
Abstract
In this work, a novel series of N-phenylacetamide-1,2,3-triazole-indole-2-carboxamide derivatives 5a–n were designed by consideration of the potent α-glucosidase inhibitors containing indole and carboxamide-1,2,3-triazole-N-phenylacetamide moieties. These compounds were synthesized by click reaction and evaluated against yeast α-glucosidase. All the newly title compounds demonstrated superior potency when compared with acarbose as a standard inhibitor. Particularly, compound 5k possessed the best inhibitory activity against α-glucosidase with around a 28-fold improvement in the inhibition effect in comparison standard inhibitor. This compound showed a competitive type of inhibition in the kinetics. The molecular docking and dynamics demonstrated that compound 5k with a favorable binding energy well occupied the active site of α-glucosidase.
Similar content being viewed by others
Introduction
α-Glucosidase is a digestive enzyme that belongs to a category of glycoside hydrolases. This enzyme liberates glucose of oligosaccharides and disaccharides by cleaving the glycosidic bond1,2,3. The most important role of this enzyme in the body is absorption of dietary carbohydrates by breaking them to glucose. Therefore, preventing the breakdown of carbohydrates by inhibition of α-glucosidase is a treatment method for type 2 diabetes4,5,6. On the other hand, α-glucosidase play an important role in maturation of glycoprotein and its folding7. This action of α-glucosidase has a special importance in treatment of other diseases such viral infection and cancer8,9,10. There are drugs with α-glucosidase inhibition mechanism for treatment of type 2 diabetes such as acarbose, emiglitate, miglitol, voglibose, but due to their low potency and digestive side effects, development of α-glucosidase inhibitor is still an important goal for medicinal chemists11.
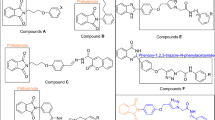
Recently, many series of α-glucosidase inhibitors containing 1,2,3-triazole ring have been reported12. Based on the construction of the 1,2,3-triazole ring by the click reaction is a valuable tool in connecting effective pharmacophores to each other, by using this property of click reaction, effective compounds against α-glucosidase were obtained12. Moreover, the modified structures based on 1,2,3-triazole ring such as benzyl-1,2,3-triazole, phenoxy-1,2,3-triazole, and 1,2,3-triazole-N-phenylacetamide moieties were also observed in the potent α-glucosidase inhibitors13,14. In addition to the latter mentioned modified structures, N-phenylacetamide-1,2,3-triazole-carboxamide moiety also was fund in potent α-glucosidase inhibitors A (Fig. 1)15. These α-glucosidase inhibitors have an acridine ring in their structures. Acridine ring and other nitrogen containing heterocycles are very popular in the design of biologically active compounds16,17. One of the most popular of these rings, especially in the design of α-glucosidase inhibitors, is indole ring18. Various types of synthetic indole-based α-glucosidase inhibitors such as compounds B had been introduced19.

Design strategy for N-phenylacetamide-1,2,3-triazole-indole-2-carboxamide scaffold as the new α-glucosidase inhibitor.
As can be seen in Fig. 1, the successful synthesis of acridine-9-carboxamide-1,2,3-triazole-N-phenylacetamide derivatives and the effectiveness of these compounds against α-glucosidase encouraged us to use indole as a popular α-glucosidase inhibitor pharmacophore instead of acridine18. As a result, N-phenylacetamide-1,2,3-triazole-indole-2-carboxamide scaffold was designed and fourteen derivatives of it were synthesized (Fig. 1). These derivatives carefully evaluated in vitro and in silico.
Results and discussion
Chemistry
In this work, we synthesized a novel series of N-phenylacetamide-1,2,3-triazole-indole-2-carboxamide derivatives 5a–n by click reaction. As outlined in Scheme 1, N-(prop-2-yn-1-yl)-1H-indole-2-carboxamide 3, as a participant component in click reaction was produced by reaction between 1H-indole-2-carboxylic acid 1 and propargylamine 2 in the presence of TBTU and Et3N. Compound 3 and azide derivatives 4a–n converted to target compounds 5a–n by a click reaction15.

Synthesis of N-phenylacetamide-1,2,3-triazole-indole-2-carboxamide derivatives 5a–n: (a) TBTU, Et3N, DMF, RT, 24 h; (b) CuSO4.5H2O, sodium ascorbate, RT, 18–24 h.
For example, 1H NMR and 13C NMR interpretations of the selected compound 5a were depicted in Scheme 2.

