
Abstract
Chronic Kidney Disease (CKD) stands as a substantial challenge within the global health landscape. The elevated metabolic demands essential for sustaining normal kidney function have propelled an increasing interest in unraveling the intricate relationship between mitochondrial dysfunction and CKD. However, the authentic causal relationship between these two factors remains to be conclusively elucidated. This study endeavors to address this knowledge gap through the Mendelian Randomization (MR) method. We utilized large-scale QTL datasets (including 31,684 eQTLs samples, 1980 mQTLs samples, and 35,559 pQTLs samples) to precisely identify key genes related to mitochondrial function as exposure factors. Subsequently, we employed GWAS datasets (comprising 480,698 CKD samples and 1,004,040 eGFRcrea samples) as outcome factors. Through a comprehensive multi-level analysis (encompassing expression, methylation, and protein quantification loci), we evaluated the causal impact of these genes on CKD and estimated glomerular filtration rate (eGFR). The integration and validation of diverse genetic data, complemented by the application of co-localization analysis, bi-directional MR analysis, and various MR methods, notably including inverse variance weighted, have collectively strengthened our confidence in the robustness of these findings. Lastly, we validate the outcomes through examination in human RNA sequencing datasets encompassing various subtypes of CKD. This study unveils significant associations between the glycine amidinotransferase (GATM) and CKD, as well as eGFR. Notably, an augmentation in GATM gene and protein expression corresponds to a diminished risk of CKD, whereas distinct methylation patterns imply an increased risk. Furthermore, a discernible reduction in GATM expression is observed across diverse pathological subtypes of CKD, exhibiting a noteworthy positive correlation with GFR. These findings establish a causal relationship between GATM and CKD, thereby highlighting its potential as a therapeutic target. This insight lays the foundation for the development of potential therapeutic interventions for CKD, presenting substantial clinical promise.
Similar content being viewed by others


Introduction
Chronic kidney disease (CKD) represents a formidable global public health challenge, affecting millions and contributing to heightened mortality rates1. Given the kidneys’ crucial role in human metabolism, maintaining their normal functions necessitates a heightened level of energy metabolism. Numerous investigations have underscored the intricate and precise nature of kidney energy metabolism, encompassing diverse mitochondria-associated metabolic pathways, including glucose, lipid, and amino acid metabolism2,3,4. The burgeoning interest in the realm of CKD is now directed towards understanding mitochondrial dysfunction, recognizing it as a significant focal point in CKD-associated research.
Mitochondria, ancient cellular powerhouses, not only function as hubs for adenosine triphosphate production but also play a pivotal role in orchestrating essential cellular functions. These functions span the regulation of oxidative stress, maintenance of calcium homeostasis, and control of cell apoptosis, thereby exerting a profound influence on cellular destinies5,6,7. Recent studies have underscored a close association between CKD and mitochondrial dysfunction. CKD patients exhibit a notable reduction in the activity of mitochondrial respiratory chain complexes8. Furthermore, mitochondrial DNA (mtDNA) damage emerges as a significant contributor to the progression of CKD9. Additional investigations have unveiled a connection between renal damage and mitochondrial biosynthesis. In tubular cells of CKD patients, there is a discernible inhibition of mitochondrial biosynthesis, potentially attributable to the abnormal activation of intracellular autophagy pathways10. Simultaneously, oxidative stress impacts mitochondria in CKD, leading to damage in mitochondrial membranes and functional impairment11. Increasingly, kidney diseases caused by mutations in mitochondrial or nuclear genes are being observed. For instance, mutations in the RMND1 gene (required for meiotic nuclear division (1) can lead to multiple mitochondrial respiratory chain deficiencies, with kidney involvement observed in about 60% of cases, sometimes necessitating renal replacement therapy12. Additionally, new mutations in the highly conserved regions of the mitochondrial tRNA (Tyr) gene have been linked to hereditary nephrotic syndrome13. In summary, the nexus between CKD and mitochondrial function has garnered substantial support and exploration, spanning various facets such as mitochondrial structure, function, and intracellular metabolic processes.
Recent research underscores the intricate interplay among mitochondrial function, dynamics, and the progression of CKD. Notably, attempts have been made to infer the causal relationship between mitochondrial dysfunction and CKD by manipulating genes associated with mitochondrial function14,15. However, conventional studies encounter certain limitations, including confounding factors, reverse causality, and measurement errors. Therefore, there is a pressing need for a reliable and novel approach that comprehensively analyzes all genes associated with CKD and mitochondrial dysfunction. This endeavor aims to elucidate the causal relationship between the two, providing clearer guidance for the development of new therapeutic approaches tailored to CKD treatment.
