
Abstract
Limited treatment options and poor prognosis present significant challenges in the treatment of lung squamous cell carcinoma (LUSC). Disulfidptosis impacts cancer progression and prognosis. We developed a prognostic signature using disulfidptosis-related long non-coding RNAs (lncRNAs) to predict the prognosis of LUSC patients. Gene expression matrices and clinical information for LUSC were downloaded from the TCGA database. Co-expression analysis identified 209 disulfidptosis-related lncRNAs. LASSO-Cox regression analysis identified nine key lncRNAs, forming the basis for establishing a prognostic model. The model’s validity was confirmed by Kaplan–Meier and ROC curves. Cox regression analysis identified the risk score (RS) as an independent prognostic factor inversely correlated with overall survival. A nomogram based on the RS demonstrated good predictive performance for LUSC patient prognosis. The relationship between RS and immune function was explored using ESTIMATE, CIBERSORT, and ssGSEA algorithms. According to the TIDE database, a negative correlation was found between RS and immune therapy responsiveness. The GDSC database revealed that 49 drugs were beneficial for the low-risk group and 25 drugs for the high-risk group. Silencing C10orf55 expression in SW900 cells reduced invasiveness and migration potential. In summary, this lncRNA model based on TCGA-LUSC data effectively predicts prognosis and assists clinical decision-making.
Similar content being viewed by others

Introduction
Lung cancer is one of the most formidable malignancies, with both incidence and mortality rates ranking first globally1. Lung squamous cell carcinoma (LUSC) and lung adenocarcinoma (LUAD) are the two major subtypes of lung cancer2. Compared to LUAD, LUSC has a poorer clinical prognosis and lacks targeted therapies. In recent years, with the development of radiotherapeutic agents and immunotherapy, the prognosis of LUSC patients has improved. However, their 5 year survival rate is still lower than 18%, and most patients eventually develop drug resistance3,4. Consequently, it is essential to identify appropriate biomarkers to forecast the outcome and treatment response in LUSC.
Targeting programmed cell death (PCD) for cancer treatment has long been a significant research focus. Genes related to PCD have played a crucial role in developing innovative cancer therapies. For example, since its introduction in 2012, ferroptosis has garnered significant attention due to its close association with various diseases, especially cancer. Ferroptosis is an iron-dependent form of regulated cell death characterized by excessive lipid peroxidation and subsequent plasma membrane rupture. Research has shown that inducing ferroptosis is an effective cancer treatment strategy, as it can reduce patient resistance to multiple drugs and enhance the efficacy of immunotherapies5,6. A recent research paper proposed disulfidptosis, a novel PCD that is triggered by glucose deprivation in cells with high SLC7A11 expression. This process results in the disruption of disulfide bonds between actin cytoskeletal proteins and actin7. Research has shown that disulfidptosis and cancer growth and progression are closely related. For example, a prognostic signature based on disulfidptosis-related genes(DRGs) can help improve bladder cancer's prognosis and drug sensitivity8. Furthermore, DRGs have the potential to be targeted for cancer therapy9. However, the effect of disulfidptosis on LUSC has not been fully investigated.
Recently, it has been found that the treatment of cancer through neoantigens of RNA vaccines, RNA immunomodulation, and RNA interference is very effective, and long noncoding RNAs (lncRNAs) have become the focus of research. LncRNAs are RNA molecules longer than 200 nucleotides that do not encode proteins but regulate gene expression at various levels, including epigenetic, transcriptional, and post-transcriptional stages. Enhancing the function or expression of lncRNAs may be a pivotal aspect in various cancer-related diseases10,11. lncRNAs influence numerous biological processes through diverse mechanisms, including cell proliferation, differentiation, migration, progression, apoptosis, sex chromosome dosage compensation, maintenance of genomic stability, and regulating stemness and metabolism12,13. Studies have demonstrated that lncRNAs can affect the development and outlook of LUSC and may be potential targets for LUSC therapy14.
Currently, a significant amount of research has focused on disulfidptosis-related lncRNAs. Models based on these genes can predict the prognosis of cervical cancer patients and their sensitivity to chemotherapeutic drugs15. Additionally, disulfidptosis-related lncRNAs can influence the prognosis, tumor microenvironment, and choices for immunotherapy and chemotherapy in colorectal adenocarcinoma16. Therefore, we believe that further investigation of disulfidptosis-related lncRNAs is of great importance for the clinical diagnosis and treatment of LUSC. And this research utilized the TCGA-LUSC database to create a lncRNA-model associated with disulfidptosis to forecast the outcome of LUSC. In addition, the model's sensitivity, specificity, and accuracy were evaluated. Finally, we delved into functional enrichment, immunomodulation, and drug sensitivity to explore the model's value and guide treating LUSC patients.
Materials and methods
Data collection and processing
We acquired the gene expression matrix, clinical information, and gene mutation information of LUSC patients from The Cancer Genome Atlas Program (TCGA) database (https://portal.gdc.cancer.gov). A total of 553 samples were obtained, including 502 tumor and 51 normal samples. There were 470 tumor samples with prognostic information and survival time greater than or equal to 30 days. We are based on these 470 samples when performing prognostic correlation analysis. Gene expression matrices were distinguished between mRNA and lncRNA expression matrices based on annotation information using the R package “SummarizedExperiment.”
Establishing a prognostic signature for disulfidptosis-related lncRNAs
We obtained 24 DRGs (ACTB, ACTN4, CAPZB, CD2AP, DSTN, FLNB, FLNA, GYS1, INF2, IQGAP1, LRPPRC, MYH10, MYH9, MYL6, NCKAP1, NDUFA11, NDUFS1, NUBPL, OXSM, PDLIM1. RPN1, SLC3A2, SLC7A11, TLN1) from the published articles17,18. We used the R package “limma” to evaluate the correlation between lncRNAs and DRGs. Using |Cor|> 0.3 and P < 0.001 as the screening criteria, we identified lncRNAs associated with disulfidptosis. Additionally, we utilized the R packages “ggalluvial” and “ggplot2” to create Sankey diagrams, visually illustrating the co-expression relationships between lncRNAs and DRGs.
Determination of TRAIN and TEST cohorts
To acquire impartial outcomes and validate the precision of the prognostic signature. We randomly divided 470 LUSC samples (ALL cohort) into two groups (TRAIN and TEST cohorts). We statistically compared multiple clinical characteristics (Age, Gender, Stage, T, M, N) of ALL, TRAIN, and TEST cohorts by chi-square test. The statistical results are shown in Table 1. The P-values of the correlations of these clinical characteristics were all > 0.05. This shows that the three cohorts are independent and can be verified against each other. The model was constructed using the TRAIN cohort. Simultaneously, the TEST and ALL cohorts were employed to validate the model.