NMR interpretation of compound 5a (unit of numbers is ppm).
Anti-α-glucosidase assay and SAR survey
Inhibitory activities of the new N-phenylacetamide-1,2,3-triazole-indole-2-carboxamide derivatives 5a–n were evaluated against yeast form of α-glucosidase and the observed data were compared to acarbose as a standard inhibitor of this enzyme. As evidenced by structures and inhibition data of title compounds 5a–n that listed in Table 1, all derivatives of N-phenylacetamide-1,2,3-triazole-indole-2-carboxamide scaffold with IC50 values ranging from 26.8 ± 0.5 to 311.3 ± 2.4 μM are excellent α-glucosidase inhibitor when compared to standard inhibitor with IC50 value of 752.0 ± 2.0 μM.
Based on the structure–activity relationship (SAR) screen, the strongest and weakest derivatives were halo-substituted compounds: 4-bromo derivative 5k as the most potent compound and 3-chloro derivative 5h as the less potent compound. Replacement of 3-chloro substituent of compound 5h with 3-nitro group as in the case of compound 5l created a significant increase in inhibition effect. Moreover, changing the position of nitro group from 3-position of compound 5l to 4-position as in the case of compound 5m, led to a moderate increase in the inhibition effect. Moreover, introduction of a methyl group on 2-position of phenyl ring of N-phenylacetamide moiety of 4-nitro derivative 5m as in the case of 2-methyl-4-nitro derivative 5n increased inhibitory activity against α-glucosidase.
The second potent compound was 2,4-dichloro derivative 5j. The inhibitory activity of this compound reduced dramatically when the 2-chloro was removed from phenyl ring of N-phenylacetamid moiety as in the case of 4-chloro derivative 5i. On the other hand, the replacement of the 4-chloro substituent of compound 5i with methoxy group did not change inhibition effect. In contrast, the replacement of 4-chloro substituent of compound 5i with ethyl or methyl group led to a significant increase in the inhibitory activity as observed in 4-ethyl derivative 5f and 4-methyl derivative 5c.
SAR study also demonstrated that un-substituted compound 5a and 3-methyl derivative 5b showed almost the same effects. Introduction of the second methyl group in 2-position of 3-methyl derivative 5b as in the case of 2,3-dimethyl derivative 5d led to a negligible decrease in inhibition effect. On the other hand, replacement of 3-methyl group in compound 5d to 6-position as in the case of 2,6-dimethyl derivative 5e created a significant decrease in anti-α-glucosidase activity.
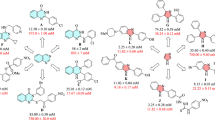
Moreover, the comparison of inhibitory activity of the substituted compounds 5b–n with un-substituted compound 5a depicted in Fig. 2. This figure showed that introduction of 4-Br, 2,4-dichloro, 4-ethyl, 4-methyl, and 2-methyl-4-nitro substituents on the phenyl ring of N-arylacetamide moiety improved inhibition effect in comparison to un-substituted compound 5a while the presence of 3-methyl, 2,3-dimethyl, 4-nitro, 4-methoxy, 4-chloro, 3-nitro, 2,6-dimethyl, and 3-chloro substituents deteriorated anti-α-glucosidase activity.

The comparison of anti-α-glucosidase activity of un-substituted compound 5a with substituted compounds 5b–n.
The comparison of IC50 values of the new N-phenylacetamide-1,2,3-triazole-indole-2-carboxamide derivatives 5 with their corresponding analogs of acridin-9-carboxamide-1,2,3-triazole-N-phenylacetamide derivatives A is showed in Fig. 315. As can be seen in this figure, all derivatives of the new compounds 5 were more potent than their analogs of A series.

α-Glucosidase inhibitory activity of the template compounds A in comparison to their corresponding analogs of new compounds 5.
Kinetics
Kinetics of α-glucosidase inhibition performed on the compound 5k as the most potent compound. As shown in Fig. 4a, the lines of Lineweaver–Burk plot with enhancement the concentration of compound 5k had a fixed intercept on the Y-intercept and X-slopes. As the result, with increasing concentration of compound 5k, Vmax values remained constant while Km values increased. The latter obtained result demonstrated that the most potent new compound was a competitive inhibitor (Fig. 4a). Furthermore, according to Fig. 4b (secondary plot of Lineweaver–Burk plots), the Ki value of compound 5k was 26.0 µM.

Kinetics of the new potent α-glucosidase inhibitor 5k.
α-Amylase assay
Inhibitory activity of the most poten new α-glucosidase inhibitors 5k, 5j, and 5f was evaluated against α-amylase (pancreatic form)15. In vitro inhibition assay of compounds 5k, 5j, and 5f demonstrated that these compounds with IC50 value > 150 μM were inactive against α-amylase when compared with positive control acarbose with IC50 of 108 ± 0.71 μM. Considering that most of the side effects associated with marketed α-glucosidase inhibitors are due to their inhibitory effect on α-amylase, this feature can be considered an advantage for our new compounds11.
Molecular docking study
In order to explain interaction modes of the newly synthesized compounds in the α-glucosidase active site and rationalize observed SAR, molecular docking study carried out. Reliability of docking process was validated by re-docking of acarbose over modeled α-glucosidase and obtained RMSD value was 1.7 Å that is in the acceptable range (< 0.2). The superposed structure of standard inhibitor (acarbose) and the most potent new compound (compound 5k) in the active site of α-glucosidase was shown in Fig. 5.

Acarbose (cyan) and the most potent compound 5k (pink) superimposed in the α-glucosidase active site.
Interaction modes of the selected compounds and acarbose were showed in the Figs. 6, 7 and 8. Acarbose as the standard α-glucosidase inhibitor with binding energy (BE) of − 4.04 kcal/mol established interactions with residues Asn241 (H-bond), Glu304 (H-bond), Ser308 (H-bond), Thr301 (H-bond), Thr307 (H-bond), Pro309 (H-bond), Arg312 (H-bond), Gln322 (H-bond), His279 (hydrophobic), Val305 (non-classical H-bond), His239 (non-classical H-bond), Thr307 (unfavorable), and Arg312 (unfavorable) in the active site of target enzyme15.

2D interaction modes of acarbose and the most potent compounds 5k and 5j in the α-glucosidase active site.

2D interaction modes of compounds 5f and 5c in the α-glucosidase active site.