Mendelian randomization (MR), rooted in genetic data, serves as a method for deducing causal relationships between genetic variations and complex traits, leveraging genetic tools as proxies for modifiable exposures. This innovative approach employs alleles as instrumental variables (IVs) to mitigate the impact of unobserved confounders such as lifestyle and environmental factors, thereby addressing potential issues related to reverse causality. This, in turn, provides more robust evidence for causal inference16,17. Genome-wide association studies (GWAS) utilize genetic associations based on single nucleotide polymorphisms (SNPs) to unveil genes associated with specific traits. The integration of GWAS data with genetic loci for gene expression and methylation quantitative trait loci (eQTL or mQTL) proves instrumental in identifying genetic variations influencing gene expression or methylation levels. These variations not only serve as indicators of gene functionality and regulatory relationships but also function as IVs in MR analysis, facilitating the exploration of causal links between gene expression or methylation and outcomes18,19. Additionally, protein quantitative trait loci (pQTL), representing genetic variations influencing protein expression levels, offer another avenue for investigating causal relationships between protein expression and outcomes20. This multifaceted approach enhances the depth and precision of exploring causal associations within the framework of MR.
The MR method encompasses various types, with two-sample MR (2SMR) and summary-data-based MR (SMR) being commonly employed approaches. 2SMR involves the utilization of summary data from distinct studies' GWAS, representing the strength of associations for the exposure factor and the outcome separately. This facilitates MR analysis, allowing for the assessment of IVs exposure-relevance and IVs exposure-outcome associations across different populations, thereby enhancing the understanding of causal relationships21. The advantage of 2SMR lies in its capability to leverage existing publicly available data without requiring access to raw data, thus addressing concerns related to sample overlap. On the other hand, SMR represents an extension of the MR method, utilizing GWAS summary data from the same study to analyze the strength of associations between genetically determined traits (such as gene expression, DNA methylation, or protein abundance at exposure) and complex traits of interest (such as disease phenotypes). SMR's strengths lie in its ability to harness more information, improve statistical efficiency, and enable heterogeneity testing and sensitivity analysis. Subsequently, the incorporation of the heterogeneity in dependent instruments (HEIDI) test proves valuable in distinguishing potential causal relationships from widespread linkage disequilibrium (LD) in the genome18.
In the context of this study, our hypothesis posits that genetic variations influencing mitochondrial function or biogenesis may play a causal role in influencing susceptibility to CKD and the estimated glomerular filtration rate (eGFR). To test this hypothesis, we employ the aforementioned methods to systematically investigate potential causal relationships within the mitochondrial pathways that impact the progression of CKD. Our aim is to identify genes associated with mitochondrial function, methylation sites, or protein expressions that are causally linked to the progression of CKD. To bolster the validity of our findings, we undertake a thorough validation process utilizing external RNA sequencing datasets. This comprehensive exploration is anticipated to furnish robust support for the development of therapeutic strategies targeting these mitochondrial pathways in the context of CKD treatment.
Methods
Data source
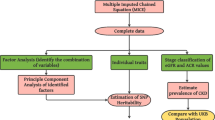
Figure 1 offers a comprehensive overview of the workflow and analytical methods applied throughout the course of this study.

Flowchart of the analyses performed.
To identify genes associated with genetic susceptibility to mitochondrial dysfunction, we extracted 1136 known mitochondrial-related genes from the human MitoCarta 3.0 database22. Subsequently, we obtained genetic variant information for the eQTL of these mitochondrial-related genes, utilizing summary statistics data provided by the eQTLGen Consortium (https://www.eqtlgen.org/cis-eqtls.html). The consortium assessed 19,250 genes expressed in blood from 31,684 individuals (excluding the 37 genes on the mtDNA and those located on the X and Y chromosomes), filtering 10,611 SNP associations related to traits based on an FDR < 0.05, MAF > 0.01 criteria. Each SNP-gene combination was within 1 Mb of the gene center and replicated in at least two cohorts23. From the cis-eQTL data, we obtained 662,968 SNPs associated with the expression of 1004 mitochondrial-related genes mentioned above. Although mtDNA was not sequenced and included in these datasets, the number of genes involved is small, and the impact on the analysis is relatively minor. Additionally, for mQTL related to mitochondrial-related genes, we extracted data from meta-analyzed summary statistics of two cohorts (total sample size of 1980)19. In this analysis, we selected 931,304 SNPs corresponding to 2,558 DNA methylation CpG sites associated with mitochondrial-related genes. We obtained plasma pQTL data from 35,559 Icelanders summarized by Ferkingstad et al. from the deCODE genetics database (https://www.decode.com/summarydata/)20, covering 232,467 SNPs associated with 298 mitochondrial-related protein expressions. All SNPs included in the analysis met the criteria Psnp-mitodys < 5.0E-8.
