
Abstract
Chikungunya virus (CHIKV) is a single-stranded RNA virus belonging to the genus Alphavirus and is responsible for causing Chikungunya fever, a type of arboviral fever. Despite extensive research, the pathogenic mechanism of CHIKV within host cells remains unclear. In this study, an in-silico approach was used to predict that CHIKV produces micro-RNAs that target host-specific genes associated with host cellular regulatory pathways. Putative micro-RNAs of CHIKV were predicted using the miRNAFold and Vmir RNA structure web servers, and secondary structure prediction was performed using RNAfold. Host-specific target genes were then predicted, and hub genes were identified using CytoHubba and module selection through MCODE. Functional annotations of hub genes revealed their association with various pathways, including osteoclast differentiation, neuroactive ligand-receptor interaction, and mRNA surveillance. We used the freely available dataset GSE49985 to determine the level of expression of host-specific target genes and found that two genes, F-box and leucine-rich repeat protein 16 (FBXL16) and retinoic acid receptor alpha (RARA), were down-regulated, while four genes, RNA binding protein with serine-rich domain 1 (RNPS1), RNA helicase and ATPase (UPF1), neuropeptide S receptor 1 (NPSR1), and vasoactive intestinal peptide receptor 1 (VIPR1), were up-regulated. These findings provide insight into novel miRNAs and hub genes associated with CHIKV infection and suggest potential targets for therapeutic intervention. Further experimental validation of these targets could lead to the development of effective treatments for CHIKV-mediated diseases.
Similar content being viewed by others


Introduction
Viruses have become a major risk to human survival and have garnered substantial attention as a cause of epidemics. CHIKV is a reemerging arboviral pathogen that poses a significant threat to public health. It causes chikungunya fever (CHIKF), a tropical disease44. CHIKV is a single-stranded RNA virus that belongs to the Togaviridae family and is primarily transmitted by Aedes aegypti and Aedes albopictus mosquitoes26,33. The first outbreak of CHIKV in humans was reported in Tanzania in 1952, and since then, the disease has spread globally30,48. In 2006, a major outbreak occurred in the Indian Ocean, affecting approximately 1.3 million people in India and resulting in 2944 deaths6,26, 33. CHIKV affects individuals of all ages but is particularly severe in elderly individuals with an average age of 79 years, children, and immunocompromised individuals5,39. The common symptoms of CHIKV infection include headache, joint swelling, fatigue, vomiting, nausea, muscle pain, and maculopapular or macular rash13,16.
The genome of CHIKV is composed of four main proteins: non-structural (nsP1, nsP2, nsP3, nsP4) and structural proteins (envelope glycoproteins capsid, E1-3, and the 6 K viroporin). The nsP1 protein exhibits both methyl- and guanyl-transferase activities, while nsP3 plays a crucial role in RNA replication. The nsP2 protein functions as a protease helicase, and the nsP4 protein is the RNA-dependent RNA polymerase. The nsP3 protein comprises three domains, including the macro-domain at the N-terminus that exhibits both RNA binding and ADP-ribose, and ADP-ribosyl hydrolase capabilities. Moreover, virus-host interactions play a vital role and may be a specific pathogenesis determinant through interactions with factors that are cell type-specific15,19.
Micro-RNAs (miRNAs) are small RNA structures that do not code for proteins53 They are involved in regulating gene expression in various metabolic pathways at the post-transcriptional level by binding to the 3'-UTR of mRNA. miRNAs are responsible for numerous biological processes, including growth, propagation, persistent swelling, fibrosis, apoptosis, and immune response11,25, 35, 36, 38, 50, 51. Due to their significant role in metabolic pathways, miRNAs have gained interest as potential targets for antiviral response.
Several studies have shown the crucial role of miRNAs in viral infections18,43. In the past, miRNAs in humans were targeted by identifying analogous genes to address disease pathogenesis24. More recent research on miRNAs has highlighted their potential to target host-specific genes20,23. In the last decade, advances in research technologies, such as bioinformatics methodologies and improvements in microarray technologies, have made the study of miRNAs a focus of intense interest for scientists, enabling them to identify potential miRNAs associated with severe viral infections.