Establishment of a prognostic signature associated with DRGs
LASSO-Cox regression is crucial in identifying prognostic key genes and constructing prognostic models. Combining LASSO regularization with the Cox proportional hazards model can automatically select genes significantly associated with survival time from high-dimensional biological data19. This method not only simplifies the model and enhances prediction accuracy but also identifies genes most relevant to the prognosis of LUSC patients, thereby constructing highly predictive models. Specifically, by sequentially performing univariate Cox regression, LASSO analysis, and multivariate Cox regression analysis on disulfidptosis related lncRNAs, lncRNAs related to LUSC prognosis were identified. We analyzed the correlation between key-lncRNAs and DRGs by using the Spearman correlation coefficient method through the “cor.test”function in R. Subsequently, we plotted the correlation heatmap through the R package “ggplot2” (https://cran.r-project.org/web/packages/ggplot2/index.html?spm=5176.28103460.0.0.5d293da2Nrdwhe). And based on these lncRNAs, risk score(RS) for each sample was calculated, leading to the construction of the prognostic signature. The RS was calculated as:
where coefi is the coefficient of the multivariate Cox regression analysis for each lncRNA, expi refers to the expression level of this lncRNA. The implication of this formula is that the RS for each sample is equal to the sum of the product of the expression of each key lncRNA and the corresponding coefficient. Meanwhile, the correlation analysis between key-lncRNAs and DRGs was performed, and the results were plotted using the R package “ggplot2”. Based on the median RS, LUSC patients were categorized into low-risk group (LRG) and high-risk group(HRG). Similarly, the TEST and ALL cohorts are divided into HRG and LRG to perform external validation.
We tested the efficacy of our model by creating survival scatter plots, lncRNA expression heatmaps, survival analysis curves, and ROC plots for various cohorts (TRAIN, TEST, and ALL). This step was done with the help of R packages “pheatmap,” “survminer,” and “timeROC”.
Prognostic analysis of the model in different subgroups
We categorized the 470 LUSC patients into subgroups based on the sample's clinical characteristics to assess the model’s applicability. For example, patients were categorized into two subgroups (≤ 65 subgroup and > 65 subgroup) using age 65 as the cut-off. Patients were categorized into Stage I and Stage II-IV subgroups according to their stage, with Stage I patients usually receiving surgical intervention. In contrast, patients with stage II-IV frequently necessitated additional treatments like chemotherapy, radiotherapy, and immunotherapy. According to the T stage, patients are divided into two subgroups: T1-T2a and T2b-T4 (before T2a, the tumor is only in the microinvasive stage). Patients were divided into two subgroups, N0-1 and N2-3, according to N staging (at the N1 stage, the tumor spread was confined to the ipsilateral lung, and at the N2 stage, the tumor had spread to the contralateral lung and tracheal lymph nodes). We examined these subgroups’ prognostic differences between HRG and LRG by K-M curves.
Construction of the nomogram
To assess the value of this model for the prognosis of LUSC patients, a nomogram was constructed based on RS and Age, Pathologic staging, Gender, T and N stage (M stage staging was not included because nearly half of the sample lacked information about it). Calibration curves verified its accuracy for the patient, 1-, 3-, and 5 year survival. This step was performed using the R packages “survival,” “regplot,” “rms,” and “ survcomp.”
Functional enrichment analysis
To explore the reasons behind the prognostic differences between the two risk groups, we screened differentially expressed genes (DEGs) in HRG and LRG using the criteria of |log2 fold change|> 1 and false discovery rate (FDR) < 0.05.” The “clusterProfiler” package (https://bioconductor.org/packages/release/bioc/html/clusterProfiler.html?spm=5176.28103460.0.0.297c3da2FRRuxX) is mainly used to perform Gene Ontology (GO) enrichment analysis and Pathway enrichment analysis. It aims to help researchers understand the biological meaning behind large-scale gene or protein data. Subsequently, we performed Gene Ontology and Kyoto Encyclopedia of Genes and Genomes (KEGG) functional enrichment analysis (P < 0.05) on DEGs by this package20,21,22. The GO functional enrichment analysis was divided into three categories: Biological Process (BP), Molecular Function (MF), and Cellular Component (CC).
The KEGG pathway gene set (c2.cp.kegg.Hs.symbols.gmt) was sourced from the MSigDB database (https://www.gsea-msigdb.org). Following this, gene set enrichment analysis (GSEA) was conducted to investigate the enrichment differences between the high-risk and low-risk groups within this gene set.
Immune microenvironment analysis
With the help of the R package”IOBR,” we explored the immune microenvironmental differences between the two risk groups using the ESTIMATE and CIBERSORT algorithms.
We obtained the immune function-related gene set (immune.gmt) from the MSigDB database. We calculated and normalized the immune function scores of all LUSC samples using the “ssGSEA algorithm” in the R package “GSVA.” The data were then merged with the risk groups, and the results were visualized as box plots using the R package “ggpubr” to show the differences in immune function scores between the different risk groups.
Analysis of TMB(Tumor mutation burden)
The TCGA website provided the mutation data for LUSC samples. The relationship between TMB and RS was analyzed using the R packages “TCGAmutations” and “maftools.” And we used the “survival” and “survminer” packages to analyze the effects of TMB and RS on the survival of the samples.
Immunotherapy analysis and drug sensitivity analysis
We downloaded data related to tumor immune dysfunction and exclusion (TIDE) in squamous lung cancer from the TIDE database (http://tide.dfci.harvard.edu). This database predicted patients’ responses to immunotherapy, particularly anti-pd -1 and anti-ctla4 immunotherapy. While the TIDE score was negatively correlated with responsiveness to immunotherapy23. We conducted a comparison of TIDE scores between the two risk groups.
The GDSC (Genomics of Drug Sensitivity in Cancer) database is a critical tool for investigating drug sensitivity in cancer. This database brings together a large amount of data on the sensitivity of tumor cells to different drugs24. We obtained the drug sensitivity data from it. By utilizing the R package “oncoPredict,” we anticipated the disparities in drug sensitivity between HRG and LRG.