The GWAS summary statistics for CKD were sourced from Wuttke M. et al., involving a meta-analysis of 480,698 individuals (41,395 cases and 439,303 controls) of European and trans-ethnic ancestry24. CKD was defined as eGFR less than 60 mL/min/1.73 m2. The GWAS summary statistics for eGFR based on creatinine (eGFRcrea) were derived from Stanzick KJ. et al., conducting a meta-analysis on the Chronic Kidney Disease Genetics Consortium (CKDGen, n = 765,348, transethnic) and UK Biobank (UKB, n = 436,581, Europeans), totaling n = 1,004,04025.
Comprehensive information for all QTL and GWAS datasets used in this study is available in Supplementary Table S1.
Statistical analysis
In the causal relationship SMR analysis, we defined several key variables. Initially, βmitodys−outcome represents the estimated effect of mitochondrial dysfunction on the outcome (CKD or eGFRcrea). Subsequently, βSNP−mitodys signifies the estimated effect of the instrumental SNP on mitochondrial dysfunction, while βSNP−outcome indicates the estimated effect of the instrumental SNP on the outcome. We established a pivotal equation:
The formulation of this equation is a crucial step in SMR analysis, aiding in understanding how the impact of the instrumental SNP on mitochondrial dysfunction correlates with the outcome (CKD or eGFRcrea).
We employed the SMR software (version 1.3.1, https://yanglab.westlake.edu.cn/software/smr/#Overview) to conduct SMR and HEIDI analyses. When calculating the odds ratio (OR) for a one-standard-deviation (SD) change in mitochondrial gene expression level concerning outcome risk, we utilized the formula:
In this equation, OR represents the estimated risk ratio for each incremental ln unit of mitochondrial gene level, and exp denotes the exponential function with the base e, the natural logarithm. To control potential type I errors, we conducted multiple testing corrections, revealing robust evidence of significant associations (FDRSMR < 0.05).
HEIDI test is a method within MR used to assess the validity of selected genetic variants and potential levels of confounding. The primary aim of this test is to detect whether there is an excessive amount of LD between genetic variants influencing exposure and those affecting the outcome. Such a scenario could indicate that the selected genetic variants are not sufficiently independent. Specifically, the HEIDI test attempts to identify a set of genetic variants whose effects are interrelated, potentially affecting the accuracy of causal inference. In this study, we employed a cutoff value of PHEIDI ≥ 0.05 to determine the presence of correlations between variants.
Posterior probability (PP) is used to assess the probability of an event or hypothesis based on both prior knowledge and observed data. In MR, PP is often utilized to evaluate the credibility or accuracy of specific hypotheses. Specifically, PP.H4 is a method within MR analysis used to assess the PP of horizontal pleiotropy (HP). HP occurs when genetic variants affecting the exposure not only influence the outcome variable but might also directly impact the outcome variable through other pathways. In our study, we employed the coloc R package (https://chr1swallace.github.io/coloc/, version 5.1.0) for PP.H4 Bayesian testing of colocalization between two traits to estimate posterior probabilities26. We set a threshold of PP.H4 > 0.8 as strong evidence of colocalization between GWAS and QTL associations.
The sensitivity analysis was conducted using the R package (version 4.1.2, TwoSampleMR, www.r-project.org) employing five additional MR analysis methods, including MR Egger, weighted median, inverse variance-weighted, simple mode, and weighted mode. These methods compute estimates of causal effects based on slightly different assumptions regarding the validity of instruments. By employing multiple methods for sensitivity analysis, we ensure the robustness of results and provide stronger evidence supporting our findings.
External dataset validation
We utilized nine CKD RNA sequencing datasets from the Nephroseq public database (www.nephroseq.org, University of Michigan, Ann Arbor, MI). Data analysis was performed using GraphPad Prism (version 9.4.0), including data visualization, t-test statistical analysis, and construction of fitted curves. For specific dataset information, please refer to Supplementary Table S2.
Results
The MR analysis of mitochondrial genome-wide cis-eQTL and CKD.
Through SMR analysis, this study identified a unique genetic locus associated with CKD from mitochondrial-related genes, as well as 6 distinct genetic loci associated with eGFRcrea (Fig. 2). It’s important to note the slight differences in how these results were presented-CKD risk was typically represented in OR format, while the risk for eGFRcrea was directly presented in the form of βmitodys−outcome. These variations stem from the distinct data types and analytical methodologies employed. These findings garnered consistent support from HEIDI tests, high-strength evidence of colocalization analysis, heterogeneity analysis, pleiotropy analysis, and the utilization of five additional MR analysis methods (Supplementary Table S3, S4). Their support by various methods and the consistency across these results enhance the credibility of these findings.

MR results for the association between the expression of mitochondrial-related genes and CKD progression outcomes.