The studies have investigated the pathobiology of CHIKV, however, there is not enough evidence to confirm the presence of miRNA in different CHIKV genomes. To address this issue, computational analysis was conducted to identify potential miRNAs in CHIKV genomes. Moreover, this recent research aimed to identify the target genes of putative miRNAs, along with their related gene ontologies and pathways in KEGG. In this study, identifying the potential CHIKV-generated miRNAs and their corresponding gene targets can facilitate understanding of the virus's pathophysiology and how the host develops long-lasting protection against the virus.
Materials and methods
Prediction of precursor miRNAs
The CHIKV genome sequences (Accession No. NC_004162.2, MW042255.1, MW042254.1, KR046234.1, MT526807.1, MT526805.1, MT526804.1, HM045821.1, HM045819.1) were obtained from NCBI in FASTA format37. To further characterize the newly identified CHIKV strain HQ846356.1, miRNAs were predicted and comparison was made among the previously identified strains and newly identified strain for the identification of conserved putative miRNAs.
Each genome was analyzed using the ab initio-based miRNAFold and Vmir software to predict the existence and location of precursor miRNAs (pre-miRNAs) in the genome. miRNAFold was used to identify potential pre-miRNA hairpin structures in the genome46. This publicly available web server, located on the EvryRNA platform (http://EvryRNA.ibisc.univ-evry.fr/miRNAFold), is highly effective for bioinformaticians and biologists working in the field of non-coding RNAs45. We used the default settings for both the general parameter (sliding window size: 150, minimum hairpin size: 0, maximum thermodynamic value of hairpins: 0) and the specific parameter (percentage of verified features: 70) to achieve the best balance between precision and sensitivity46.
To ensure high reliability of pre-miRNA predictions, we also used Vmir22, which has default settings of minimum hairpin size of 60 nt, maximum hairpin size of 220 nt, and minimum hairpin score of 11522. To obtain highly reliable pre-miRNA predictions, we selected pre-miRNAs predicted by both miRNAFold and Vmir.
Secondary structure prediction
The RNA structure and RNAfold web servers (http://rna.urmc.rochester.edu/RNAstructureWeb, http://rna.tbi.univie.ac.at/) were utilized to determine the actual secondary structure of the miRNA3,27. Both algorithms were used to identify the pre-miRNA with a minimum hairpin size of 50 nt. To minimize the likelihood of obtaining false-negative results, we employed stringent filtering criteria to ensure that only highly conserved pre-miRNAs were retained.
Recognition of mature miRNA sequences and host specific target genes
Accurately identifying mature miRNAs is crucial for discovering their target host genes. We used two web servers, MatureBayes and Duplex SVM, to predict the mature miRNAs. MatureBayes is a Naive Bayes Classifier (NBC) (http://mirna.imbb.forth.gr/MatureBayes.html)21 while Duplex SVM is available at http://139.91.162.64/duplexsvm/31. MatureBayes is used to predict the start position of the mature miRNA on human and mouse miRNA precursors while Duplex SVM is useful to predicte the miRNA. miRNA ∗ duplex is a first step towards identifying the mature miRNA, suggesting possible miRNA targets and ultimately, reducing experimentation effort, time, and cost. Both tools provide information about the position and strands of mature miRNAs. To identify the potential host-specific target genes of miRNAs, we employed RNAhybrid and psRNATarget with a slightly relaxed threshold of Expectation ≤ 328,32. The MFE percentage in RNA hybrid was set to 75% to identify the host-specific target genes of mature miRNAs.