2D interaction modes of compounds 5n and 5m in the α-glucosidase active site.
The most active compound 5k established a hydrogen bond with Thr301 and two non-classical hydrogen bonds with His279 and Ser308 (Fig. 6). The latter amino acid also formed an unfavorable interaction with compound 5k. Three π-interactions between compound 5k and the active site residues were observed: a π-anion interaction with Glu304, a π-cation interaction with His239, and a π–π interaction with Phe157. Furthermore, compound 5k formed several hydrophobic interactions with residues Val316, Val305, Pro309, and Arg312. BE of compound 5k was − 8.49 kcal/mol.
The second potent compound 5j established two hydrogen bonds with residues Ser308 and Thr307 and a non-classical hydrogen bond with Arg439 (Fig. 6). This compound formed two π-anion interactions with Glu304. Compound 5j also established several hydrophobic interactions with residues Tyr313, Pro309, Phe311, and Arg312 and BE of this compound was -9.01 kcal/mol.
4-Ethyl derivative 5f as the third potent compound created a classical hydrogen bond with residue Phe157 and a non-classical hydrogen bond with residue Arg312 (Fig. 7). Two π-anion interactions observed between compound 5f and Glu304. The latter compound also formed hydrophobic interactions with residues Phe300 and Pro309. BE of compound 5f was − 8.73 kcal/mol. Replacement of 4-ethyl substituent with 4-methyl substituent as in the case of compound 5c, created a negligible decrease in the inhibition effect (Table 1). As can be seen in the interaction mode of compound 5c, this compound with BE of − 8.18 kcal/mol established three hydrogen bonds with Thr307 (two H-bond) and Glu304, a non-classical hydrogen bond with residue His239, and several hydrophobic interactions with Phe158, Arg312, Pro309, and Val305. The comparison of the BEs of compound 5f with 5c showed that the third potent compound 5f with BE value lower than the fourth potent compound 5c attached to active site. This finding is in agreement with in vitro obtained data (Table 1).
As mentioned in SAR study (Sect. 2.2), the addition of a methyl group on position 2 on phenyl ring of N-phenylacetamide moiety of 4-nitro derivative 5m, as in the case of compound 5n, the inhibitory activity increased to about two fold. This observation was confirmed by docking study in two ways:
-
1.
Compound 5n interacted with a greater number of important amino acids (Val305, His279, His239, Asn241, Pro309, and Arg312) in the active site in comparison to compound 5m (Gly306, Pr0309, Glu304, and Arg312).
-
2.
BE of compound 5n (BE = − 8.17 kcal/mol) was lower than compound 5m (BE = − 8.08 kcal/mol).
Molecular dynamics
It is difficult to grasp the interaction between a ligand and a receptor or assess the stability and flexibility of the resultant complex without resorting to molecular dynamic simulation, given that all atoms in the natural environment are in perpetual motion. To achieve this goal, the protein–ligand complex is confined within a box, after which water molecules and ions are introduced. The dynamics of the ligand-receptor complex are then simulated within this controlled environment. According to the in vitro studies, compound 5k demonstrates the highest potential for inhibiting α-glucosidase. The stability and flexibility of the α-glucosidase-5k complex were examined by simulating its molecular dynamics in an explicit hydration environment. To provide a basis for comparison, a molecular dynamics simulation was conducted on the α-glucosidase-acarbose complex, which serves as a standard inhibitor. This study used a two-step simulation approach. Initially, a 10 ns simulation was conducted to assess the stability of ligands (5k and acarbose) at their binding site on α-glucosidase. Upon confirming the ligands' stability, the simulation duration was extended by another 10 ns to further understand the behavior of these compounds within the enzyme's active site. This extended simulation reaffirmed the stability of 5k and acarbose at their binding site on α-glucosidase. In the subsequent analysis, various tools were utilized to examine the trajectory file from the molecular dynamic simulation.
To measure the stability of the complexes, calculations of root-mean-square deviation (RMSD) and radius of gyration (Rg) were performed on all structures within the trajectory, and corresponding graphs were plotted. Additionally, to evaluate the residual flexibility of α-glucosidase and the flexibility of ligand atoms, the root mean square fluctuation (RMSF) of the backbone atoms in α-glucosidase and the heavy atoms in the ligands were computed. The results of the RMSD calculations are depicted in Fig. 9. According to this figure, the RMSD of the backbone atoms of α-glucosidase in complex with 5k and acarbose exhibits minimal changes over time, never exceeding 0.3 nm. This indicates a stable protein structure. The average RMSD values for α-glucosidase in the complex with acarbose and/or 5k were 0.18 nm and 0.21 nm, respectively. Similarly, the RMSD values of acarbose and/or 5k in the complex with α-glucosidase remained consistently below 0.3 nm over time. The average RMSD values for acarbose and/or 5k in complex with α-glucosidase were 0.12 nm and 0.16 nm, respectively. These observations collectively affirm the stability of both the enzyme and the ligands throughout the simulation period. The radius of gyration (Rg) of α-glucosidase was computed to assess protein compactness during the simulation, as depicted in Fig. 9. The average Rg of α-glucosidase was measured at 2.50 nm and 2.48 nm in the complexes with acarbose and 5k, respectively. In the presence of both acarbose and 5k, the Rg value of α-glucosidase remained within the narrow range of 2.38 to 2.54 nm, showing no significant upward or downward trend throughout the simulation period. This consistent Rg range indicates the stable compactness of α-glucosidase in these complexes.