For CKD, an increase of one SD in gene expression of rs7164602-glycine amidinotransferase (rs7164602-GATM) is associated with a 12.1% decrease in the risk of CKD (OR = 0.879, 95% CI 0.848, 0.910, FDR = 5.25E−10, PHEIDI = 6.07E−02, PP.H4 = 0.815). The inverse variance weighted analysis yielded the same results (OR = 0.895, 95% CI 0.822, 0.975, P = 1.10E−02). Meanwhile, there was no heterogeneity (PCochran's Q = 3.67E−01) or pleiotropy (PIntercept = 8.27E−01).
Moreover, gene expressions of rs12706818-SND1, rs3752493-MCRIP2, rs12983363-ATP5F1D, rs479572-ATP5IF1, rs3864285-HINT1, and rs3769698-COA5 were also causally associated with eGFRcrea, but their contribution were relatively small (the absolute value of β < 0.03).
The MR analysis of mitochondrial genome-wide cis-mQTL and CKD
For the causal relationship between DNA methylation of mitochondrial-related genes and CKD, we discovered a total of 4 association signals across 3 unique genetic loci associated with CKD and 15 association signals across 13 unique genetic loci for eGFRcrea (Fig. 3, Supplementary Table S5). The sensitivity analysis corroborated these associations (Supplementary Table S6).

MR results for the association between the DNA methylation of mitochondrial-related genes and CKD progression outcomes.
Here, we identified a total of 2 unique loci that regulate the methylation level of 3 different CpG sites in GATM, positively associated with CKD risk. A one SD increase in methylation at rs56850226-GATM by cg00767496 was linked to a 20.1% higher risk of CKD (OR = 1.201, 95% CI 1.132, 1.275, FDR = 3.51E−07, PHEIDI = 1.25E−01, PP.H4 = 0.815) and a 2.4% higher risk of decreased eGFRcrea (β = − 0.024, 95% CI − 0.028, − 0.02, FDR = 3.22E−24, PHEIDI = 6.82E−02, PP.H4 = 0.814). Additionally, a one SD increase in methylation at rs56850226-GATM by cg14910265 corresponded to an 18.9% higher risk of CKD (OR = 1.189, 95% CI 1.125, 1.256, FDR = 2.15E−07, PHEIDI = 9.86E−02, PP.H4 = 0.815). Furthermore, a one SD increase in methylation at rs12593371-GATM by cg24328539 was associated with an 11.3% higher risk of CKD (OR = 1.113, 95% CI 1.079, 1.148, FDR = 8.84E−09, PHEIDI = 5.80E−02, PP.H4 = 0.815). The inverse variance weighted analysis yielded the same results (OR = 1.124, 95% CI 1.076, 1.175, P = 1.97E−07). Meanwhile, there was no heterogeneity (PCochran's Q = 1.36E-01) or pleiotropy (PIntercept = 8.06E−01). Lastly, a one SD increase in methylation at rs2233956-MCCD1 by cg04048339 was linked to a 25.8% higher risk of CKD (OR: 1.258, 95% CI 1.114, 1.420, FDR = 3.30E−02, PHEIDI = 8.24E−01, PP.H4 = 0.978). The wald ratio analysis yielded the same results (OR = 1.258, 95% CI 1.140, 1.388, P = 4.64E−06).
The methylation at various loci of genes including rs2058389-STYXL1, rs13246286-SND1, rs322817-SND1, rs140291167-SND1, rs35660964-SND1, rs2242517-GCDH, rs75461554-CASP9, rs2276731-ALDH1L1, rs34293138-GPX1, rs55663021-SLC25A29, as well as rs11235548-PDE2A and rs148038373-PDE2A, were also found to be causally associated with eGFRcrea, but their contributions were relatively small (the absolute value of β < 0.01).
The MR analysis of mitochondrial genome-wide cis-pQTL and CKD
Concerning protein expression of mitochondrial-related genes, we identified a unique genetic locus significantly associated with CKD and 3 unique genetic loci associated with eGFRcrea (Fig. 4, Supplementary Table S7, S8). Notably, an increase of one standard deviation in protein expression of rs1153858-GATM reduced the risk of CKD occurrence by 33.9% (OR = 0.661, 95%CI 0.587, 0.744, FDR = 9.26E−10, PHEIDI = 8.91E−02, PP.H4 = 0.027). The inverse variance weighted analysis yielded the same results (OR = 0.733, 95% CI 0.640, 0.840, P = 7.12E−06). Meanwhile, there was no pleiotropy (PIntercept = 5.89E−01), and the only heterogeneity had little impact on the results (PCochran's Q = 3.60E−02). Furthermore, protein expression of rs1486308-NIT2, rs4699179-PPA2, and rs10887871-PRXL2A genes exhibited causative relationships with eGFRcrea. Unfortunately, their PP.H4 were all less than 0.8.