Cell culture and quantitative real-time PCR(qRt-PCR)
BEAS-2B (lung epithelial cell line) and H520 (lung squamous carcinoma cell line) were procured from Procell Life Science & Technology Co., Ltd. In contrast, two lung squamous carcinoma cell lines(H596 and SW900) were acquired from Nanjing Cobioer Biosciences Co., Ltd. All four cell lines were cultivated in DMEM medium (Gibco, USA) supplemented with 10 ml of fetal bovine serum (FBS) and maintained in a 37 ℃ incubator with 5% CO2. Total RNA was extracted using TRIzol reagent (Invitrogen, China), and cDNA was synthesized using a reverse transcription kit (Takara Bio, Shanghai, China). LncRNA expression levels were assessed using a PCR machine under the following conditions: 95 ℃for 60 s, 95 ℃for 15 s, and 60 ℃for 60 s for 40 cycles. GAPDH was used as the internal control, and relative gene expression levels were calculated using the 2−ΔΔCT method. Primer sequences are detailed in Table 2.
Plasmid transfection experiment
To validate our study further, we selected C10orf55 as the target for knockdown experiments for several reasons. Firstly, C10orf55 has been shown to play a significant role in the progression of head and neck cancer and esophageal cancer25,26. C10orf55 is also associated with ferroptosis, an essential form of programmed cell death.
In our experiments, we selected three interference sequences to knock down C10orf55 expression in SW900 cells, with NC plasmid and GAPDH as controls (gene sequences are provided in Table 3). We validated these sequences through PCR experiments and selected the one with the highest interference efficiency. Subsequently, SW900 cells were divided into three groups for the experiments: Control group (SW900 cells), NC group (SW900 cells with NC plasmid), and siRNA group (SW900 cells with C10orf55 knockdown).
Transwell invasion and scratch tests
Matrigel matrix gel (Corning, USA) was thawed at 4 ℃ and mixed with a serum-free medium at a 1:7.5 ratio. The thawed matrix gel was evenly applied to the Transwell chamber membrane at 60 µL/well. Three cells (Control group, NC group, siRNA group) were added to the upper chamber at a concentration of 5.0 × 105 cells/mL (150 µL per well), while 500 µL of medium containing 10% FBS was added to the lower chamber. After 48 h of incubation, the Transwell chambers were removed, fixed with anhydrous ethanol, and stained with crystal violet for 30 min. Cell counts were performed under a microscope at 100 × magnification, counting the membrane’s top, bottom, left, right, and center areas, and the average was taken. Each experiment was repeated 5 times to reduce error.
The three groups of cells were suspended at a density of 5.0 × 105 cells/mL and seeded into 6-well plates. When cell confluence reached over 80%, a sterile pipette tip was used to vertically and uniformly scratch the surface of the wells. The cells were then incubated in a 37 ℃, 5% CO2 environment for 48 h. Cell migration was observed, and migration rates were calculated. Each experiment was repeated three times.
Statistical analysis
The version of R software used in this study is 4.3.0. We used the t-test (Student's t-test) for the analysis of continuous variables that follow a normal distribution. We used the WilCoxon-Mann–Whitney test for data that did not meet the normal distribution criteria or for two independent samples. The Kruskal–Wallis test was also employed to compare more than two independent samples. The Spearman correlation coefficient was used to measure the strength and direction of the nonlinear relationship between two variables. A significance level of P < 0.05 was considered statistically significant. The significance levels are indicated: * for P < 0.05, ** for P < 0.01, and *** for P < 0.001.
Ethics approval and consent to participate
The patient data in this work is from publicly available patients consent to a complete data set without ethical approval.
Results
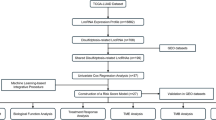
The research flow of this paper is demonstrated in Fig. 1A.

(A) The study flow chart of the article. (B) Sankey diagram showing the lncRNAs that co-expressed with disulfidptosis-related genes (P < 0.05).
Identification of kEY-lncRNAs.
Using |R|> 0.3 and P < 0.001 as screening criteria, we identified 1,432 lncRNAs that were co-expressed with DRGs. The co-expression relationships between DRGs and lncRNAs were visualized using a Sankey diagram (Fig. 1B). These genes were then integrated with the TCGA-LUSC-lncRNA expression data, ultimately identifying 209 disulfidptosis-related lncRNAs (Supplementary Table S1).
The 209 genes were further analyzed by univariate Cox regression in the TRAIN cohort, and a total of 21 lncRNAs related to LUSC prognosis were obtained (P < 0.05) and plotted as a forest plot (Fig. 2A).

Identification of key-lncRNAs. (A) Forest plot showing lncRNAs associated with LUSC prognosis. The red color represents the poor factors of LUSC prognosis. Green represents favorable factors for LUSC prognosis. (B-C) LASSO regression with tenfold cross-validation and trajectories for each independent variable. (D) Results and coefficients of multivariate Cox regression analysis (P < 0.05). (E) Heatmap showing the relationship between KEY-lncRNA and DRGs. (F) Differential expression of KEY-lncRNA in TCGA-LUSC dataset.
LASSO analysis could control overfitting through L1 regularization, select biologically significant and explanatory key genes, and improve model interpretability27,28. We uses LASSO analysis to ascertain the presence of 16 lncRNAs (Fig. 2B, C). Finally, multivariate Cox regression analysis was performed to screen out nine lncRNAs with the highest correlation with LUSC (LINC01311, AC010285.3, C10orf55, AL021368.3, AC009509.4, AL606489.1, AL133410.1, AC130462.2. AC005618.4) are called Key-lncRNAs. The name of Key-lncRNAs and the coefficients of the corresponding multivariate Cox regression analysis are shown in Fig. 2D.
Expression of key-lncRNAs in tumor tissues and their correlation with DRGs
According to the correlation heatmap of key-lncRNAs and DRGs (Fig. 2E), we can find that NDUFA11 presents a strong correlation with Key-lncRNAs, which is also consistent with the Sankey plot results (Fig. 1B). In comparison, AC009509.4 and C10orf55 presented a strong correlation with multiple DRGs. Figure 2F demonstrates the difference in expression of Key-lncRNAs in TCGA-LUSC. The results show that AL021368.3, AC009509.4, AL606489.1, and AL133410.1 were expressed higher in normal samples than in the tumor samples (P < 0.001). The expression of LINC01311, AC010285.3, C10orf55, AC130462.2, and AC005618.4 was higher in the tumor samples than in the normal sample (P < 0.001).
Construction and validation of the prognostic signature.