Protein–protein interaction network
To identify the functional interactions between the various targeted genes, we utilized protein–protein interaction (PPI) networks. To achieve this, we employed the Search Tool for the Retrieving of Interaction between Genes/Proteins (STRING) (Mering et al., 2003). The genes that were discovered were then analyzed using Cytoscape_v3.8.242. The Molecular Complex Detection (MCODE) plugin in Cytoscape was used to identify the module that revealed the best clusters of target genes12. In addition, we employed the CytoHubba plugin in Cytoscape to differentiate between the hub genes and specific target genes in the host. Among the 12 available topological analysis methods, we selected CytoHubba, MCC, MNC, and DEGREE for the identification of hub genes. We then selected the top 50 genes ranked by MCC, MNC, and DEGREE for further analysis.
Gene enrichment analysis
The targeted genes were analyzed using DAVID14 and FunRich49 for annotations, visualization, and combined discovery. Gene Ontology (GO) enrichment analysis was used to identify the molecular function (MF), biological process (BP), and cellular component (CC) of the targeted genes. KEGG was used for pathway analysis14, Linbang49. FunRich is a standalone software that provides functional enrichment analysis, interaction network analysis, and gene annotation for different species14.
Microarray analysis of gene expression
The search term "CHIKV" was utilized to retrieve microarray datasets (GSE49985) from the Gene Expression Omnibus (GEO) database in the NCBI database2. The acquired datasets were then analyzed using a multi-step approach to predict miRNA and their target genes. The overall methodology used to predict miRNA and their target genes is displayed in (Fig. 1). All the parameters of the sofwares and tools mentioned in the Fig. 1 were used by using default setting.

A graphic illustration for demonstrating the entire procedural approach of this analysis.
Results
Identification of precursor miRNAs
The complete genomes of CHIKV were obtained from NCBI using accession numbers NC_004162.2, MW042255.1, MW042254.1, KR046234.1, MT526807.1, MT526805.1, MT526804.1, HM045821.1, HM045819.1, and HQ846356.1. Each genome of CHIKV consisted of 11,826, 11,826, 11,754, 12,003, 11,797, 11,785, 11,787, 11,823, and 11,860, and 11,746 nucleotides, with a linear topology. We then subjected the resulting genomes to the miRNAFold and Vmir tools to predict pre-miRNAs.
Upon searching the CHIKV genomes for their precursor miRNAs, we obtained 200 pre-miRNA predictions from each tool. To identify the most reliable predictions, we selected pre-miRNAs that were commonly predicted by both miRNAFold and Vmir (Table S1).
Prediction of secondary structure
To predict the secondary structure of pre-miRNAs, we utilized the RNA structure and RNAfold web servers. Based on the minimum free energy (MFE) and nucleotide length, pre-miRNAs were selected to predict mature miRNAs. From the full genome of CHIKV and newly identified strain, a total of 181 and 170 pre-miRNAs were identified To ensure the reliability of the predicted mature miRNA sequences and to removing the redundant seqeunce, we discarded sequences with MFE values ranging from -1 to -15 or those with a length of over 50 nucleotides. Therefore, the validation of the sequence used to identify mature miRNA was based on both the MFE values and nucleotide length.
Prediction of mature miRNA sequences and host specific target genes
A single or both real pre-miRNA sequences can impart the function of the mature miRNA. Using the MatureBayes and Duplex SVM tools, we identified 19 mature miRNAs. Out of these 19, 3 mature miRNAs were found to be located on the stem-loop hairpin structure at the 5′ arm, while 16 occupied the stem loop-hairpin at the 3′ arm. Out of these 19 miRNAs, 5 were conserved with the newly identified strain and those These five predicted miRNAs range in length from 22 nucleotides and exhibit diverse sequences, suggesting they may target a variety of host cellular pathways to facilitate viral replication and survival, as shown in Table 1. To predict the host-specific targeted genes of CHIKV's miRNAs, we employed RNAhybrid and psRNATarget. Collectively, we identified 569 targeted genes via these tools. Among these, 391 genes were found to be distinctive to humans, and thus, were selected for further investigation. As the aim of this study is to discover the targeted gene of CHIKV in humans, these 391 genes were subjected to further analysis.
