
Abstract
True morels (Morchella) are globally renowned medicinal and edible mushrooms. White mold disease caused by fungi is the main disease of Morchella, which has the characteristics of wide incidence and strong destructiveness. The disparities observed in the isolation rates of different pathogens indicate their varying degrees of host adaptability and competitive survival abilities. In order to elucidate its potential mechanism, this study, the pathogen of white mold disease from Dafang county, Guizhou Province was isolated and purified, identified as Pseudodiploöspora longispora by morphological, molecular biological and pathogenicity tests. Furthermore, high-quality genome of P. longisporus (40.846 Mb) was assembled N50 of 3.09 Mb, predicts 7381 protein-coding genes. Phylogenetic analysis of single-copy homologous genes showed that P. longispora and Zelopaecilomyces penicillatus have the closest evolutionary relationship, diverging into two branches approximately 50 (44.3–61.4) MYA. Additionally, compared with the other two pathogens causing Morchella disease, Z. penicillatus and Cladobotryum protrusum, it was found that they had similar proportions of carbohydrate enzyme types and encoded abundant cell wall degrading enzymes, such as chitinase and glucanase, indicating their important role in disease development. Moreover, the secondary metabolite gene clusters of P. longispora and Z. penicillatus show a high degree of similarity to leucinostatin A and leucinostatin B (peptaibols). Furthermore, a gene cluster with synthetic toxic substance Ochratoxin A was also identified in P. longispora and C. protrusum, indicating that they may pose a potential threat to food safety. This study provides valuable insights into the genome of P. longispora, contributing to pathogenicity research.
Similar content being viewed by others


Introduction
Morels are globally rare and precious edible mushrooms containing a variety of bioactive substances1. Modern medical research shows that Morchella has significant antioxidant, anti-inflammatory, immune regulation and liver protection effects, in addition to lowering blood sugar and blood lipids2,3,4,5. These medicinal values are mainly attributed to the active ingredients such as polysaccharides, phenolic compounds, amino acids and carotenoids6,7,8. The indoor cultivation of Morchella was not successful in the United States until the 1990s9,10. The cultivation area of Morchella in Guizhou province increased year by year from 2015 to 2100 hectares by 202011. However, it is seriously infected by diseases during the cultivation process, including white mold disease (P. longispora, Z. penicillatus)12,13, cobweb disease (C. mycophilum, C. protrusum)14,15, stipe rot disease (Fusarium incarnatum-F. equiseti)16, white rot disease (Aspergillus sp.)17, stalk rot (F. nematophilum)18 and rot disease (Lecanicillium aphanocladii, Clonostachys rosea, Trichoderma atroviride)19,20,21, which has potential risks to food safety and has become one of the important issues facing sustainable Morchella cultivation.
White mold disease (known as pileus rot disease) caused by Ascomycetes is the most destructive and highest incidence disease on Morchella9. The disease is characterized by white, velvety pili on infected fruiting bodies, with rapid expansion at high temperature (> 25 °C) and high humidity (> 90%), leading to wilting, rotting, deformity, and significant damage, potentially causing death in Morchella12. In 2017, the first report of white mold disease caused by Z. penicillatus was reported in China, with an estimated incidence of 10–80% on M. importuna22. Then, the disease caused by P. longispora was first reported in Chongqing in 2018, it can infect all cultivated Morchella varieties, mainly infecting their cap part of fruiting bodies, and very few are stipes12. Furthermore, in 2022, an investigation of Morchella disease in China found that P. longispora was the main pathogen, with a separation rate of 75.48%2. In addition, cobweb disease caused by Cladobotryum is the second fungal disease of Morchella, The symptoms of the disease are a small amount of white flocculation aerial hyphae on the surface of the stipe, thick white hyphae covering the fruiting body, and rotted in the later stage14,15. White mold and cobweb disease were the main disease of Morchella, but the isolation rates of pathogens were quite different. The main pathogen reported is P. longispora, while Z. penicillatus and Cladobotryum sp. were only reported in a few areas.