We determined the riskscore for each sample by analyzing the expression of the nine Key-lncRNAs. The calculation formula is expLINC01311*(−0.548372490461769) + expAC010285.3*(−1.18133243514436) + expC10orf55*0.351310706169649 + expAL021368.3*(−0.473280791062113) + expAC009509.4*0.511059938402485 + expAL606489.1*0.284340017375496 + expAL133410.1*0.776155666213932 + expAC130462.2*1.16718718964378 + expAC005618.4*(−0.618559418880687). The TRAIN cohort was categorized into HRG and LRG based on the median-RS. The other two cohorts (TEST and ALL) were divided into two risk groups using the same formula and grouping method. Figure 3A–C shows the risk stratification and survival status of HRG and LRG across three cohorts (TRAIN, TEST, and ALL), along with heatmaps depicting the expression of key lncRNAs in the two risk groups. In Fig. 3A, part (a) represents the risk stratification, where red dots indicate patients in the HRG group and blue dots indicate patients in the LRG group. Part (b) features a dashed line representing the median risk score, with patients to the left of the line in the low-risk group and those to the right in the high-risk group. Here, red dots indicate deceased samples and blue dots indicate non-deceased samples, with the vertical axis representing survival time. We observe more deceased samples and shorter survival times in the high-risk group. Part (c) indicates that LINC01311, AC010285.3, AL021368.3, and AC005618.4 are more highly expressed in the LRG, whereas the other five lncRNAs are more highly expressed in the HRG. This pattern is consistent across all three cohorts. Kaplan–Meier curves show that HRG has a poorer prognosis compared to LRG in all three cohorts (P < 0.05) (Fig. 4A–C).

Construction of prognostic signature. Riskscore distribution, survival status, and key-lncRNA expression in the (A)TRAIN, (B)TEST, and (C) ALL cohorts.

Validation of prognostic signature. (A) K-M survival curve analysis of two risk groups in the (A)TRAIN, (B)TEST, and (C) ALL cohorts. ROC curves for 1-, 3-, and 5-year survival for the (D) TRAIN, (E) TEST, and (F) ALL cohorts. ROC curves for risk score, age, gender, and staging in (G) TRAIN, (H) TEST, and (I) ALL cohorts.
Additionally, the accuracy of the prognostic value was evaluated using time-dependent ROC curves (Fig. 4D). The areas under the Curves (AUCs) for 1 year, 3 year, and 5 year survival in the TRAIN cohort were 0.694, 0.788, and 0.834, respectively. These results underscore the potential predictive value of the prognostic model for patients with LUSC. Similar ROC curves were created using the TEST cohort (Fig. 4E) and the ALL cohorts (Fig. 4F), with both the TEST and ALL cohorts showing relatively high AUC values, consistent with the results from the TRAIN cohort. ROC curves were also used to compare the predictive ability of the RS with other clinical features. In the TRAIN (Fig. 4G), TEST (Fig. 4H), and the ALL cohorts (Fig. 4I), the AUC values for the RS were 0.788, 0.643, and 0.702, indicating a robust predictive capability.
The K-M curve analysis consistently suggested that the HRG exhibited a more unfavorable prognosis when categorized by risk for various clinical subgroups (P < 0.05) (Figs. 5A–H). These results indicate that our prognostic model has a broad application scope, even in the presence of various clinical features.

K-M curves for two risk groups in multiple clinical subgroups. (A) The graph shows subgroups with age < 65. (B) The subgroup with age > 65. (C) and (D) are stage I and stage II-IV subgroups. (E) and (F) are subgroups of T1-T2a and T2b-T4. (G) and (H) for subgroups N0-1 and N2-3.
Establishment and validation of the nomogram
To determine whether RS is an independent predictor of survival in LUSC, we conducted univariate and multivariate Cox regression analyses (Fig. 6A, B). The analyses considered factors such as Age, Gender, Stage, and RS. The results indicate that both RS and Stage can independently predict the prognosis of LUSC (P < 0.05). Subsequently, a nomogram was created based on the RS and the aforementioned clinical-pathological indicators to generate an accurate tool for assessing the survival of LUSC patients (Fig. 6C). Taking TCGA-21–1080 as an example, a 66 year-old male patient in Stage I with T2, N0, and a low-risk score, the patient’s total score was 194, resulting in 1-, 3-, and 5 year survival rates of 0.911, 0.734, and 0.626, respectively. Calibration curves were established to assess the predictive capability of the prognostic model. The gray line in the graph represents the ideal line (predicted survival rate), and the calibration curve shows that the actual survival rates at 1, 3, and 5 years closely align with the ideal line (Fig. 6D). The findings suggest that the prognostic signature exhibits predictive solid capabilities.

Creation and validation of Nomogram. (A-B) Results of univariate Cox regression analysis and multivariate Cox regression analysis of clinical characteristics (Age, Sex, Stage) and RS. (C) The nomogram is built based on RS. (D) Validation of the nomogram using calibration curves.
Functional enrichment analysis
To explore the mechanisms by which risk models predict patient prognosis, we explored the differences in biological and immune-related functions between the two groups through gene enrichment analysis. Through differential analysis, we first identified 187 DEGs from the high and low-risk groups (Supplementary Table 2). Subsequently, we conducted GO and KEGG analyses on these 187 DEGs. We visualized the top six enriched terms from each of the three categories (BP, CC, MF) in the GO analysis results (P < 0.05). The results indicate that, in terms of BP, DEGs are primarily enriched in immune and inflammatory responses (cellular response to biotic stimulus, cellular response to lipopolysaccharide, cellular response to molecule of bacterial origin, antimicrobial humoral immune response mediated by antimicrobial peptide). In CC and MF, DEGs are mainly enriched in membrane composition and various activities related to cytokines and receptors (receptor ligand activity, cytokine activity, CXCR chemokine receptor binding, etc.) (Fig. 7A).

Differences in functional enrichment across two risk groups. (A) Results of GO enrichment analysis of differential genes. The innermost circle (first circle) is the Rich Factor. The second circle is the number of select genes. The third circle indicates the number of genes, and the outermost circle indicates the serial number of GO items. (B) KEGG analysis results of differential genes. (C-D) GSEA analysis results of HRG and LRG.
The KEGG analysis indicates that DEGs are mainly concentrated in multiple immune response signaling pathways (Cytokine-cytokine receptor interaction, IL-17 signaling pathway, TNF signaling pathway), amino acid metabolism (Arginine and proline metabolism, Arginine biosynthesis), and transcriptional misregulation in cancer (Fig. 7B). The significance of these enrichment results will be detailed in the discussion section.
Additionally, the GSEA analysis findings for the two risk groups suggest that the HRG is predominantly concentrated in pathways associated with cytokines and chemokines (KEGG-CYTOKINE-CYTOKINE-RECEPTOR-INTERACTION, KEGG-CHEMOKINE-SIGNALING-PATHWAY) (Fig. 7C). In contrast, the LRG is primarily enriched in the PPAR signaling and cytochrome P450 metabolism pathways (Fig. 7D). Some studies suggest that the PPAR signaling pathway can have anti-cancer effects. This pathway is typically associated with inhibiting cancer cell growth and promoting cell differentiation. It can suppress angiogenesis in non-small cell lung cancer by inhibiting the production of ELR + CXC chemokines29,30. This may provide evidence for a better prognosis in the LRG. To better explain this difference, further exploration was conducted to investigate the disparities in the immune microenvironment between the two risk groups.