Superimposed RMSD of Cα atoms of α-glucosidase in complex with 5k (violet) and acarbose (orange) (A). Superimposed RMSD of 5k (violet) and acarbose (orange) in complex with α-glucosidase (B). Time dependence of the radius of gyration (Rg) graph of α-glucosidase in complex with 5k (violet) and acarbose (orange) (C).
Figure 10 illustrates the root mean square fluctuation (RMSF) of α-Glucosidase residues in complex with acarbose and 5k. α-Glucosidase is comprised of various domains, each with distinct structures and functions. As depicted in Fig. 10, different parts of this protein exhibit dissimilar fluctuation patterns. Notably, the fluctuation of α-glucosidase residues in the complexes with acarbose and 5k does not significantly differ; they follow the same pattern. Within α-glucosidase, there exists a cleft between the A domain and B domain, housing the enzyme's active site. Residues from these domains situated in this cleft, which contribute to non-bonded interactions with ligands, display lower fluctuations. This phenomenon aligns with the common observation in proteins, where loops typically exhibit the highest fluctuations. In α-glucosidase, the residues forming the B domain loop and the active site lid also demonstrate significant fluctuations. In Fig. 10, the fluctuation of heavy atoms in acarbose and 5k is depicted. It is evident that the RMSF of all heavy atoms in these ligands is less than 2 nm. This minimal fluctuation serves as an indicator of their stable complex with α-glucosidase, suggesting that intermolecular interactions effectively limit their movements.

RMSF graph of the Cα atoms of α-glucosidase in complex with 5k (violet) and acarbose (orange). Close-up representation of α-glucosidase active site (B). RMSF graph of the heavy atoms of 5k (C) and acarbose (D) in complex with α-glucosidase. Structure of these compounds are illustrated.
Binding free energy analysis
The assessment of ligand binding energy to a receptor can be conducted through the molecular mechanic/Poisson–Boltzmann surface area (MM/PBSA) method, providing insights into the predominant interactions within a ligand–receptor complex. Molecular docking, relying on singular structural snapshots, lacks precision compared to molecular dynamics (MD) simulations, which offer multiple snapshots over time, thus enhancing the accuracy of binding energy estimation. Results from the free binding energy analysis are delineated in Table 2. In this investigation, both acarbose and 5k exhibited negative binding energies. Acarbose, a recognized inhibitor, demonstrated an average MM/PBSA free binding energy of − 115.7 kJ/mol with α-glucosidase, while 5k manifested a binding free energy of − 159.4 kJ/mol. Figure 8 illustrates the binding energy changes over the 20 ns of MD simulation. Both complexes displayed consistent negative binding energies, indicative of stability. 5k showed a binding energy even higher than that of acarbose, which indicates its high affinity to bind to α-glucosidase. Further investigation of free energy components revealed that the molecular mechanics interaction energy (van der Waals energy + electrostatic energy) favored complex formation, while solvation energy (polar solvation energy + solvent accessible surface area energy) impeded it (Fig. 11).

Diagram of binding energy changes during the last 20 ns of simulation time. α-glucosidase in complex with acarbose (orange), and 5k (violet).
The contribution of individual protein residues to binding energy were calculated too. Most residues deemed significant in ligand-receptor interaction via docking studies exhibited negative values in dynamic simulations, though some had minimal impact. Additionally, new residues with substantial binding energy contributions emerged, highlighting the dynamic nature of macromolecules and ligands, which can reveal previously unnoticed intermolecular interactions. Notably, four residues including Lys155, Phe157, Glu276 and Asp349 consistently played a significant role across all complexes with the most negative energies while Asp232 and Arg312 had the most positive energies (Figs. 12 and 13).

Residues with the largest and smallest contribution to the binding energy (KJ/mol) of α-glucosidase-5k complex.