The genome-wide study of pathogenic fungi is conducive to improving the identification of pathogenicity-related genes such as carbohydrate-active enzymes database (CAZy), secondary metabolites (SMs), and pathogen-host interaction (PHI) genes, and other virulence factors, revealing the molecular mechanism of its pathogenesis23,24. CAZy in fungi can participate in the pathogenicity of pathogenic fungi and promote the attachment, invasion, colonization and nutrient absorption of pathogens in host cells, and the SMs helps pathogenic fungi invade the host to compete for nutrients or niches25. SMs include some toxins, which can be used as pathogenic factors for fungal pathogens invading hosts, and can also be used as toxins to contaminate food and feed, posing a threat to human and animal health26. For example, glycoside hydrolase family 18 (GH18) chitinases have been shown to participate in many physiological processes including tissue degradation, pathogenicity and immune defense27. For Morchella disease, previous studies have shown that Z. penicillatus could secrete cell wall degrading enzymes such as glucanase and mannanase to degrade the cell wall of Morchella during the infection of Morchella28,29,30. The peptaibols were confirmed to be a virulence factor involved in the invasion of P. longispora into Morchella25. In addition, Genome analysis of the Morchella pathogen C. protrusum and C. mycophilum has predicted a large number of secretory proteins and carbohydrate enzyme genes related to pathogenesis such as Glycosyl Transferases (GTs) gene families14,31. Unfortunately, the whole genome information with P. longispora is missing. Therefore, we want to analyze the genome of P. longispora and compare it with related pathogen genomes to understand why it has strong host adaptability and competitive survival abilities.
In the present study, we isolated and identified the white mold of Morchella from Dafang county, Guizhou province, and sequenced the whole genome of the pathogen. In addition, a genome-wide phylogenetic tree was constructed to clarify its evolutionary relationship. In order to understand the potential pathogenic factors, comparative analysis with Morchella related pathogen genomes was performed to predict potential pathogenic genes. The results of this study will lay a foundation for further revealing the function of white mold pathogenic genes and formulating effective strategies for white mold control.
Materials and methods
Pathogen isolation, maintenance and pathogenicity tests
The diseased fruiting bodies of M. esculenta were collected from Dafang county, Guizhou province (N: 27° 08′ 29.80″; E: 105° 36′ 46.80″) in May 2022. Pathogens were isolated using the spread plate and tissue isolation method32. Purified strains were transferred to PDA (Potato Dextrose Agar: potato 200 g/L, glucose 20 g/L, agar 20 g/L) for growth for 7 days, and then maintained in 25% (v/v) glycerol at − 80 °C for long-term storage in the Culture Collection of the Department of Plant Pathology, Agriculture College, Guizhou University33. All strains were prepared into spore suspension (1 × 106 spores mL−1) using sterile water for pathogenicity test34. The 15 μL spore suspension was noculated on the fruiting body of healthy Morchella with three replicates, and the control was treated with sterile water35. After five days, the pathogen was re-isolated from the infected fruiting body and identified by morphology and phylogenetic analysis to fulfill Koch’s postulates13.