Protein–protein interaction network
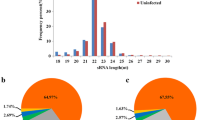
The top 40 genes were selected and ranked using MCC, MNC, and DEGREE methods using CytoHubba. The resulting genes were visualized and analysed using a Venn diagram to identify any overlaps. From these analyses, a total of 25 genes were identified as hub genes, as shown in Fig. 2.

(A) Common target genes identified by three methods in cytoHubba using Venn diagrams. (B) Information on 25 commonly identified genes.
To examine the network of interactions between the centre genes, a PPI network was generated using STRING for the targeted genes (Fig. 3 and Figure S1) and analysed using the MCODE plugin of Cytoscape. This analysis revealed four important modules, and one cluster was selected based on the cut-off criteria of node and score of > 4 and ≥ 4, respectively, as shown in Fig. 4. The resulting network represents the interactions between these selected genes.

Construction of PPI network of hub genes.

One module was selected having cut off criteria node > 4 and the score is ≥ 4. The module was constructed from MCODE and comprised 9 genes. In Fig (A) pink color shows the total number of genes which is 25, while in Fig (B) yellow color is shown genes that participated in the construction module.
Gene set enrichment analysis
The functions of the identified significant genes were analysed in terms of their biological processes, molecular function, and cellular component. In terms of biological processes, the genes were found to be associated with leukocyte migration, activation of adenylate cyclase activity, phosphatidylinositol-3-phosphate biosynthetic process, phosphatidylinositol phosphorylation, T cell co-stimulation, and positive regulation of cell proliferation.
Furthermore, in terms of molecular function, the genes were found to be involved in kinase activity, protein phosphate binding, phosphatidylinositol-4, 5-bisphosphate 3 kinase activity, platelet activation, bacterial invasion of epithelial cells, ephrin receptor binding, RNA, and protein binding.
In the case of cellular components, the genes were found to be enriched in the plasma membrane, endosome, neuron projection, and integral component of the plasma membrane. Additionally, KEGG pathway analysis revealed that the genes are involved in neuroactive ligand-receptor interaction, gap junction, mRNA surveillance pathways, RNA transport, transmembrane transport of small molecules, as well as SMAD2 signalling, as shown in Fig. 5.

(A) in KEGG pathways genes show their related pathways including percentage, -Log10 (p-value), and reference p-value. (B) Gene ontology in terms of MF of hub genes shows the percentage of the genes, –Log10 (p-value) and p-value in blue, green, and purple color. (C) In terms of CC green color shows the percentage of the genes, the sea green color shows a p-value of 0.05 reference, while grey shows the p-value of participating genes. (D) a p-value of genes that are correlated with BP is shown horizontally while the percentage of genes is shown vertically.
Microarray analysis of gene expression
To further investigate the hub genes, we analysed the differentially expressed genes (DEGs) in CHIKV-infected subjects using a microarray dataset (GSE49985). The hub genes were identified as distinctive genes based on the criteria of adjusted P-value < 0.05. Using these criteria, a total of six genes were identified as significant genes, as shown in Table 2.
Discussion
Over the past decade, CHIKV has received significant attention and has become a universal threat due to its potential to cause epidemics. CHIKV has been found to persist in joints. However, various research studies have reported transmission from person to person, from mother to child, and even death. Although extensive research has been done on CHIKV, the detailed pathogenesis procedure within the host is still missing. Therefore, in this analysis, our first aim was to predict novel miRNAs encoded by CHIKV. In recent years, miRNAs have emerged as a principal biomedical target due to their association with many biological phenomena. Identification of miRNAs encoded by the virus is, therefore, of incredible importance for developing better therapeutic inventions29.