Contribution of α-glucosidase residues to the binding energy (KJ/mol) of α-glucosidase-5k complex.
Conclusion
In conclusion, we designed, synthesized, and screened highly potent α-glucosidase inhibitors from the connection of indole to N-phenylacetamide-1,2,3-triazole-carboxamide moiety. In this regard, 14 derivatives 5a–n were synthesized by alteration of substituents on phenyl ring of N-phenylacetamide moiety. All the synthesized compounds 5a–n were more potent than standard inhibitor and selected compound for their design. Moreover, SAR study demonstrated that 4-bromo derivative 5k exhibited the best activity against the title enzyme. This compound showed 28 times better inhibitory effect than acarbose. Molecular modeling and molecular dynamics demonstrated that studied new compounds with acceptable binding energies interacted with important amino acids of the α-glucosidase active site. These data strongly correlated with the observed in vitro anti-α-glucosidase activity of our new compounds. Thus, presented new scaffold could be a good candidate for initiating a lead anti-diabetes drug discovery program.
Experimental
Synthesis of N-(prop-2-yn-1-yl)-1H-indole-2-carboxamide 3
To a mixture of 1H-indole-2-carboxylic acid 1 (10 mmol) and propargylamine 2 (10 mmol) in DMF (50 mL), TBTU (13 mmol) and Et3N (13 mmol) were added and the obtained mixture was stirred for 24 h at room temperature (RT). After completion of the reaction, water was added to this reaction mixture and the observed participate was filtered to give pure N-(prop-2-yn-1-yl)-1H-indole-2-carboxamide 3.
General procedure for the synthesis of N-phenylacetamide-1,2,3-triazole-indole-2-carboxamide derivatives 5a–n
In the next step, N-(prop-2-yn-1-yl)-1H-indole-2-carboxamide 3 (1 mmol), sodium ascorbate (15 mol %, 0.13 g), and CuSO4 (7 mol%) were added to a stirred mixture of the freshly prepared azide derivatives 4a–n in H2O and t-BuOH (10 ml, 1:1) at RT. After 20–24 h, this reaction mixture was diluted with a mixture of ice and water, precipitated products 5a–n were filtered off, washed with water, and purified by recrystallization in ethanol.
N-((1-(2-oxo-2-(phenylamino)ethyl)-1H-1,2,3-triazol-4-yl)methyl)-1H-indole-2-carboxamide (5a)
Yield: 75%. Light brown solid. M.p. 216–218 °C. 1H NMR (301 MHz, DMSO-d6) δ 11.65 (s, 1H), 10.48 (s, 1H), 9.11 (s, 1H), 8.07 (s, 1H), 7.80–6.86 (m, 10H), 5.34 (s, 2H), 4.78–4.44 (m, 2H). 13C NMR (76 MHz, DMSO-d6) δ 164.70, 161.55, 138.89, 136.93, 131.94, 129.38, 127.55, 125.17, 124.22, 123.81, 122.00, 120.19, 120.16, 119.66, 112.78, 103.23, 52.65, 34.87. Anal. Calcd for C20H18N6O2: C, 64.16; H, 4.85; N, 22.45; Found: C 63.91; H 5.02; N 22.29.
N-((1-(2-oxo-2-(m-tolylamino)ethyl)-1H-1,2,3-triazol-4-yl)methyl)-1H-indole-2-carboxamide (5b)
Yield: 81%. Light brown solid. M.p. 224–226 °C. 1H NMR (301 MHz, DMSO-d6) δ 11.68 (s, 1H), 10.43 (s, 1H), 9.15 (d, J = 5.6 Hz, 1H), 8.09 (s, 1H), 7.65 (d, J = 7.8 Hz, 1H), 7.57–7.35 (m, 3H), 7.22 (d, J = 6.6 Hz, 3H), 7.06 (t, J = 7.4 Hz, 1H), 6.92 (d, J = 7.4 Hz, 1H), 5.36 (s, 2H), 4.83–4.44 (m, 2H), 2.29 (s, 3H). 13C NMR (76 MHz, DMSO-d6) δ 164.65, 161.61, 138.83, 138.62, 136.97, 131.97, 129.21, 127.59, 125.25, 124.96, 123.84, 122.02, 120.24, 120.21, 116.89, 112.81, 103.29, 52.71, 34.90, 21.63. Anal. Calcd for C21H20N6O2: C, 64.94; H, 5.19; N, 21.64; Found: C 64.80; H 5.41; N 21.76.
N-((1-(2-oxo-2-(p-tolylamino)ethyl)-1H-1,2,3-triazol-4-yl)methyl)-1H-indole-2-carboxamide (5c)
Yield: 81%. Light brown solid. M.p. 231–233 °C. 1H NMR (301 MHz, DMSO-d6) δ 11.64 (s, 1H), 10.39 (s, 1H), 9.11 (d, J = 5.7 Hz, 1H), 8.04 (s, 1H), 7.75–7.32 (m, 4H), 7.35–6.89 (m, 5H), 5.31 (s, 2H), 4.60 (d, J = 5.4 Hz, 2H), 2.26 (s, 3H). 13C NMR (76 MHz, DMSO-d6) δ 164.44, 161.53, 136.92, 136.38, 133.19, 131.93, 129.75, 127.54, 125.13, 123.80, 122.00, 120.19, 119.75, 119.65, 112.78, 103.21, 52.61, 34.85, 20.92. Anal. Calcd for C21H20N6O2: C, 64.94; H, 5.19; N, 21.64; Found: C 64.77; H 4.98; N 21.45.
N-((1-(2-((2,3-dimethylphenyl)amino)-2-oxoethyl)-1H-1,2,3-triazol-4-yl)methyl)-1H-indole-2-carboxamide (5d)
