit. 12, e12044 (2020).
Gutierrez, J. et al. Brain perivascular spaces as biomarkers of vascular risk: Results from the northern manhattan study. Am. J. Neuroradiol. 38, 862–867 (2017).
Gutierrez, J., Rundek, T., Ekind, M., Sacco, R. L. & Wright, C. B. Perivascular spaces are associated with atherosclerosis: An insight from the Northern Manhattan study. Am. J. Neuroradiol. 34, 1711–1716 (2013).
Klunk, W. E. et al. The Centiloid Project standardizing quantitative amyloid plaque estimation by PET. Alzheimers Dement. 11(1), 1–15. https://doi.org/10.1016/j.jalz.2014.07.003 (2015).
Tudorascu, D. L. et al. The use of Centiloids for applying [(11)C]PiB classification cutoffs across region-of-interest delineation methods. Alzheimer’s Dement. 10, 332–339. https://doi.org/10.1016/j.dadm.2018.03.006 (2018).
Davies, R. B. Hypothesis testing when a nuisance parameter is present only under the alternative: Linear model case. Biometrika 64(2), 247–254 (2002).
Orlhac, F. et al. A guide to ComBat harmonization of imaging biomarkers in multicenter studies. J. Nucl. Med. 63, 172–179 (2022).
Koscik, R. L. et al. Amyloid duration is associated with preclinical cognitive decline and tau PET. Alzheimer’s Dement.: Diagn. Assess. Dis. Monit. 12, 63–72 (2020).
Acknowledgements
The Alzheimer’s Biomarkers Consortium-Down Syndrome (ABC-DS) is funded by the National Institute on Aging and the National Institute for Child Health and Human Development (U01 AG051406, U01 AG051412, U19 AG068054). The work contained in this publication was also supported through the following National Institutes of Health Programs: The Alzheimer’s Disease Research Centers Program (P50 AG008702, P30 AG062421, P50 AG16537, P50 AG005133, P50 AG005681, P30 AG062715, and P30 AG066519), the Eunice Kennedy Shriver Intellectual and Developmental Disabilities Research Centers Program (U54 HD090256, U54 HD087011, and P50 HD105353), the National Center for Advancing Translational Sciences (UL1 TR001873, UL1 TR002373, UL1 TR001414, UL1 TR001857, UL1 TR002345), the National Centralized Repository for Alzheimer Disease and Related Dementias (U24 AG21886), and DS-Connect® (The Down Syndrome Registry) supported by the Eunice Kennedy Shriver National Institute of Child Health and Human Development (NICHD). In Cambridge, UK this research was supported by the NIHR Cambridge Biomedical Research Centre and the Windsor Research Unit, CPFT, Fulbourn Hospital Cambridge, UK. The authors are grateful to the ABC-DS study participants, their families and care providers, and the ABC-DS research and support staff for their contributions to this study. This manuscript has been reviewed by ABC-DS investigators for scientific content and consistency of data interpretation with previous ABC-DS study publications. The content is solely the responsibility of the authors and does not necessarily represent the official views of the NIH, the CPFT, the NIHR or the UK Department of Health and Social Care. Alzheimer’s Biomarker Consortium- Down Syndrome (ABC-DS) Investigators: Howard J. Aizenstein, MD PhD; Beau M. Ances, MD PhD; Howard F. Andrews, PhD; Karen Bell, MD; Rasmus M. Birn, PhD; Adam M. Brickman, PhD; Peter Bulova, MD; Amrita Cheema, PhD; Kewei Chen, PhD; Bradley T. Christian, PhD; Isabel Clare, PhD; Lorraine Clark, PhD; Ann D. Cohen, PhD; John N. Constantino, MD; Eric W. Doran, MS; Anne Fagan, PhD; Eleanor Feingold, PhD; Tatiana M. Foroud, PhD; Benjamin L. Handen, PhD; Jordan Harp, PhD; Sigan L. Hartley, PhD; Elizabeth Head, PhD; Rachel Henson, MS; Christy Hom, PhD; Lawrence Honig, MD; Milos D. Ikonomovic, MD; Sterling C Johnson, PhD; Courtney Jordan, RN; M. Ilyas Kamboh, PhD; David Keator, PhD; William E. Klunk, MD PhD; Julia K. Kofler, MD; William Charles Kreisl, MD; Sharon J. Krinsky-McHale, PhD; Florence Lai, MD; Patrick Lao, PhD; Charles Laymon, PhD; Joseph Hyungwoo Lee, PhD; Ira T. Lott, MD; Victoria Lupson, PhD; Mark Mapstone, PhD; Chester A. Mathis, PhD; Davneet Singh Minhas, PhD; Neelesh Nadkarni, MD; Sid O’Bryant, PhD; Melissa Parisi, MD PhD; Deborah Pang, MPH; Melissa Petersen, PhD; Julie C. Price, PhD; Margaret Pulsifer, PhD; Michael S. Rafii, MD PhD, Eric Reiman, MD; Batool Rizvi, MS; Herminia Diana Rosas, MD; Laurie Ryan, PhD; Frederick Schmitt, PhD; Nicole Schupf, PhD; Wayne P. Silverman, PhD; Dana L. Tudorascu, PhD; Rameshwari Tumuluru, MD; Benjamin Tycko, MD PhD; Badri Varadarajan, PhD; Desiree A. White, PhD; Michael A. Yassa, PhD; Shahid Zaman, MD PhD; Fan Zhang, PhD. AVID Pharmaceuticals supplied AV1451, but did not provide financial support for this study nor did it have a role in designing the study or interpreting the results.
Funding
This work was supported by US National Institutes of Health grants RF1 AG079519, U19 AG068054, U01 AG051412, U01 AG051406, and R00 AG065506.
Author information
Authors and Affiliations
Contributions
All authors contributed to the drafting of the work. P.J.L. contributed to conception, design, analysis, and interpretation of the work. N.E., L.F.A., and B.R. contributed to interpretation of the work. M.A. and D.T. contributed to analysis of the work. D.T., H.D.R., M.Y., B.T.C., M.M., B.H., M.E.Z., J.G., D.W., E.H., and A.M.B. contributed to the design, acquisition, and interpretation of the work. E.H. and A.M.B. additionally contributed to the conception of the work.
Corresponding author
Ethics declarations
Competing interests
Michael A. Yassa has received consulting fees from Eisai Cognito Therapeutics, LLC, CuraSen Therapeutics, Inc, and Enthorin Therapeutics, LLC. Adam Brickman receives compensation for consultation to Cognition Therapeutics and Cognito Therapeutics and for his role on the Scientific Advisory Board of CogState. He is an inventor a patent for white matter hyperintensity quantification (US Patent US9867566B2) and serves on a Data Safety Monitoring Board for University of Illinois, Urbana-Champaign. All other authors do not have competing interests to declare.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Lao, P., Edwards, N., Flores-Aguilar, L. et al. Cerebrovascular disease emerges with age and Alzheimer’s disease in adults with Down syndrome. Sci Rep 14, 12334 (2024). https://doi.org/10.1038/s41598-024-61962-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-024-61962-y
- Springer Nature Limited