In another case, several antiviral molecules are currently being employed to inhibit the entry of CHIKV and its replication within the body. These molecules include monoclonal antibodies, Rabrivin, inhibitors such as furin, chloroquine (which is being used to cure malarial infection), 20–50 oligoadenylate synthase, and arbidol7. Following this article, we have focused our attention on drug discovery in CHIKV, involving miRNA to inhibit the replication of CHIKV. Several experimental pieces of evidence have shown that miRNAs produced by those viral genes that are targeted in humans work as mediators like antiviral agents for the suppression of the pathogenesis of the virus29. A similar strategy was used in a related research study performed on the Zika virus9,41.
Overall, the six genes upregulated and downregulated genes having > 0.05 cut-off LogFC value have various functions not only post viral infections but also key factors of the human physiology. Brief description of function of these genes is as follows. Hepatitis C and gastric leiomyoma are two conditions linked to the Gs-protein-coupled receptor family known as vasoactive intestinal peptide receptor 1 (VIPR1). Vasoactive intestinal peptide (VIP), a neuropeptide that controls a number of bodily processes, including smooth muscle relaxation, immune system regulation, and hormone production, binds to and activates VIPR14. Although its precise roles may change depending on the virus and the environment of the infection, retinoic acid receptor alpha (RARA) can play a role in the immune response during viral infections. In the nucleus, RNA binding proteins (RNPS1) are necessary to stabilise developing transcripts. These proteins may bind to viral genomes and transcriptomes during the flu or other viral infections. Human UPF1 is a cytosolic protein that binds RNA and exhibits a nucleic acid-dependent ATPase as well as a helicase activity. The best-characterized function of UPF1 is in the nonsense-mediated decay (NMD) pathway, of which it is an essential component and UPF1 helicase promotes an early postentry step in HIV-1 replication. The overall function of an F-box/LRR protein is to recognize specific target proteins, usually phosphorylated or otherwise post-translationally modified, and facilitate their ubiquitination8,40.
Overall, this analysis highlights the importance of identifying miRNAs encoded by CHIKV for the development of better therapeutic interventions. Additionally, several antiviral molecules are currently being employed to inhibit the entry of CHIKV and its replication within the body. These findings have important implications for the development of effective treatments against CHIKV.
The prevention of T cells and their combined stimulation can be linked with miRNA, and functional enrichment analysis has clarified this fact. Based on this analysis, it is clear that CHIKV miRNAs target genes associated with leukocyte migration and activation of adenylate cyclase activity. Additionally, most of the genes were involved in the phosphatidylinositol-3-phosphate biosynthetic process and phosphatidylinositol phosphorylation. Some of these genes function in viral mRNA translation. Therefore, viral miRNA might target these genes to facilitate its replication. These findings support our hypothesis that miRNAs of CHIKV may target host genes associated with T-cell co-stimulation and leukocyte migration.
MiRNAs encoded by CHIKV have been observed to target host-specific pathways, such as mRNA surveillance, transmembrane transport of small molecules, and gap junctions, which could facilitate the defence escape of CHIKV1,17, 52. Protein kinases are involved in a broad range of eukaryotic cellular functions, including cell cycle control, cell metabolism, hormone response, translation control, and transcription10. Understanding viral protein-kinase may lead to comprehending the viral replication mechanism and its cell interaction10. CHIKV affects protein expression involved in mRNA processing, cyclin-dependent kinase 1 (CDK1), and host metabolic machinery47. as well as the regulation of cytoplasmic and nuclear SMAD2 signalling34. These discoveries support our theory that CHIKV miRNAs may focus on host qualities related to T cell co-stimulation and leukocyte migration guidelines. Given these discoveries, we propose an instrument for CHIKV pathogenesis through miRNA-mediated gene silencing.