MR results for the association between the protein expression of mitochondrial-related genes and CKD progression outcomes.
Bi-directional MR analysis of mitochondrial dysfunction and CKD
The mtDNA copy number variation is considered a surrogate biomarker for mitochondrial dysfunction. Currently, a GWAS dataset, based on exome sequencing results from 415,422 individuals of European descent within the UK Biobank, has identified genetic variations associated with mtDNA copy27. We utilized this dataset to investigate whether mitochondrial dysfunction contributes to CKD. Excitingly, our results suggested that CKD or eGFRcrea didn’t impact changes in mtDNA copy number (Supplementary Table S9). The direction of causality suggests a specificity of mitochondrial dysfunction predisposing towards CKD susceptibility.
External dataset validation of the GATM expression and CKD.
Additionally, we validated our findings across multiple external RNA sequencing datasets. The results demonstrated that the expression levels of GATM were significantly reduced in nine CKD conditions, including diabetic nephropathy, vasculitis, focal segmental glomerulosclerosis, membranous glomerulonephritis and so on (Fig. 5). Further analysis revealed a significant positive correlation between the expression levels of GATM and GFR (calculate using MDRD method) across multiple datasets, as indicated by the fitted curve equation and associated P-values in Fig. 6.

Comparison of GATM expression levels between various types of CKD and healthy control patients. **, P < 0.01; ****, P < 0.0001.

Correlation analysis between GFR and GATM expression levels in different datasets.
Ethics approval and consent to participate
This study was conducted following the Strengthening the Reporting of Observational studies in Epidemiology—Molecular Epidemiology (STROBE-ME): An extension of the STROBE statement38. The summary statistics data utilized in the MR analysis were obtained from publicly available databases or previous studies, all of which had received individual ethical approvals.
Discussion
In this investigation, we have substantiated the causal nexus between genetic susceptibility-induced mitochondrial dysfunction and the advancement of CKD. Moreover, our scrutiny has pinpointed GATM as a seminal mitochondrial-related gene pertinent to this phenomenon.
The GATM gene encodes the enzyme glycine amidinotransferase, classified within the amidinotransferase family. This enzyme plays a pivotal role in the biosynthesis of creatine by facilitating the transfer of the guanido group from L-arginine to glycine, resulting in the formation of guanidinoacetic acid—the immediate precursor of creatine28. This enzymatic process represents a crucial step in creatine production, a compound vital for rapid energy release in muscular activity. GATM exhibits noteworthy expression in tissues characterized by high energy demand, particularly in skeletal muscle, heart, brain, and kidneys, serving as a source of creatine for ATP generation29. In the kidneys, GATM manifests prominent expression in proximal tubule cells30, which are known for their heightened mitochondrial density, thus fulfilling the energy requirements essential for their intensive transport functions.
Research findings suggest that mutations in the GATM gene can give rise to latent creatine deficiency syndromes and autosomal dominant renal Fanconi syndrome, ultimately culminating in renal failure29. Homozygous missense mutations in GATM, characterized by complete penetrance, lead to the aggregation of GATM within proximal tubule cells. This aggregation is accompanied by heightened levels of reactive oxygen species, activation of the NLRP3 inflammasome, increased expression of the pro-fibrotic factor IL-18, and augmented cell death. Consequently, these molecular events contribute to kidney fibrosis and functional impairment31.
Previous investigations have not definitively elucidated the relationship between GATM gene polymorphisms and susceptibility to CKD. Notably, the allele A of rs2453533-GATM exhibits a higher prevalence among non-dialysis-dependent CKD patients (FDR = 0.020)32. Conversely, rs2467853-GATM/SPATA5L1 and rs13230509-GATM are associated with eGFRcrea but do not exert an impact on CKD susceptibility33,34,35. In the current study, we have observed significant causal relationships between the GATM gene and the occurrence of CKD at the levels of expression regulation (eQTL), methylation regulation (mQTL), and protein expression regulation (pQTL). For instance, an increase in the expression of rs7164602-GATM per standard deviation is associated with a 12.1% reduction in the risk of CKD occurrence, while an increase in the protein expression of rs1153858-GATM per standard deviation is linked to a 33.9% reduction in CKD occurrence risk. Additionally, elevated methylation levels at different loci show positive associations with CKD occurrence. Specifically, methylation at the cg00767496 locus of rs56850226-GATM increases CKD risk by 20.1% and augments the risk of a decrease in eGFRcrea by 2.4%. Similarly, methylation at the cg14910265 locus raises CKD risk by 18.9%, and methylation at the cg24328539 locus of rs12593371-GATM elevates CKD risk by 11.3%.