Relationship between RS and tumor immune microenvironment (TIME)
The ESTIMATE algorithm is a computational method leveraging gene expression data to assess the scores of stromal and immune cells in tumor tissues31. The outcomes illustrate that HRG exhibits significantly elevated StromalScore (indicating the extent of tumor stromal components), ImmuneScore (reflecting the infiltration level of immune cells), and ESTIMATEScore (the composite score) compared to LRG(P < 0.001) (Fig. 8A). This finding implies that the immune microenvironment of HRG patients is more complex, characterized by a greater infiltration of both tumor stromal cells and a heightened level of immune cell infiltration.

Differences in the immune microenvironment of different risk subgroups. (A-C) Results of ESTIMATE analysis, CIBERSORT analysis, and immune function difference analysis for HRG and LRG.
CIBERSORT is a computational method based on gene expression data to estimate the relative abundance of various immune cell subtypes in tissue samples32. The results indicate that CD8 + T cells, resting dendritic cells, and eosinophils have higher enrichment scores in LRG while resting CD4 memory T cells and neutrophils are more enriched in HRG (Fig. 8B). This also suggests that different risk groupings lead to differences in tumor cell infiltration.
ssGSEA has been widely used in the analysis of gene expression data, especially in cancer research, to reveal biological differences and clinical features between different tumor subtypes33. We delved deeper into the distinctions in immune function between the HRG and LRG using ssGSEA. This exploration aimed to elucidate the relationship between RS and the immune system. The results caught our attention. On 29 immune-related functions, 27 immune functions (Th1_cells, CD8 + _T_cells, NK_cells, Treg, T_helper_cells, T_fh, Th2_cells, TIL, Cytolytic_activity, iDCs, pDCs, DCs, Macrophages, Neutrophils, Mast_cells, B_cells, APC_co_stimulation, T_cell_co-stimulation, T_cell_co-inhibition, Check-point, HLA, MHC_class_I, Type_IFN_Response, Type_II_IFN_Response, Inflammation-promoting, Parainflammation, CCR) all scored higher in the HRG ( Fig. 8C). The significance of this difference will be explained in detail in the discussion section.
Correlation of RS and TMB.
TMB refers to the number of mutations in tumor cells, typically expressed as mutations per megabase (must/Mb). TMB is a significant biomarker that can be employed to evaluate the genetic variant load of a tumor, thereby aiding in the prediction of the treatment response and prognosis of patients. Numerous studies have demonstrated that TMB can be a marker for immunotherapy in various cancers34,35,36,37. Consequently, we decided to investigate the inherent connection between TMB and RS. Figure 9A–B demonstrates the top 15 genes with the highest mutation rates in the species of HRG and LRG, with no significant differences. Additionally, the LRG exhibited a considerably greater total TMB value than the HRG’s (Fig. 9C). We subdivided the samples into two groups, H-TMB and L-TMB, based on the median TMB values of all samples to compare the prognosis. K-M curves showed that the OS of the L-TMB group was higher than that of the H-TMB (P = 0.002; Fig. 9D). Moreover, the sample was further divided into “H-TMB + H-RS,” “H-TMB + L-RS,” “L-TMB + H-RS,” and “L-TMB + L-RS” groups to compare OS. The results showed that the L-TMB + H-RS had the worst prognosis, while the H-TMB + H-RS was also worse. This outcome confirms RS’s superior predictive power for patient prognosis and implies that our model can be used with TMB to forecast patient prognosis (Fig. 9E).

Correlation analysis of RS and TMB. (A-B)Tumor mutation waterfall plots of HRG and LRG. (C) Violin plot of the difference in TMB values between two risk groups. (D) Kaplan–Meier curves showing survival differences between high and low tumor mutation burden groups. (E) Patient survival curve for LUSC based on two variables (TMB and RS).
Prognostic modeling for prediction of clinical treatment
We calculated the TIDE scores for the high and low-risk groups based on the TIDE database. The results showed that the LRG had lower TIDE scores, indicating that the LRG may have better sensitivity to immunotherapy, particularly anti-PD-L1 and anti-CTLA-4 treatments (Fig. 10A). Additionally, using drug sensitivity data from the GDSC database, we calculated the IC50 (half-maximal inhibitory concentration) values for various drugs in both risk groups. And the lower the IC50, the higher the sensitivity. We found that 49 drugs were more sensitive to LRG, and 25 were more sensitive to HRG (Supplementary Table S3). We selected the drugs commonly used in clinical oncology treatment (Selumetinib, Paclitaxel, Trametinib, Afatinib, Erlotinib, Gefitinib, Osimertinib, Cisplatin, AZD4547, BI-2536, MK-2206) to be visualized (Fig. 10B–L). Among them, Selumetinib, Paclitaxel, and Trametinib were highly sensitive to HRG, while the other eight drugs were more sensitive to patients in the LRG. These two results can effectively help clinicians treat lung cancer for drug selection.

Immunotherapy and drug sensitivity differences between HRG and LRG. (A) Comparison of immunotherapy predictions between HRG and LRG according to the TIDE algorithm. (B-L) Differences in sensitivity to multiple drugs (Selumetinib, Paclitaxel, Trametinib, Afatinib, Erlotinib, Gefitinib, Osimertinib, Cisplatin, AZD4547, BI-2536, MK-2206) between the high and low-risk groups, respectively.
Results of the experiment
Figure 11A shows that the expression of C10orf55 was higher in three lung squamous carcinoma cell lines (H520, H596, and SW900) than in normal cell lines (BEAS-2B). Figure 11B showed the highest efficiency of SW900-687 interference, so we chose this interference sequence for subsequent analysis. The results of the Transwell assay showed that the invasive migration ability of SW900 cells in the siRNA group was decreased (Fig. 11C–D). According to Fig. 11E–F, we learned that after 48 h of 6-well plate culture, the siRNA group SW900 cells had the weakest ability to move.

(A) PCR results of C10orf55 in BEAS-2B, H520, H596, and SW900. (B) Expression levels in SW900 cells + NC plasmid, GAPDH, and three interfering plasmids after interference in SW900 cells. (C-D) Invasion results in three groups of SW900, SW900 + NC, and siRNA. (E–F) Scratch results of the three groups of SW900, SW900 + NC, and siRNA.