Conclusion
The study of viral miRNAs is still in its early stages, but it has the potential to become a valuable tool for modulating viral and gene expression. This research sheds light on the pathogenesis of CHIKV by proposing a mechanism involving the virus's miRNAs targeting genes that play important roles in osteoclast differentiation, chronic myeloid leukaemia, and neuroactive ligand-receptor interaction. Our analysis has revealed several new miRNAs and hub genes produced by CHIKV, and the genes targeted by these miRNAs are associated with gap junction and mRNA surveillance. It is worth noting that there is currently no vaccine available for CHIKV treatment. However, this project has identified novel miRNAs and their corresponding targets, which could potentially lead to the development of new therapeutic approaches to combat CHIKV. These findings offer exciting possibilities for future research in this field and underscore the importance of continuing to explore the role of viral miRNAs in disease pathogenesis.
Data availability
The complete genomes of CHIKV were obtained from NCBI (https://www.ncbi.nlm.nih.gov/) using Accession Numbers NC_004162.2, MW042255.1, MW042254.1, KR046234.1, MT526807.1, MT526805.1, MT526804.1, HM045821.1, and HM045819.1. Gene expression microarray datasets were downloaded from Gene Expression Omnibus (GEO) database (http://www.ncbi.nlm.nih.gov/geo/).
References
Abdelnabi, R., Neyts, J. & Delang, L. Towards antivirals against chikungunya virus. Antivir. Res. 121, 59–68 (2015).
Barrett, T. et al. NCBI GEO: Archive for functional genomics data sets—update. Nucleic Acids Res. 41(D1), D991–D995 (2012).
Bellaousov, S., Reuter, J. S., Seetin, M. G. & Mathews, D. H. RNAstructure: Web servers for RNA secondary structure prediction and analysis. Nucleic Acids Res. 41(W1), W471–W474 (2013).
Branch, D. R. et al. VPAC1 is a cellular neuroendocrine receptor expressed on T cells that actively facilitates productive HIV-1 infection. AIDS. 16, 309–319 (2002).
Brito, C. AAd. Alert: Severe cases and deaths associated with Chikungunya in Brazil. Revista da Sociedade Brasileira de Medicina Tropical 50(5), 585–589 (2017).
Caglioti, C. et al. Chikungunya virus infection: An overview. New Microbiol. 36(3), 211–227 (2013).
Chaaithanya, I. K. et al. Role of proinflammatory cytokines and chemokines in chronic arthropathy in CHIKV infection. Viral immunol. 24(4), 265–271 (2011).
Chakravarty, A. & Yang, P. L. Targeted protein degradation as an antiviral approach. Antivir. Res. 210, 105480 (2023).
Chandak, N. H. et al. Neurological complications of Chikungunya virus infection. Neurol. India 57(2), 177 (2009).
Chen, M.-R., Chang, S.-J., Huang, H. & Chen, J.-Y. A protein kinase activity associated with Epstein-Barr virus BGLF4 phosphorylates the viral early antigen EA-D in vitro. J. Virol. 74(7), 3093–3104 (2000).
Cho, W. C. S. OncomiRs: The discovery and progress of microRNAs in cancers. Mol. Cancer 6(1), 60 (2007).
Cline, M. S. et al. Integration of biological networks and gene expression data using Cytoscape. Nat. protoc. 2(10), 2366 (2007).
Deeba, I. M. et al. Manifestations of atypical symptoms of chikungunya during the dhaka outbreak (2017) in Bangladesh. Am. J. Trop. Med. Hyg. 100(6), 1545–1548 (2019).
Dennis, G. et al. DAVID: Database for annotation, visualization, and integrated discovery. Genome boil. 4(9), 1–11 (2003).
Eckei, L. et al. The conserved macrodomains of the non-structural proteins of Chikungunya virus and other pathogenic positive strand RNA viruses function as mono-ADP-ribosylhydrolases. Sci. Rep. 7(1), 1–18 (2017).
Economopoulou, A. et al. Atypical Chikungunya virus infections: Clinical manifestations, mortality and risk factors for severe disease during the 2005–2006 outbreak on Reunion. Epidemiol. Infect. 137(4), 534–541 (2009).
Eugenin, E. A., Clements, J. E., Zink, M. C. & Berman, J. W. Human immunodeficiency virus infection of human astrocytes disrupts blood–brain barrier integrity by a gap junction-dependent mechanism. J. Neurosci. 31(26), 9456–9465 (2011).