Upon validation with external datasets, we discerned a pervasive reduction in the expression levels of GATM across diverse types of CKD patients. Significantly, the expression levels of GATM exhibited a noteworthy positive correlation with the GFR, thereby providing additional support and strengthening the robustness of the findings derived from our study.
Indeed, disparities in GATM gene polymorphisms are evident across various continents and ethnicities. Notably, the association signal at the GATM gene locus from the Uganda Genomic Prostate Cancer (GPC) GWAS, indicated by the lead SNP rs2433603 with a significance level of P = 1.0 E−8, diverges from previously reported signals at this locus36. This genetic variation is monomorphic in European populations but exhibits rarity in East Asian populations. In the Uganda GPC cohort, the minor allele frequency for this variation is 48%, whereas it stands at 44% among Kenyan Luhya and 37% among Nigerian Yoruba, as per data from Phase 3 of the 1000 Genomes Project37. These findings underscore the imperative of amalgamating diverse GWAS data sources, particularly through the undertaking of large-scale studies tailored to local populations. Such endeavors are essential for gaining profound insights into the polymorphisms of the GATM gene associated with susceptibility to CKD.
The primary strength of this study lies in its comprehensive approach, particularly in leveraging mitochondrial-related gene expressions at eQTL, mQTL, and pQTL levels for conducting MR analysis. This multifaceted strategy enables the exploration of causal relationships between mitochondrial dysfunction and CKD, as well as eGFRcrea. The integrated utilization of these diverse data levels imparts substantial credibility to our findings. Noteworthy is the commendable performance exhibited by GATM across these three levels, affirming a clear causal relationship between genetic determinants of mitochondrial dysfunction and CKD susceptibility. Furthermore, the bidirectional MR analysis conducted in our study validates the specificity of the directionality of this causal relationship. Additionally, the augmentation of SMR with five other MR methods and colocalization analysis serves to further fortify the robustness of our study results. This combined analytical rigor contributes to the depth and reliability of the insights derived from our investigation.
This study is not without limitations. One notable constraint arises from the absence of specific QTL datasets tailored to the mitochondrial genome, posing a limitation to the scope of our analysis. The genetic variations associated with the entire mitochondrial genome primarily derive from nuclear genes related to mitochondria, rather than the mitochondrial genes themselves. Developing specific QTL datasets tailored to the mitochondrial genome to address this gap will be a goal in our next phase of research. Additionally, the PP.H4 value for the relationship between rs1153858-GATM protein expression and CKD falls below 0.8, indicating a weak co-localization relationship between the two. This may be attributed to the limited collection of integrated pQTL data, covering only 298 mitochondrial-related protein expressions and not encompassing the majority of SNP information. Our next research phase aims to include more SNP information associated with mitochondrial-related protein expressions, which may uncover more meaningful differential results. Furthermore, our reliance on mtDNA copy number as the sole metric for assessing the directionality of causal relationships in bidirectional MR analysis is constrained by the absence of GWAS datasets directly reflecting mitochondrial dysfunction. This limitation restricts our ability to conduct a more nuanced analysis, emphasizing the need for further advancements in GWAS datasets specifically targeting mitochondrial dysfunction. Lastly, all GWAS and SNP data involved in this study are derived from European populations. Therefore, the findings on the causal relationships and associations between mitochondrial-related genes and CKD are only applicable to this population. To extend the applicability of our results broadly, it is essential to develop databases for different ancestral backgrounds, such as Asian and African populations, and to perform specific analyses and validations. We plan to incorporate more diverse population data in the future as databases become more comprehensive, which will enhance our understanding of genetic variations associated with CKD.
Conclusions
This study employed MR analysis as a valuable tool to investigate potential causal relationships between mitochondrial-related genes and CKD, along with the eGFRcrea. Notably, the study places a particular emphasis on elucidating the pivotal role of GATM in the pathogenesis of CKD. The identification of GATM's significance in this context not only provides valuable insights for potential drug targets in the realm of CKD treatment but also holds promise for cancer treatment and prevention. Subsequent research endeavors could further delve into the underlying biological mechanisms governing the observed associations, thereby advancing our understanding and paving the way for more targeted therapeutic interventions.
Data availability
The study’s summary datasets are freely accessible and can be obtained in publications that are mentioned (Methods, Supplementary Table S1 and S2). cis-eQTL: eQTLGen Consortium (https://eqtlgen.org/cis-eqtls.html). cis-mQTL: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC6279736/. cis-pQTL: https://www.nature.com/articles/s41588-021–00,978-w#additional-information. CKD and eGFRcrea: CKDGen (https://ckdgen.imbi.uni-freiburg.de/). CKD RNA sequencing datasets from the Nephroseq public database (www.nephroseq.org, University of Michigan, Ann Arbor, MI). cis-eQTL: eQTLGen Consortium (https://eqtlgen.org/cis-eqtls.html). cis-mQTL: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC6279736/. cis-pQTL: https://www.nature.com/articles/s41588-021-00978-w#additional-information. CKD and eGFRcrea: CKDGen (https://ckdgen.imbi.uni-freiburg.de/). CKD RNA sequencing datasets from the Nephroseq public database (www.nephroseq.org, University of Michigan, Ann Arbor, MI).