Discussion
LUSC has a poor prognosis and limited treatment options. It is necessary to construct reliable predictive models to determine the prognosis of LUSC patients. PCD is a crucial feature of tumorigenesis and a breakthrough in cancer treatment38. For example, Ferroptosis-related genes can be used to guide the treatment of lung cancer39. The occurrence of disulfidptosis, a recently discovered form of PCD, has been associated with the growth and progression of cancer40.
Furthermore, Some studies propose that lncRNAs play a critical regulatory role in lung cancer. They can influence the progression of non-small cell lung cancer (NSCLC) by modulating cyclin-dependent kinase 1 (CDK1), suggesting that lncRNAs could serve as potential biomarkers and therapeutic targets for lung cancer41. Notably, a lncRNA-associated prognostic signature is critical for multiple diseases42,43 . We, therefore, constructed a disulfidptosis-based lncRNAs model to predict the prognosis of LUSC patients.
Twenty-four DRGs were identified from previous studies. Subsequently, co-expression analysis and LASSO-Cox analysis were performed sequentially, and 9 key lncRNAs were identified (LINC01311, AC010285.3, C10orf55, AL021368.3. AC009509.4, AL606489.1, AL133410.1, AC130462.2, AC005618.4). Moreover, the prognostic signature was constructed based on nine lncRNAs. Among them, C10orf55 is involved in the prognostic prediction of esophageal adenocarcinoma and head and neck squamous carcinoma25,26. In addition, several other genes have been shown to play a role in re-cancer. For example, LINC01311 inhibits thyroid cancer progression44. These studies suggest that these critical lncRNAs may play a role in the progression and prognosis of LUSC. Based on the key-lncRNAs, we calculated the RS of the samples and categorized the samples into HRG and LRG according to the median of RS.
Moreover, heatmap, ROC, and K-M curves of different clinical subgroups all suggested that our prognostic signature had an excellent discriminatory and predictive ability for two risk groups of LUSC. Moreover, Cox regression analysis showed that RS was an independent poor prognostic indicator prognostic indicator for LUSC and disease stage. Furthermore, an advantage of our model over tumor staging is that it can differentiate between high- and low-risk patients at the same disease stage for individualized treatment.
In order to explore the reasons for the different prognoses between the HRG and LRG, we performed GO and KEGG enrichment analysis of DEGs between the two risk groups. The enrichment analysis showed that the DEGs were enriched for immune and inflammatory responses and cancer regulation. Among them, IL-17 and TNF signaling pathways were noteworthy for their key roles in regulating inflammation and immune responses. These pathways also exert regulatory influence on the development and progression of various cancers45,46. Moreover, it has been shown that IL-17 promotes metastasis and lung cancer progression47. TNF signaling pathways also play a vital role in cancer and are potential immunotherapy treatments for cancer48,49. The results of the GSEA analysis also showed different immune pathways enriched by different risk groups. The enrichment analysis results help us understand the biological differences between high and low-risk groups and provide potential ideas for treating high and low-risk groups.
The tumor immune microenvironment plays a pivotal role in cancer progression and prognosis. In our study, the ESTIMATE algorithm suggested that HRG tumor tissues harbor a richer population of stromal cells and infiltrating immune cells than LRG. Stromal cells contribute to tumor growth and advancement50, implying that HRG tumors may progress more rapidly and exhibit higher aggressiveness, aligning with their poorer prognosis. Moreover, the elevated immune scores in HRG indicate a more intricate cellular infiltration within their immune microenvironment.
We employed CIBERSORT to analyze the infiltration patterns of immune cells in both risk groups. Notably, CD8 + T cells, resting dendritic cells, and eosinophils were more enriched in LRG, whereas HRG featured higher enrichment of resting CD4 memory T cells and neutrophils. Given that CD8 + T cells are pivotal in directly eliminating tumor cells51, their enrichment in LRG potentially signifies a more robust immune surveillance capability in these patients, facilitating tumor cell recognition and clearance. Conversely, the proliferation of resting CD4 memory T cells, key regulators of immune tolerance and response, might suppress effective anti-tumor immunity in HRG, facilitating tumor immune evasion52. Neutrophil enrichment, associated with enhanced inflammatory responses and potential immune suppression, further underscores an immunosuppressive environment in HRG53.
Of the 29 immune-related functions assessed by ssGSEA, 27 functions, including key immune activities such as Th1 responses, CD8 + T-cell function, natural killer cell activity, and regulatory T-cell modulation, were more active in HRG, which seems to contradict the results of the CIBERSORT algorithm. However, this apparent discrepancy can be reconciled by understanding the different emphases of CIBERSORT and ssGSEA: the former quantifies the relative proportions of immune cell types, whereas the latter reflects functional enrichment and activity levels. Thus, while CD8 + T cell infiltration in HRG may be lower, their functional activation and antitumor activity is increased. Of note, several of these functions, such as Treg cell activity linked to immune tolerance in lung squamous cell carcinoma and checkpoint molecules involved in both immune regulation and tumor immune escape, are pivotal in cancer therapies54. Understanding the dynamics of these checkpoints can illuminate opportunities for immunotherapy. This discovery underscores the intricate relationship between lung squamous cell carcinoma and the immune response, paving the way for immunotherapeutic strategies. It establishes a foundation for personalized treatment and future immunological research, promising patient management and prognosis advancements. Our findings hold the potential to enhance our comprehension of the immune characteristics of LUSC and facilitate the development of more efficacious therapeutic interventions.
TMB is frequently regarded as a pivotal immunotherapy biomarker in many tumors, including lung and bladder cancer55,56. This study discloses a higher TMB in the LRG, implying a richer landscape of potential neoantigen targets for the immune system. Elevated TMB is theoretically correlated with enhanced responsiveness to immunotherapy, presaging a more robust immune response against tumors. Immunotherapy has become an indispensable and efficacious component of cancer treatment, notably through its mechanisms of relieving T-cell suppression via anti-PD-1 therapy and amplifying early immune response activation via anti-CTLA-4 treatment. The synergistic action of these two therapies successfully reinvigorates and bolsters the anti-tumor immune reaction. Their combined application has substantially improved clinical outcomes across various cancer therapies57,58.
Furthermore, the TIDE scoring system indicates a potentially better therapeutic response trend to PD-1 and CTLA-4 inhibitors in the LRG, aligning with previous analyses. And Differential drug sensitivity profiles between risk groups provide a scientific basis for medication selection for HRG and LRG patients, aiming to optimize treatment efficacy and survival prospects through precision medicine strategies. This highlights the clinical significance of personalized treatment approaches.