Morphological and molecular characterization
For morphological identification, the pathogen isolates were cultured on PDA medium at 25 °C for 10 days. Photographs of colonies were taken using an ultra-depth of field stereomicroscope (Digital microscope system Keyence VHX-7000). Photomicrographs of isolates were taken with a compound light microscope (Zeiss Scope 5 with camera AxioCam 208 color), including mycelium, conidia, conidiophore and chlamydospore, including conidiophore and conidia size36. Total genomic DNA was extracted by SDBS (sodium dodecyl benzene sulfonate) method and quantified by agarose gel electrophoresis and Qubit® 2.0 fluorescence (Thermo Scientific)37,38. The internal transcribed spacer gene region (ITS), large subunit rRNA gene region (LSU), small subunit ribosomal RNA gene region (SSU), partial translation elongation factor 1-α (TEF), and RNA polymerase II second largest subunit (RPB2) were amplified with the primer pairs ITS5/ITS4, LROR/LR5, NS1/NS4, 983F/2218R, and fRPB2-5F/fRPB2-7cR, respectively38. The PCR was conducted in a Bio-Rad, T100 Thermal Cycler (Bio-Rad Laboratories Inc., California, USA). The total reaction volume of the PCR reaction was 25 µL consisting of 1.6 µL of dNTP mix (2.5 mM µL−1), 0.2 µL of Taq polymerase (5 U µL−1), 1 µL of genomic DNA (50 ng µL−1), 2 µL of polymerase buffers (10 × µL−1, Takara, Japan), and 1 µL of each primer (25 mM µL−1)39. The following amplification conditions were used: 1 cycle at 95 °C for 3 min; 34 cycles at 95 °C for 30 s, 55 °C for 30 s, and 72 °C for 30 s; and 1 cycle at 72 °C for 7 min40. Amplification products were detected by gel electrophoreses on 1.0% agarose gels stained with Gel Green. The PCR products were sequenced using the same PCR primers used in the amplification reaction of Qingke Biotech (Chengdu) Co., Ltd. The reference related sequences used in this study were obtained from GenBank database (https://www.ncbi.nlm.nih.gov/). The multi-gene phylogenetic tree was constructed using OFPT “a one-stop software for fungal phylogeny”41.
De novo genome sequencing and assembly
The whole genome of GUCCX-091 was sequenced using PacBio Sequel Single Molecule Real-Time (SMRT) at the Beijing Novogene Bioinformatics Technology Co., Ltd42. For library construction, 1 µg DNA sample was fragmented by sonication to a size of 350 bp, then DNA fragments were end-polished, A-tailed, and ligated with the full-length adaptor for Illumina sequencing with further PCR amplification. At last, PCR products were purified (AMPure XP system) and libraries were analysed for size distribution by Agilent 2100 Bioanalyzer and quantified using real-time PCR. The genome sequence was assembled using SOAP de novo software43.
The raw data is filtered to obtain valid data. Data filtering includes deleting more than 40% of bases with mass value ≤ 20, more than 10% of N numbers, and more than 15 bp of adapter overlap and less than 3 mismatched reads. In the assembly stage, the SOAP de novo software is used for assembly, and the optimal K-mer (The default parameters are 95, 107, 119) and bracket parameters and other parameters are adjusted according to the project type44. The gap closure software was used to fill the gap of the preliminary assembly results and eliminate the errors. Fragments below 500 bp were filtered to obtain the final assembly results.
Gene prediction and annotation
Genome component prediction included the prediction of the coding gene, repetitive sequences and non-coding RNA. Repeat predictions and tandem repeats were predicted using the RepeatMasker (http://www.repeatmasker.org/) and TRF (Tandem Repeats Finder)45. Gene models were predicted with a combination of ab initio Augustus, SNAP, Glimmer, GeneMarkHMM and FGENESH. Finally, PASA was used to update the EVM consensus predictions, adding UTR annotations and models for alternatively spliced isoforms, we obtained the final gene sets46,47. Transfer RNA (tRNA) genes were predicted by the tRNAscan-SE48
Multiple databases, including GO (Gene Ontology)49, KEGG (Kyoto Encyclopedia of Genes and Genomes)50, KOG (Clusters of Orthologous Groups)51, NR (Non-Redundant Protein Database databases)52, TCDB (Transporter Classification Database)53, P450 and Swiss-Prot54 were used to make functional annotations for the predicted gene models, using BlastP with E-values ≤ 1e−555,56.
We also used the Signal P database to predict secreted proteins57. For pathogenic fungi, we conducted pathogenicity and drug resistance analyses. To carry out these analyses, we used Pathogen Host Interactions (PHI)58.