Yield: 83%. Light brown solid. M.p. 205–207 °C. 1H NMR (301 MHz, DMSO-d6) δ 11.66 (s, 1H), 9.87 (s, 1H), 9.12 (s, 1H), 8.06 (s, 1H), 7.78–6.77 (m, 8H), 5.39 (s, 2H), 4.62 (s, 2H), 2.26 (s, 3H), 2.12 (s, 3H). 13C NMR (76 MHz, DMSO-d6) δ 164.95, 161.57, 137.61, 136.95, 135.73, 131.95, 131.50, 127.74, 127.56, 125.77, 125.15, 123.81, 123.72, 122.00, 120.23, 120.20, 112.79, 103.25, 52.36, 34.89, 20.60, 14.48. Anal. Calcd for C22H22N6O2: C, 65.66; H, 5.51; N, 20.88; Found: C 65.38; H 5.37; N 20.72.
N-((1-(2-((2,6-dimethylphenyl)amino)-2-oxoethyl)-1H-1,2,3-triazol-4-yl)methyl)-1H-indole-2-carboxamide (5e)
Yield: 85%. Light brown solid. M.p. 208–210 °C. 1H NMR (301 MHz, DMSO-d6) δ 11.64 (s, 1H), 9.77 (s, 1H), 9.11 (d, J = 5.8 Hz, 1H), 8.05 (s, 1H), 7.63 (d, J = 7.7 Hz, 1H), 7.46 (d, J = 8.0 Hz, 1H), 7.31–6.92 (m, 6H), 5.38 (s, 2H), 4.70–4.45 (m, 2H), 2.17 (s, 6H). 13C NMR (76 MHz, DMSO-d6) δ 164.55, 161.55, 136.93, 135.55, 134.68, 131.94, 128.22, 127.54, 127.22, 124.98, 123.81, 122.00, 120.21, 120.20, 112.78, 103.22, 52.04, 34.87, 18.50. Anal. Calcd for C22H22N6O2: C, 65.66; H, 5.51; N, 20.88; Found: C 65.41; H 5.74; N 20.98.
N-((1-(2-((4-ethylphenyl)amino)-2-oxoethyl)-1H-1,2,3-triazol-4-yl)methyl)-1H-indole-2-carboxamide (5f)
Yield: 82%. Light brown solid. M.p. 203–205 °C. 1H NMR (301 MHz, DMSO-d6) δ 11.65 (s, 1H), 10.41 (s, 1H), 9.12 (s, 1H), 8.06 (s, 1H), 7.71–7.38 (m, 4H), 7.31–7.00 (m, 5H), 5.32 (s, 2H), 4.62 (s, 2H), 2.71–2.37 (m, 2H), 1.16 (t, J = 7.2 Hz, 3H). 13C NMR (76 MHz, DMSO-d6) δ 164.44, 161.55, 139.64, 136.93, 136.57, 131.94, 128.56, 127.55, 125.07, 123.81, 122.00, 120.20, 119.84, 119.74, 112.78, 103.23, 52.63, 34.87, 28.06, 16.11. Anal. Calcd for C22H22N6O2: C, 65.66; H, 5.51; N, 20.88; Found: C 65.43; H 5.32; N 21.01.
N-((1-(2-((4-methoxyphenyl)amino)-2-oxoethyl)-1H-1,2,3-triazol-4-yl)methyl)-1H-indole-2-carboxamide (5g)
Yield: 81%. Light brown solid. M.p. 209–211 °C. 1H NMR (301 MHz, DMSO-d6) δ 11.66 (s, 1H), 10.37 (s, 1H), 9.12 (s, 1H), 8.07 (s, 1H), 7.80–7.33 (m, 4H), 7.32–6.82 (m, 5H), 5.31 (s, 2H), 4.62 (s, 2H), 3.73 (s, 3H). 13C NMR (76 MHz, DMSO-d6) δ 164.17, 161.56, 155.98, 136.94, 131.98, 131.94, 127.55, 125.20, 123.82, 122.01, 121.32, 121.23, 120.21, 114.47, 112.78, 103.25, 55.61, 52.60, 34.87. Anal. Calcd for C21H20N6O3: C, 62.37; H, 4.98; N, 20.78; Found: C 62.14; H 5.21; N 20.63.
N-((1-(2-((3-chlorophenyl)amino)-2-oxoethyl)-1H-1,2,3-triazol-4-yl)methyl)-1H-indole-2-carboxamide (5h)
Yield: 70%. Light brown solid. M.p. 211–213 °C. 1H NMR (301 MHz, DMSO-d6) δ 11.64 (s, 1H), 10.07 (s, 1H), 9.11 (s, 1H), 8.06 (s, 1H), 7.77–7.02 (m, 9H), 5.44 (s, 2H), 4.60 (s, 2H). 13C NMR (76 MHz, DMSO-d6) δ 165.41, 161.53, 136.92, 134.62, 131.92, 130.09, 128.03, 127.53, 127.16, 126.69, 126.30, 124.95, 123.80, 121.99, 120.21, 120.19, 112.77, 103.20, 52.36, 34.84. Anal. Calcd for C20H17ClN6O2: C, 58.76; H, 4.19; N, 20.56; Found: C 58.54; H 3.98; N 20.69.
N-((1-(2-((4-chlorophenyl)amino)-2-oxoethyl)-1H-1,2,3-triazol-4-yl)methyl)-1H-indole-2-carboxamide (5i)
Yield: 74%. Light brown solid. M.p. 225–227 °C. 1H NMR (301 MHz, DMSO-d6) δ 11.64 (s, 1H), 10.63 (s, 1H), 9.12 (s, 1H), 8.06 (s, 1H), 7.76–6.84 (m, 9H), 5.34 (s, 2H), 4.61 (s, 2H).
13C NMR (76 MHz, DMSO-d6) δ 164.91, 161.55, 137.83, 136.92, 131.92, 129.30, 127.82, 127.54, 125.23, 123.81, 121.99, 121.33, 121.23, 120.20, 112.78, 103.23, 52.63, 34.86.
Anal. Calcd for C20H17ClN6O2: C, 58.76; H, 4.19; N, 20.56; Found: C 58.59; H 4.37; N 20.33.
N-((1-(2-((2,4-dichlorophenyl)amino)-2-oxoethyl)-1H-1,2,3-triazol-4-yl)methyl)-1H-indole-2-carboxamide (5j)
Yield: 74%, Light brown solid. M.p. 233–235 °C. 1H NMR (301 MHz, DMSO-d6) δ 11.66 (s, 1H), 10.17 (s, 1H), 9.14 (s, 1H), 8.38–6.71 (m, 10H), 5.48 (s, 2H), 4.64 (s, 2H).
13C NMR (76 MHz, DMSO-d6) δ 165.59, 161.60, 136.95, 133.84, 131.92, 130.22, 129.52, 128.14, 127.56, 127.48, 127.17, 125.12, 123.84, 122.01, 120.26, 120.22, 112.80, 103.31, 52.45, 34.91.
Anal. Calcd for C20H16Cl2N6O2: C, 54.19; H, 3.64; N, 18.96; Found: C 53.96; H 3.82; N 19.18.
N-((1-(2-((4-bromophenyl)amino)-2-oxoethyl)-1H-1,2,3-triazol-4-yl)methyl)-1H-indole-2-carboxamide (5k)