Fani, M. et al. The role of microRNAs in the viral infections. Curr. Pharm. Des. 24(39), 4659–4667 (2018).
Gao, Y., Goonawardane, N., Ward, J., Tuplin, A. & Harris, M. Multiple roles of the non-structural protein 3 (nsP3) alphavirus unique domain (AUD) during Chikungunya virus genome replication and transcription. PLoS Pathog. 15(1), e1007239 (2019).
Ghosh, Z., Mallick, B. & Chakrabarti, J. Cellular versus viral microRNAs in host–virus interaction. Nucleic Acids Res. 37(4), 1035–1048 (2009).
Gkirtzou, K., Tsamardinos, I., Tsakalides, P. & Poirazi, P. MatureBayes: A probabilistic algorithm for identifying the mature miRNA within novel precursors. PloS One 5(8), e11843 (2010).
Grundhoff, A. Computational prediction of viral miRNAs. Antiviral RNAi 143–152 (Springer, UK, 2011).
Grundhoff, A. & Sullivan, C. S. Virus-encoded microRNAs. J. Virol. 411(2), 325–343 (2011).
Hariharan, M., Scaria, V., Pillai, B. & Brahmachari, S. K. Targets for human encoded microRNAs in HIV genes. Biochem. Biophys. Res. Commun. 337(4), 1214–1218 (2005).
Hennino, M.-F. et al. miR-21-5p renal expression is associated with fibrosis and renal survival in patients with IgA nephropathy. J. Sci. Rep. 6, 27209 (2016).
Her, Z. et al. Active infection of human blood monocytes by Chikungunya virus triggers an innate immune response. J. Immunol. 184(10), 5903–5913 (2010).
Hofacker, I. L. Vienna RNA secondary structure server. Nucleic Acids Res. 31(13), 3429–3431 (2003).
Islam, M. S. & Khan, M.A.-A.-K. Computational analysis revealed miRNAs produced by Chikungunya virus target genes associated with antiviral immune responses and cell cycle regulation. Comput. Biol. Chem. 92, 107462 (2021).
Islam, M. S., Khan, M. A. A. K., Murad, M. W., Karim, M. & Islam, A. B. M. M. K. In silico analysis revealed Zika virus miRNAs associated with viral pathogenesis through alteration of host genes involved in immune response and neurological functions. J. Med. Virol. 91(9), 1584–1594 (2019).
Josseran, L. et al. Chikungunya disease outbreak, Reunion island. Emerg. Infect. Dis. https://doi.org/10.3201/eid1212.060710 (2006).
Karathanasis, N., Tsamardinos, I. & Poirazi, P. MiRduplexSVM: A high-performing miRNA-duplex prediction and evaluation methodology. PloS One 10(5), e0126151 (2015).
Krüger, J. & Rehmsmeier, M. RNAhybrid: microRNA target prediction easy, fast and flexible. J. Nucleic Acids Res. 34(suppl_2), W451–W454 (2006).
Lewthwaite, P. et al. Chikungunya virus and central nervous system infections in children, India. Emerg. Infect. Dis. 15(2), 329 (2009).
Liu, L. et al. Smad2 and Smad3 have differential sensitivity in relaying TGFβ signaling and inversely regulate early lineage specification. Sci. Rep. 6(1), 1–14 (2016).
Lu, L. F. & Liston, A. MicroRNA in the immune system, microRNA as an immune system. J. Immunol. 127(3), 291–298 (2009).
Manni, I. et al. The microRNA miR-92 increases proliferation of myeloid cells and by targeting p63 modulates the abundance of its isoforms. J. FASEB J. 23(11), 3957–3966 (2009).
Pruitt, K. D. & Maglott, D. R. RefSeq and LocusLink: NCBI gene-centered resources. Nucleic Acids Res. 29(1), 137–140 (2001).