Abbreviations
- CKD:
-
Chronic kidney disease
- MR:
-
Mendelian randomization
- IVs:
-
Instrumental variables
- GWAS:
-
Genome-wide association studies
- SNPs:
-
Single nucleotide polymorphisms
- eQTL:
-
Expression quantitative trait loci
- mQTL:
-
Methylation quantitative trait loci
- pQTL:
-
Protein quantitative trait loci
- SMR:
-
Summary-data-based MR
- 2SMR:
-
Two-sample MR
- HEIDI:
-
Heterogeneity in dependent instruments
- LD:
-
Linkage disequilibrium
- eGFR:
-
Estimated glomerular filtration rate
- eGFRcrea:
-
EGFR based on creatinine
- OR:
-
Odds ratio
- SD:
-
Standard-deviation
- HP:
-
Horizontal pleiotropy
- PP:
-
Posterior probability
- mtDNA:
-
Mitochondrial DNA
References
Saran, R. et al. US renal data system 2019 annual data report: Epidemiology of kidney disease in the United States. Am J Kidney Dis. 75(1 Suppl 1), A6-a7 (2020).
Wen, L. et al. Glucose metabolism in acute kidney injury and kidney repair. Front. Med. 8, 744122 (2021).
Pan, X. Cholesterol metabolism in chronic kidney disease: Physiology, pathologic mechanisms, and treatment. Adv. Exp. Med. Biol. 1372, 119–143 (2022).
Li, X., Zheng, S. & Wu, G. Amino acid metabolism in the kidneys: Nutritional and physiological significance. Adv. Exp. Med. Biol. 1265, 71–95 (2020).
Pan, R., Ryan, J., Pan, D., Wucherpfennig, K. W. & Letai, A. Augmenting NK cell-based immunotherapy by targeting mitochondrial apoptosis. Cell. 185(9), 1521–38.e18 (2022).
Johnson, S. C., Rabinovitch, P. S. & Kaeberlein, M. mTOR is a key modulator of ageing and age-related disease. Nature. 493(7432), 338–345 (2013).
Chandel, N. S. Mitochondria. Cold Spring Harb. Perspect. Biol. 13(3), 40543 (2021).
Sharma, K. et al. Metabolomics reveals signature of mitochondrial dysfunction in diabetic kidney disease. J. Am. Soc. Nephrol. JASN. 24(11), 1901–1912 (2013).
Wei, Z. et al. Urinary mitochondrial DNA level as a biomarker of tissue injury in non-diabetic chronic kidney diseases. BMC Nephrol. 19, 1–7 (2018).
Liu, L., Li, Y., Chen, G. & Chen, Q. Crosstalk between mitochondrial biogenesis and mitophagy to maintain mitochondrial homeostasis. J. Biomed. Sci. 30(1), 86 (2023).
Granata, S. et al. Mitochondrial dysregulation and oxidative stress in patients with chronic kidney disease. BMC Genomics. 10(1), 388 (2009).
Ng, Y. S. et al. The clinical, biochemical and genetic features associated with RMND1-related mitochondrial disease. J. Med. Genet. 53(11), 768–775 (2016).
Scaglia, F. et al. Novel homoplasmic mutation in the mitochondrial tRNATyr gene associated with atypical mitochondrial cytopathy presenting with focal segmental glomerulosclerosis. Am. J. Med. Genet. A. 123a(2), 172–178 (2003).
Zhang, X., Agborbesong, E. & Li, X. The role of mitochondria in acute kidney injury and chronic kidney disease and its therapeutic potential. Int. J. Mol. Sci. 22(20), 11253 (2021).
Takemura, K., Nishi, H. & Inagi, R. Mitochondrial dysfunction in kidney disease and uremic sarcopenia. Front. Physiol. 11, 565023 (2020).
Zhu, Z. et al. Causal associations between risk factors and common diseases inferred from GWAS summary data. Nat. Commun. 9(1), 224 (2018).
Davies, N. M., Holmes, M. V. & Smith, G. D. Reading Mendelian randomisation studies: A guide, glossary, and checklist for clinicians. BMJ. 362, k601 (2018).
Zhu, Z. et al. Integration of summary data from GWAS and eQTL studies predicts complex trait gene targets. Nat. Genetics. 48(5), 481–487 (2016).
McRae, A. F. et al. Identification of 55,000 replicated DNA methylation QTL. Sci. Rep. 8(1), 17605 (2018).