Despite the significant findings of this study, several limitations exist. Firstly, the analysis was primarily based on a dataset with a limited sample size, and future research needs to validate these findings with larger samples. Secondly, although we conducted several cell experiments to verify the results, further analysis using animal models or tissue samples is necessary to consolidate and extend our findings.
Conclusion
In this study, we formulated a prognostic signature based on disulfidptosis-related lncRNAs for LUSC. We analyzed to explore the associations between RS and TMB, immune cells, immunotherapy considerations, and drug sensitivity. The results of this study provide new insights into predicting the prognosis of patients with LUSC and offer new ideas for patient treatment.
Data availability
All data used in this work can be acquired from the The Cancer Genome Atlas Program (TCGA; https://portal.gdc.cancer.gov).
Abbreviations
- LUSC:
-
Lung squamous cell carcinoma
- LUAD:
-
Lung adenocarcinoma
- PCD:
-
Programmed cell death
- DRGs:
-
Disulfide bond pathology-related genes
- lncRNA:
-
Long non-coding RNA
- TCGA:
-
The cancer genome atlas
- GO:
-
Gene ontology
- BP:
-
Biological process
- CC:
-
Cellular component
- MF:
-
Molecular function
- KEGG:
-
Kyoto encyclopedia of genes and genomes
- GSEA:
-
Gene set enrichment analysis
- TMB:
-
Tumor mutation burden
- ssGSEA:
-
Single-sample gene set enrichment analysis
- HRG:
-
High-risk group
- LRG:
-
Low-risk group
- K-M:
-
Kaplan–Meier
- RS:
-
Risk Score
- DEGs:
-
Differentially expressed genes
- AUC:
-
Area under the curve
- ROC:
-
Receiver operating characteristic
- CDK1:
-
Cyclin-dependent kinase 1
- FDR:
-
False discovery rate
- IC50:
-
Half-maximal inhibitory concentration
- TIME:
-
Tumor immune microenvironment
- GDSC:
-
Genomics of drug sensitivity in cancer
- TIDE:
-
Tumor Immune dysfunction and exclusion
- FBS:
-
Fetal bovine serum
- qRT-PCR:
-
Quantitative real-time polymerase chain reaction
- NSCLC:
-
Non-small cell lung cancer
References
Duffy, M. J. & O’Byrne, K. Tissue and blood biomarkers in lung cancer: A review. Adv. Clin. Chem. 86, 1–21 (2018).
Li, Y. et al. Transcriptomic and functional network features of lung squamous cell carcinoma through integrative analysis of GEO and TCGA data. Sci. Rep. 8(1), 15834 (2018).
Zappa, C. & Mousa, S. A. Non-small cell lung cancer: Current treatment and future advances. Transl. Lung Cancer Res. 5(3), 288–300 (2016).
Lin, J. J. & Shaw, A. T. Resisting resistance: Targeted therapies in lung cancer. Trends Cancer 2(7), 350–364 (2016).
Dixon, S. J. et al. Ferroptosis: An iron-dependent form of nonapoptotic cell death. Cell 149(5), 1060–1072 (2012).
Chen, X. et al. Broadening horizons: The role of ferroptosis in cancer. Nat. Rev. Clin. Oncol. 18(5), 280–296 (2021).
Liu, X. et al. Actin cytoskeleton vulnerability to disulfide stress mediates disulfidptosis. Nat. Cell. Biol. 25(3), 404–414 (2023).
Chen, H. et al. Leveraging a disulfidptosis-based signature to improve the survival and drug sensitivity of bladder cancer patients. Front. Immunol. 14, 1198878 (2023).
Zheng, P. et al. Disulfidptosis: A new target for metabolic cancer therapy. J. Exp. Clin. Cancer Res. 42(1), 103 (2023).
Xue, V. W. et al. Promising RNA-based cancer gene therapy using extracellular vesicles for drug delivery. Expert Opin. Biol. Ther. 20(7), 767–777 (2020).
Chen, R. et al. Cascades between miRNAs, lncRNAs and the NF-kappaB signaling pathway in gastric cancer (review). Exp. Ther. Med. 22(1), 769 (2021).
Wu, Y. et al. m(6)A-induced lncRNA RP11 triggers the dissemination of colorectal cancer cells via upregulation of Zeb1. Mol. Cancer 18(1), 87 (2019).
Wang, P. et al. An interferon-independent lncRNA promotes viral replication by modulating cellular metabolism. Science 358(6366), 1051–1055 (2017).
Li, C. & Zhang, Y. Construction and validation of a cuproptosis-related five-lncRNA signature for predicting prognosis, immune response and drug sensitivity in breast cancer. BMC Med. Genom. 16(1), 158 (2023).
Liu, L. et al. Disulfidptosis-associated LncRNAs index predicts prognosis and chemotherapy drugs sensitivity in cervical cancer. Sci. Rep. 13(1), 12470 (2023).
Xue, W. et al. Disulfidptosis-associated long non-coding RNA signature predicts the prognosis, tumor microenvironment, and immunotherapy and chemotherapy options in colon adenocarcinoma. Cancer Cell Int. 23(1), 218 (2023).
Yang, Z. et al. Characterization and prognosis of biological microenvironment in lung adenocarcinoma through a Disulfidptosis-related lncRNAs signature. Genet. Res. (Camb) 2023, 6670514 (2023).
Ni, L. et al. The expression and prognostic value of disulfidptosis progress in lung adenocarcinoma. Aging (Albany NY) 15(15), 7741–7759 (2023).
Wang, Q. et al. Nomogram established on account of Lasso-Cox regression for predicting recurrence in patients with early-stage hepatocellular carcinoma. Front. Immunol. 13, 1019638 (2022).
Kanehisa, M. & Goto, S. KEGG: Kyoto encyclopedia of genes and genomes. Nucl. Acids Res 28(1), 27–30 (2000).
Kanehisa, M. Toward understanding the origin and evolution of cellular organisms. Protein Sci. 28(11), 1947–1951 (2019).
Kanehisa, M. et al. KEGG for taxonomy-based analysis of pathways and genomes. Nucl. Acids Res. 51(D1), D587–D592 (2023).
Jiang, P. et al. Signatures of T cell dysfunction and exclusion predict cancer immunotherapy response. Nat. Med. 24(10), 1550–1558 (2018).
Yang, W. et al. Genomics of drug sensitivity in cancer (GDSC): a resource for therapeutic biomarker discovery in cancer cells. Nucl. Acids Res. https://doi.org/10.1093/nar/gks1111 (2013).
Paszkowska, A. et al. C10orf55, CASC2, and SFTA1P lncRNAs are potential biomarkers to assess radiation therapy response in head and neck cancers. J. Pers. Med. https://doi.org/10.3390/jpm12101696 (2022).