Yield: 81%. Light brown solid. M.p. 236–238 °C. 1H NMR (301 MHz, DMSO-d6) δ 11.63 (s, 1H), 10.63 (s, 1H), 9.11 (s, 1H), 8.11–7.04 (m, 10H), 5.34 (s, 2H), 4.60 (s, 2H). 13C NMR (76 MHz, DMSO-d6) δ 164.88, 161.55, 138.23, 136.92, 132.21, 131.90, 127.53, 125.29, 123.82, 121.99, 121.71, 121.62, 120.21, 115.87, 112.78, 103.25, 52.76, 34.91. Anal. Calcd for C20H17BrN6O2: C, 52.99; H, 3.78; N, 18.54; Found: C 52.79; H 4.01; N 18.31.
N-((1-(2-((3-nitrophenyl)amino)-2-oxoethyl)-1H-1,2,3-triazol-4-yl)methyl)-1H-indole-2-carboxamide (5l)
Yield: 79%. Brown solid. M.p. 254–256 °C. 1H NMR (301 MHz, DMSO-d6) δ 11.65 (s, 1H), 10.99 (s, 1H), 9.12 (t, J = 5.6 Hz, 1H), 8.61 (s, 1H), 8.09 (s, 1H), 8.01–7.84 (m, 2H), 7.65 (t, J = 7.9 Hz, 2H), 7.47 (d, J = 8.2 Hz, 1H), 7.13 (dt, J = 44.7, 6.9 Hz, 3H), 5.41 (s, 2H), 4.63 (d, J = 5.6 Hz, 2H). 13C NMR (76 MHz, DMSO-d6) δ 165.63, 161.58, 148.44, 139.97, 136.94, 131.93, 130.87, 127.56, 125.66, 125.15, 123.82, 122.00, 121.83, 120.20, 118.77, 113.85, 112.78, 103.26, 52.65, 34.88. Anal. Calcd for C20H17N7O4: C, 57.28; H, 4.09; N, 23.38; Found: C 56.99; H 4.31; N 23.59.
N-((1-(2-((4-nitrophenyl)amino)-2-oxoethyl)-1H-1,2,3-triazol-4-yl)methyl)-1H-indole-2-carboxamide (5m)
Yield: 69%. Brown solid. M.p. 259–261 °C. 1H NMR (301 MHz, DMSO-d6) δ 11.64 (s, 1H), 11.09 (s, 1H), 9.12 (s, 1H), 8.43–7.71 (m, 5H), 7.62 (d, J = 7.7 Hz, 1H), 7.45 (d, J = 8.0 Hz, 1H), 7.12 (d, J = 44.2 Hz, 3H), 5.43 (s, 2H), 4.61 (s, 2H). 13C NMR (76 MHz, DMSO-d6) δ 165.87, 161.55, 144.98, 143.03, 136.92, 131.91, 127.53, 125.58, 125.21, 123.82, 121.99, 120.20, 119.52, 119.48, 112.78, 103.23, 52.75, 34.86. Anal. Calcd for C20H17N7O4: C, 57.28; H, 4.09; N, 23.38; Found: C 57.02; H 4.28; N 23.17.
N-((1-(2-((2-methyl-4-nitrophenyl)amino)-2-oxoethyl)-1H-1,2,3-triazol-4-yl)methyl)-1H-indole-2-carboxamide (5n)
Yield: 73%. Brown solid. M.p. 229–231 °C. 1H NMR (301 MHz, DMSO-d6) δ 11.63 (s, 1H), 10.04 (s, 1H), 9.11 (s, 1H), 8.35–7.79 (m, 5H), 7.62 (d, J = 7.6 Hz, 1H), 7.45 (d, J = 7.9 Hz, 1H), 7.27–6.93 (m, 3H), 5.49 (s, 2H), 4.61 (s, 2H), 2.41 (s, 3H). 13C NMR (76 MHz, DMSO-d6) δ 165.77, 161.54, 143.91, 142.59, 136.92, 131.92, 131.73, 127.53, 126.00, 124.82, 123.81, 123.67, 122.32, 121.99, 120.21, 120.19, 112.77, 103.22, 52.61, 34.88, 18.30. Anal. Calcd for C21H19N7O4: C, 58.19; H, 4.42; N, 22.62; Found: C 58.01; H 4.66; N 22.35.
In vitro α-glucosidase and α-amylase inhibition assays
In vitro assays (inhibition effects and kinetics) of the new compounds 5a–n performed according to pervious reported work15 (Supplementary Information S1).
Docking study
Molecular modeling of the selected compounds 5c, 5f, 5j, and 5k, and molecular dynamics of the most potent compound 5k done on modeled α-glucosidase based on our pervious reported works15,20. Coordinates of active site of the modeled α-glucosidase was placed as follows:
Center of the grid box: x = 12.5825, y = − 7.8955, and z = 12.519 Å
Dimensions of the active site box: 40 × 40 × 40 Å.
Free binding energy calculations
The evaluation of the binding free energy of the protein–ligand complex was conducted utilizing the g_mmpbsa program21,22,23. Developed specifically for this purpose, g_mmpbsa facilitates the computation of the various constituents contributing to the binding free energy employing the molecular mechanics/Poisson-Boltzmann surface area (MM/PBSA) methodology. This computational tool enables the determination of the binding energy components pertinent to the protein–ligand complex which can be described as:
Data availability
The datasets used or analyzed during the current study are available from the corresponding author on reasonable request.
References
Li, X. et al. Food-derived non-phenolic α-amylase and α-glucosidase inhibitors for controlling starch digestion rate and guiding diabetes-friendly recipes. Lwt. 153, 112455 (2022).
Chiba, S. Molecular mechanism in α-glucosidase and glucoamylase. Biosci. Biotechnol. Biochem. 61, 1233 (1997).
Baron, A. D. Postprandial hyperglycaemia and α-glucosidase inhibitors. Diabetes Res. Clin. Pract. 40, S51 (1998).
Piero, M. N. et al. Diabetes mellitus-A devastating metabolic disorder. Asian J. Biomed. Pharm. 5, 1 (2015).
Buchanan, D. R. et al. Effectiveness of acarbose, an alpha-glucosidase inhibitor, in uncontrolled non-obese non-insulin dependent diabetes. Eur. J. Clin. Pharmacol. 34, 51–53 (1998).