Analysis of homologous gene families, phylogenetics and synteny
The gene family was analyzed by OrthoMCL (Evalue ≤ 1e−5)59, reference species include Neurospora crassa (GCA_000182925.2), Escovopsis weberi (GCA_003055145.1), Tolypocladium inflatum (GCA_003945575.1), C. protrusum (GCA_004303015.1), Z. penicillatus (GCA_005765155.1), Cordyceps militaris (GCA_008080495.1), Fusarium oxysporum (GCA_013085055.1), Clonostachys rosea (GCA_015832225.1), Calonectria ilicicola (GCA_020809705.1), F. avenaceum (GCA_025948275.1), Akanthomyces muscarius (GCA_028009165.1), Simplicillium aogashimaense (GCA_028455935.1), Trichoderma breve (GCA_028502605.1), T. pseudokoningii (GCA_943193705.1). Based on the analysis of homologous gene families, a single-copy homologous gene group was selected for phylogenetic analysis. Use Gblocks (using default parameters) to remove divergent and unclearly aligned blocks from the comparison to obtain a better CDS file. The maximum likelihood tree was constructed using RaxML, and the phylogenetic relationship between P. longispora and the above 14 fungi were inferred using the gtrgamma model and 100 Bootstrap replicates60. The differentiation time was estimated using Mcmctree in the PAML software package. Genome-wide homology analysis was performed using MUMmer software, and homologous point maps were generated using mummerplot61.
Annotation of CAZymes analysis
The protein sequences of P. longispora and Z. penicillatus were subjected to BLASTP search (E-value ≤ 1e−5), and the annotated proteins were annotated by the tool dbCan3, and the sequences that could be annotated by Hmmer, Diamond and eCAMI were selected as the final annotation set62. The predicted CAZy genes were then classified into different families based on their functional domains, including Auxiliary Activities (AAs), Glycosyl Transferases (GTs), Carbohydrate Esterases (CEs), Polysaccharide Lyases (PLs), Glycoside Hydrolases (GHs), and Carbohydrate-Binding Modules (CBMs)63. A comparison of the carbohydrate-active enzyme genes between the three fungi was conducted to identify shared and unique enzyme genes. This comparison involved sequence alignment and analysis using the BLAST tool or comparative genomics analysis software. Subsequently, functional annotation was performed on the compared enzyme genes to understand their roles in carbohydrate metabolism.
Secondary metabolism genes and clusters analysis
AntiSMASH 7.0 was used to predict and annotate the putative secondary metabolism gene clusters (BGCs) such as polyketide synthase (PKS), nonribosomal peptide synthase (NRPS) genes, PKS-NRPS hybrids and their modules64. The default setting was, and the parameter setting was "strict". Based on homology analysis, the key enzymes of secondary metabolism (parameter 1E-5,75% coverage) were analyzed by BLASTn and search results were listed in Table S4 of additional file 265.
Results
Pathogen isolation and identification
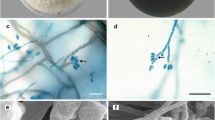
Ten strains of fungi were isolated from five diseased fruiting bodies, and they were identified as the same species by colony and microscopic morphology. Strain GUCCX-091 grew slowly on PDA plate, colonies were 3–4 cm in diameter at 25 °C for 8 days, convex, white, with a smooth margin, and aerial mycelia were sparse to moderate (Fig. 1C). Hyphae were hyaline, smooth, septate, branched, and 2.16–4.03 μm in diameter. Conidiophores were micronematous to semi-macronematous, hyaline, and indistinguishable from vegetative hyphae. Conidia were ellipsoidal or citriform, 4.22–5.18 × 15.84–18.47 μm, 1–3-septate (predominantly 1-septate) (Fig. 1F,G). Conidiogenous cells were blastic or thallic, acropetal or intercalary. Light—brown chlamydospores formed from vegetative hyphal cells in terminal or intercalary clusters of 2–10 cells. They were spherical, with a smooth or verrucose surface, 6.53–11.09 μm in diameter, and released mostly in clusters or in short or long chains, and sometimes in pairs or individually (Fig. 1D–F). The cultural and morphological characteristics were consistent with those of P. longispora.