Ferkingstad, E. et al. Large-scale integration of the plasma proteome with genetics and disease. Nat. Genet. 53(12), 1712–1721 (2021).
Lawlor, D. A. Commentary: Two-sample Mendelian randomization: Opportunities and challenges. Int. J. Epidemiol. 45(3), 908–915 (2016).
Rath, S. et al. MitoCarta3.0: An updated mitochondrial proteome now with sub-organelle localization and pathway annotations. Nucl. Acids Res. 49(D1), D1541–D1547 (2020).
Võsa, U. et al. Large-scale cis- and trans-eQTL analyses identify thousands of genetic loci and polygenic scores that regulate blood gene expression. Nat. Genet. 53(9), 1300–1310 (2021).
Wuttke, M. et al. A catalog of genetic loci associated with kidney function from analyses of a million individuals. Nat. Genet. 51(6), 957–972 (2019).
Stanzick, K. J. et al. Discovery and prioritization of variants and genes for kidney function in >1.2 million individuals. Nat. Commun. 12(1), 4350 (2021).
Giambartolomei, C. et al. Bayesian test for colocalisation between pairs of genetic association studies using summary statistics. PLOS Genet. 10(5), e1004383 (2014).
Pillalamarri, V. et al. Whole-exome sequencing in 415,422 individuals identifies rare variants associated with mitochondrial DNA copy number. HGG Adv. 4(1), 100147 (2023).
DesRoches, C. L., Bruun, T., Wang, P., Marshall, C. R. & Mercimek-Mahmutoglu, S. Arginine–glycine amidinotransferase deficiency and functional characterization of missense variants in GATM. Hum. Mutat. 37(9), 926–932 (2016).
Sinn, M. et al. Guanidino acid hydrolysis by the human enzyme annotated as agmatinase. Sci. Rep. 12(1), 22088 (2022).
Bastian, F. B. et al. The Bgee suite: Integrated curated expression atlas and comparative transcriptomics in animals. Nucl. Acids Res. 49(D1), D831–D847 (2021).
Reichold, M. et al. Glycine amidinotransferase (GATM), renal fanconi syndrome, and kidney failure. J. Am. Soc. Nephrol. JASN. 29(7), 1849–1858 (2018).
Šalamon, Š, Bevc, S., Ekart, R., Hojs, R. & Potočnik, U. Polymorphism in the GATM locus associated with dialysis-independent chronic kidney disease but not dialysis-dependent kidney failure. Genes. 12(6), 834 (2021).
Köttgen, A. et al. Multiple loci associated with indices of renal function and chronic kidney disease. Nat Genet. 41(6), 712–717 (2009).
Hellwege, J. N. et al. Mapping eGFR loci to the renal transcriptome and phenome in the VA million veteran program. Nat Commun. 10(1), 3842 (2019).
Kintu, C. et al. Meta-analysis of African ancestry genome-wide association studies identified novel locus and validates multiple loci associated with kidney function. BMC Genomics. 24(1), 496 (2023).
Fatumo, S. et al. Discovery and fine-mapping of kidney function loci in first genome-wide association study in Africans. Hum. Mol. Genet. 30(16), 1559–1568 (2021).
Auton, A. et al. A global reference for human genetic variation. Nature. 526(7571), 68–74 (2015).
Gallo, V. et al. STrengthening the reporting of observational studies in epidemiology—Molecular epidemiology (STROBE-ME): An extension of the STROBE statement. Eur. J. Clin. Invest. 42(1), 1–16 (2012).
Acknowledgements
We sincerely thank the reviewers and editors of scientific reports for providing valuable suggestions on this paper.
Funding
This work was supported by the National Natural Science Foundation of China (No. 82270785, 81501381), the Jilin Province Medical and Health Talent Special Project (No. JLSWSRCZX2023-27) and Natural Science Foundation of Jilin Province (No. YDZJ202301ZYTS046).
Author information
Authors and Affiliations
Contributions
B.L. Conceptualization, Formal analysis, Investigation, and Writing—original draft; X.G. Data curation, Methodology, and Writing—original draft; H.T. Investigation, Validation, Visualization, and Writing—review & editing; H.Z. Funding acquisition, Investigation, and Writing—review & editing; B.G. Funding acquisition, and Writing—review & editing; F.L. Funding acquisition, Project administration, Supervision, and Writing—review & editing. This manuscript has been approved in its final form by all authors.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Liu, B., Gao, X., Teng, H. et al. Association between GATM gene polymorphism and progression of chronic kidney disease: a mitochondrial related genome-wide Mendelian randomization study. Sci Rep 14, 20346 (2024). https://doi.org/10.1038/s41598-024-68448-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-024-68448-x
- Springer Nature Limited