Rauen, T. & Floege, J. Inflammation in IgA nephropathy. J. Pediatr. Nephrol. 32(12), 2215–2224 (2017).
Sarkar, J., Chatterjee, S. & Chakravarty, S. Haemorrhagic fever in Calcutta: Some epidemiological observations. Indian J. Med. Res. 52, 651–659 (1964).
Serquiña, A. K. et al. UPF1 is crucial for the infectivity of human immunodeficiency virus type 1 progeny virions. J. Virol. 87, 8853–8861 (2013).
Sergon, K. et al. Seroprevalence of chikungunya virus (CHIKV) infection on Lamu Island, Kenya, October 2004. Am. J. Trop. Med. Hyg. 78(2), 333–337 (2008).
Shannon, P. et al. Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome Res. 13(11), 2498–2504 (2003).
Sullivan, C. S. & Ganem, D. MicroRNAs and viral infection. J. Mol. Cell 20(1), 3–7 (2005).
Tahir Ul Qamar, M. et al. Peptide vaccine against chikungunya virus: Immuno-informatics combined with molecular docking approach. J. Transl. Med. 16(1), 298. https://doi.org/10.1186/s12967-018-1672-7 (2018).
Tav, C., Tempel, S., Poligny, L. & Tahi, F. miRNAFold: A web server for fast miRNA precursor prediction in genomes. Nucleic Acids Res. 44(W1), W181–W184 (2016).
Tempel, S. & Tahi, F. A fast ab-initio method for predicting miRNA precursors in genomes. Nucleic Acids Res. 40(11), e80–e80 (2012).
Thio, C.L.-P., Yusof, R., Abdul-Rahman, P. S. A. & Karsani, S. A. Differential proteome analysis of chikungunya virus infection on host cells. PLoS One 8(4), e61444 (2013).
Valamparampil, J. J., Chirakkarot, S., Letha, S., Jayakumar, C. & Gopinathan, K. Clinical profile of Chikungunya in infants. Indian J. Pediatr. 76(2), 151–155 (2009).
Wang, L., Wang, B. & Quan, Z. Identification of aberrantly methylated-differentially expressed genes and gene ontology in prostate cancer. Mol. Med. Rep. 21(2), 744–758 (2020).
Wang, Y. & Lee, C. G. MicroRNA and cancer–focus on apoptosis. J. Cell Mol. Med. 13(1), 12–23 (2009).
Wienholds, E., Koudijs, M. J., van Eeden, F. J., Cuppen, E. & Plasterk, R. H. The microRNA-producing enzyme Dicer1 is essential for zebrafish development. J. Nat. Genet. 35(3), 217–218 (2003).
Wilen, C. B., Tilton, J. C. & Doms, R. W. Molecular mechanisms of HIV entry. In Viral Molecular Machines (eds Rossmann, M. G. & Rao, V. B.) 223–242 (Springer US, Boston, MA, 2012).
Zhang, B., Pan, X., Wang, Q., Cobb, G. P. & Anderson, T. A. Computational identification of microRNAs and their targets. J. Comput. Boil. 30(6), 395–407 (2006).
Acknowledgements
The authors are thankful to the Researchers Supporting Project number (RSPD2024R1035), King Saud University, Riyadh, Saudi Arabia.
Author information
Authors and Affiliations
Contributions
SS, MS, and MK and UAA conceived the research and designed experiments. BA, HK, NAA, AA AND MA conducted experiments. NAA, AA, MA and MAT analyzed the data. SS, BA, AA and MA wrote the manuscript. MAT, MK and UAA edited and revised the manuscript. All authors read and approved the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Ashraf, S., Sufyan, M., Aslam, B. et al. Uncovering chikungunya virus-encoded miRNAs and host-specific targeted genes associated with antiviral immune responses: an integrated bioinformatics approach. Sci Rep 14, 18614 (2024). https://doi.org/10.1038/s41598-024-67436-5
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-024-67436-5
- Springer Nature Limited