Colony and microscopic characteristics of GUCCX-091. (A,B) Field symptoms of white mold disease on M. sextelata; (C) the colony on PDA after 8 days and the reverse side, respectively; (D,E) conidiophores with conidiogenous cells and conidia (F–J) conidiophores; (K) chlamydospores. Scale bars: B = 1 mm; D = 30 μm; E = 20 μm; F = 50 μm; G-K = 25 μm.
The ITS, LSU, SSU and TEF of two strains were amplified by PCR and sequenced. The ITS fragments of strains GUCCX-090 and GUCCX-091 were 509 and 514 bp (GenBank accession numbers OR889858, OR889859), the LSU fragments were 859 and 840 bp (GenBank accession numbers OR889860, OR889861), the SSU fragments were 1051 and 1059 bp (GenBank accession numbers OR889862, OR889863), and the TEF fragments were 913 and 909 bp (GenBank accession numbers OR908784, OR908785), respectively. After searching and comparing the sequences with the NCBI database BLAST, it was found that the similarity of these sequences with the accession numbers OR770478.1 (ITS), KY765315.1 (LSU), OP231754.1 (SSU) and OP265137.1 (TEF) was higher than 99.80%, respectively. The phylogenetic tree showed that the sequences of strain GUCCX-090 and GUCCX-091 were found to be in the same branch as P. longispora (Fig. 2) and divided into two branches with Z. penicillatus CBS 448.69. The morphology of GUCCX-090 and GUCCX-091 was significantly different from that of Z. penicillatus. So they were identified as P. longispora based on morphological and molecular phylogenetic analyses.

ML phylogenetic tree based on the ITS-LSU-SSU-TEF sequences. mlikelihood (ML) values > 50%. Tindicates the type. The tree is rooted with two Protocrea species. The strains in this study in red.
Pathogenicity tests
White mold disease mainly occurs at the cap of the fruiting body of Morchella (Fig. 3A). In the early stage of the disease, irregular lesions and white villous mycelium were formed on the cap, with the continuous infection of the pathogen, the mold layer continued to expand, resulting in shrinkage and dryness at the infection site. The cap rotted and even perforated, which seriously affected the growth and development of the fruiting body and eventually caused the fruiting body to wither and die.

Field symptoms and pathogenicity of M. sextelata white mold disease. (A) Field symptoms of white mold disease on M. sextelata; (B) control, the five days of inoculation of sterile water; (C) inoculation of spore suspension (1 × 106 spores mL−1 ) on the first day; (D) a small amount of white mold layer appeared at the inoculation site. after three days of inoculation with spore suspension; (E) five days after a large number of white mold layers appeared and the lesions expanded; (F) normal M. sextelata fruiting bodies by SEM; (G) early stage of infection, hyphae (red arrow) of P. longispora invaded into the fruiting body of M. sextelata along the gaps, no infection structure was observed. conidia (yellow arrow).
Strain of GUCCX-091 was randomly selected for pathogenicity test. Pathogenicity results showed that the typically dense white, coarse mycelia were visible three days at the base of the fruiting body after inoculation with the spore suspension (Fig. 3D). Five days after a large number of white mold layers appeared and the lesions expanded (Fig. 3E), while the control group was abnormal (Fig. 3B). These symptoms from artificial inoculation were similar to those observed in the field. To fulfill Koch’s postulates, re-isolation from infected fruiting bodies were done, the pathogens was confirmed to be consistent with the inoculated isolates based on morphological characteristics.