Matsuo, T. et al. Effect of an intestinal disaccharidase inhibitor (AO-128) on obesity and diabetes. Am. J. Clin. Nutr. 55, 314S-S317 (1992).
Hakamata, W. et al. Design and screening strategies for α-glucosidase inhibitors based on enzymological information. Curr. Top. Med. Chem. 9, 3 (2009).
Chapel, C. et al. Antiviral effect of α-glucosidase inhibitors on viral morphogenesis and binding properties of hepatitis C virus-like particles. J. Gen. Virol. 87, 861 (2006).
Pili, R. The α-glucosidase I inhibitor castanospermine alters endothelial cell glycosylation, prevents angiogenesis, and inhibits tumor growth. Cancer Res. 55, 2920 (1995).
Mehta, A. et al. α-Glucosidase inhibitors as potential broad based anti-viral agents. FEBS Lett. 430, 17 (1998).
Dong Y. et al. Intervention of prediabetesby flavonoids from Oroxylum indicum. In Bioactive food as dietary interventions for diabetes. Academic Press. 559–575 (2019).
Fallah, Z. et al. A review on synthesis, mechanism of action, and structure-activity relationships of 1, 2, 3-triazole-based α-glucosidase inhibitors as promising anti-diabetic agents. J. Mol. Struct. 1255, 132469 (2022).
Emadi, M. et al. Indole-carbohydrazide linked phenoxy-1, 2, 3-triazole-N-phenylacetamide derivatives as potent α-glucosidase inhibitors: design, synthesis, in vitro α-glucosidase inhibition, and computational studies. BMC Chem. 17, 1 (2023).
Emadi, M. et al. Design, synthesis, in vitro anti-α-glucosidase evaluations, and computational studies of new phthalimide-phenoxy-1, 2, 3-triazole-N-phenyl (or benzyl) acetamides as potential anti-diabetic agents. Sci. Rep. 13, 10030 (2023).
Sepehri, N. et al. New acridine-9-carboxamide linked to 1, 2, 3-triazole-N-phenylacetamide derivatives as potent α-glucosidase inhibitors: Design, synthesis, in vitro, and in silico biological evaluations. Med. Chem. Res. 29, 1836 (2020).
Henary, M. et al. Benefits and applications of microwave-assisted synthesis of nitrogen containing heterocycles in medicinal chemistry. RSC Adv. 10, 14170 (2020).
Ebenezer, O. et al. An overview of the biological evaluation of selected nitrogen-containing heterocycle medicinal chemistry compounds. Int. J. Mol. Sci. 23, 8117 (2022).
Wang, J. et al. Structure-activity relationships of natural and synthetic indole-derived scaffolds as α-glucosidase inhibitors: A mini-review. Mini-Rev. Med. Chem. 20, 1791 (2020).
Solangi, M. et al. Indole acrylonitriles as potential antihyperglycemic agents: Synthesis, α-glucosidase inhibitory activity and molecular docking studies. Bioorg. Med. Chem. 28, 115605 (2020).
Noori, M. Design, synthesis, in vitro, and in silico enzymatic evaluations of thieno [2, 3-b] quinoline-hydrazones as novel inhibitors for α-glucosidase. Bioorg. Chem. 127, 105996 (2022).
Abraham, M. J. et al. GROMACS: High performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX. 1–2, 19–25 (2015).
Zoete, V. et al. SwissParam: A fast force field generation tool for small organic molecules. J. Comput. Chem. 32, 11 (2011).
Kumari, A. et al. g_mmpbsa—A GROMACS tool for high-throughput MM-PBSA calculations. J. Chem. Inf. Model. 54, 1951 (2014).
Acknowledgements
The presented work was financially supported by the Deanship of Research and Graduate Studies at King Khalid University (the Grant Number: RGP.2/491/44).
Author information
Authors and Affiliations
Contributions
M.H.S, B.L., and M.M. conceived the idea and designed the experiments, S.Z. and A.M. synthesized the designed compounds, M.M-Kh and M.H. performed in silico studies and wrote the main manuscript text, M.A. and H.R. contributed in the analysis of data, and S.M., MA.F., P.T., E.H.I, and H.A.G. performed in vitro biological assay. All authors reviewed the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Sayahi, M.H., Zareei, S., Halimi, M. et al. Design, synthesis, in vitro, and in silico anti-α-glucosidase assays of N-phenylacetamide-1,2,3-triazole-indole-2-carboxamide derivatives as new anti-diabetic agents. Sci Rep 14, 15791 (2024). https://doi.org/10.1038/s41598-024-66201-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-024-66201-y
- Springer Nature Limited