Zhang, D. et al. Development of a novel miR-3648-related gene signature as a prognostic biomarker in esophageal adenocarcinoma. Ann. Transl. Med. 9(22), 1702 (2021).
Zhu, A. et al. Comprehensive analysis to identify a novel diagnostic marker of lung adenocarcinoma and its immune infiltration landscape. Front. Oncol. 13, 1199608 (2023).
Friedman, J., Hastie, T. & Tibshirani, R. Regularization paths for generalized linear models via coordinate descent. J. Stat. Softw. 33(1), 1–22 (2010).
Wagner, N. & Wagner, K. D. PPAR beta/delta and the hallmarks of cancer. Cells 9(5), 1133 (2020).
Keshamouni, V. G. et al. PPAR-gamma activation inhibits angiogenesis by blocking ELR+CXC chemokine production in non-small cell lung cancer. Neoplasia 7(3), 294–301 (2005).
Yoshihara, K. et al. Inferring tumour purity and stromal and immune cell admixture from expression data. Nat. Commun. 4, 2612 (2013).
Chen, B. et al. Profiling tumor infiltrating immune cells with CIBERSORT. Methods Mol. Biol. 1711, 243–259 (2018).
Pisanti, F. A. et al. Influence of acetyl homocysteine thiolactone on erythrocyte superoxide dismutase activity. Biochem. Med. Metab. Biol. 37(2), 265–267 (1987).
Kohli, K. & Pillarisetty, V. G. Dendritic cells in the tumor microenvironment. Adv. Exp. Med. Biol. 1273, 29–38 (2020).
Chan, T. A. et al. Development of tumor mutation burden as an immunotherapy biomarker: Utility for the oncology clinic. Ann. Oncol. 30(1), 44–56 (2019).
Zhang, L. et al. The prognostic value of TMB and the relationship between TMB and immune infiltration in head and neck squamous cell carcinoma: A gene expression-based study. Oral Oncol. 110, 104943 (2020).
Addeo, A. et al. TMB or not TMB as a biomarker: That is the question. Crit. Rev. Oncol. Hematol. 163, 103374 (2021).
Peng, F. et al. Regulated cell death (RCD) in cancer: Key pathways and targeted therapies. Signal Transduct. Target Ther. 7(1), 286 (2022).
Tian, Q. et al. Development and validation of a ferroptosis-related gene signature for overall survival prediction in lung adenocarcinoma. Front. Cell Dev. Biol. 9, 684259 (2021).
Zhao, S. et al. Crosstalk of disulfidptosis-related subtypes, establishment of a prognostic signature and immune infiltration characteristics in bladder cancer based on a machine learning survival framework. Front. Endocrinol. (Lausanne) 14, 1180404 (2023).
Wu, F. et al. IncRNA KTN1-AS1 silencing inhibits non-small-cell lung cancer cell proliferation and KTN1-AS1 expression predicts survival. Crit. Rev. Eukaryot. Gene Expr. 32(2), 39–46 (2022).
Huang, L., Zhang, L. & Chen, X. Updated review of advances in microRNAs and complex diseases taxonomy, trends and challenges of computational models. Brief Bioinform. https://doi.org/10.1093/bib/bbac358 (2022).
Chen, X. et al. Long non-coding RNAs and complex diseases: from experimental results to computational models. Brief Bioinform. 18(4), 558–576 (2017).
Liu, M. et al. LINC01311 exerts an inhibitory effect in thyroid cancer progression by targeting the miR-146b-5p/IMPA2 axis. Transl. Oncol. 27, 101588 (2023).
Coffelt, S. B. et al. IL-17-producing gammadelta T cells and neutrophils conspire to promote breast cancer metastasis. Nature 522(7556), 345–348 (2015).
Gundel, H. Annual scientific meeting of german psychosomatic medicine and psychotherapy. Psychother. Psychosom. Med. Psychol 66(2), 55–56 (2016).
Liu, L. et al. The role of IL-17 in lung cancer growth. Cytokine 169, 156265 (2023).
Xu, H. et al. Roburic acid targets TNF to inhibit the NF-kappab signaling pathway and suppress human colorectal cancer cell growth. Front Immunol 13, 853165 (2022).
Josephs, S. F. et al. Unleashing endogenous TNF-alpha as a cancer immunotherapeutic. J. Transl. Med. 16(1), 242 (2018).
Quail, D. F. & Joyce, J. A. Microenvironmental regulation of tumor progression and metastasis. Nat. Med. 19(11), 1423–1437 (2013).
van der Leun, A. M., Thommen, D. S. & Schumacher, T. N. CD8(+) T cell states in human cancer: Insights from single-cell analysis. Nat. Rev. Cancer 20(4), 218–232 (2020).
Wherry, E. J. T cell exhaustion. Nat. Immunol. 12(6), 492–499 (2011).
Xiao, Y. et al. Cathepsin C promotes breast cancer lung metastasis by modulating neutrophil infiltration and neutrophil extracellular trap formation. Cancer Cell. 39(3), 423–437 (2021).
Duan, M. C. et al. The Treg/Th17 paradigm in lung cancer. J. Immunol. Res. 2014, 730380 (2014).
Fusco, M. J., West, H. J. & Walko, C. M. Tumor mutation burden and cancer treatment. JAMA Oncol. 7(2), 316 (2021).
McGrail, D. J. et al. High tumor mutation burden fails to predict immune checkpoint blockade response across all cancer types. Ann. Oncol. 32(5), 661–672 (2021).
Han, Y., Liu, D. & Li, L. PD-1/PD-L1 pathway: Current researches in cancer. Am. J. Cancer. Res. 10(3), 727–742 (2020).
Rowshanravan, B., Halliday, N. & Sansom, D. M. CTLA-4: A moving target in immunotherapy. Blood 131(1), 58–67 (2018).
Funding
This study was funded by Henan Provincial Health Commission (LHGJ20190422), Henan Provincial Health Commission (LHGJ20210486) and the Key Scientific Item of Henan Province Education Department (21A320037).
Author information
Authors and Affiliations
Contributions
Ankang Zhu and Yan Zong designed this work. Ankang Zhu performed the validation of the experiments. Yan Zong integrated and analyzed the data. Ankang Zhu wrote this manuscript. Xingcai Gao edited and revised the manuscript. All authors approved this manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Zhu, A., Zong, Y. & Gao, X. Development of a disulfidptosis-related lncRNA prognostic signature for enhanced prognostic assessment and therapeutic strategies in lung squamous cell carcinoma. Sci Rep 14, 17804 (2024). https://doi.org/10.1038/s41598-024-68423-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-024-68423-6
- Springer Nature Limited