General genomic structure of Pseudodiploöspora longispora GUCCX-091
The raw data obtained by sequencing of P. longispora GUCCX-091 has a certain proportion of low-quality data, and the clean data obtained after processing is 7,341,219 kb, 458,883 reads number, with a maximum read length of 48,174 bp and a minimum read length of 424 bp with a sequencing depth of approximately 94 × . SOAP de novo software was used to assemble the sequenced clean data to obtain a genome of 40.846 Mb, the genome consists of 24 contigs, with N50 of 3,090,083 bp, and 43.42% G+C content, the length of the largest contig is 7.58 Mbp (Fig. 4). We predicted 7381 genes with an average length of 1449 bp, and accounting for 26.18% of the total genome length (Table 1).

Overview of the Pseudodiploöspora longispora GUCCX-091 genome assembly. The outermost circle is the position coordinates of the genome sequence, and the color block is 12 sequences with coding sequences. (A) GC content per window. (B) Genome GC skew value. (C) Gene density per window. (D) rRNA, snRNA and tRNA density per window. (A–C) The window (genome/1000) bp, step (genome/1000) bp. Links within and between chromosomes represent collinear blocks generated from MCScanX and JCVI software. The plot was visualized using Circos software.
The repeated sequences were predicted by RepeatMasker software, and the tandem repeats in the DNA sequence were searched by TRF, and the repeated sequences were identified, accounting for 4.84% of the P. longispora genome, (Interspersed Repeats 1.271%, Tandem Repeat, TR 3.577%). The majority of Interspersed Repeats sequences were LTR retrotransposons (0.96%), where 0.2% and 0.149% of the repeat element types were DNA transposons and LINE (Table S1). The majority of Tandem Repeat were TR (1.925%) and Minisatellite DNA (1.514%).
Gene assembly and annotation of Pseudodiploöspora longispora GUCCX-091
Annotation was performed with the NCBI nr, KEGG, GO, KOG, TCDB, PHI-base and P450 databases (Table 2). There were 6792 Non-redundant proteins found in P. longispora 2022051405-3. They matched closest with T. arundinaceum (818), T. paradoxum (581), T. harzianum (401), T. ophioglossoides (386), Purpureocillium lilacinum (385), T. asperellum (292), Pochonia chlamydosporia (290), and T. virens (270) which accounted for 50.39% of total nr predicted genes (Fig. S1). Predicted genes were mapped to the KEGG database and assigned functional classifications to 6684 gene models (90.56% of the total gene models, 7381). Some translation related to protein families and metabolism were highly enriched including “Global and overview maps” (858), “Translation” (298), “Carbohydrate metabolism” (277), “Transport and catabolism” (252), “Amino acid metabolism” (251), “Signal transduction” (233), “Folding, sorting and degradation” (223) etc. (Fig. 5).

KEGG pathway annotation of Pseudodiploöspora longispora GUCCX-091.
There were 1901 proteins assigned to NCBI KOG categories (Fig. S2). The “Posttranslational modification, protein turnover, chaperones” category had the most enriched genes (223), followed by “General function prediction only” (214), “ranslation, ribosomal structure and biogenesis” (204), “Energy production and conversion” (155), and “Amino acid transport and metabolism” (154). TCDB search was to perform protein domain analysis and assigned 290 putative transport proteins to 7 functional classes including “Accessory factors involved in transport” (53), “Channels/pores” (75), “Electrochemical potential-driven transporters” (169), “Group translocators” (8), “Incompletely characterized transport systems” (73), “Primary active transporters” (175), and “Transmembrane electron carriers” (4) (Fig. S3). The top two enriched categories were “The Major Facilitator Superfamily (MFS)” (48) and “The Nuclear Pore Complex (NPC) Family” (36). Mino acid sequences were mapped with PHI-base and identified 1341 candidate pathogenicity-related proteins (Fig. S4). The “Reduced virulence” category had the most enriched proteins (533), followed by “Unaffected pathogenicity” (491), “Loss of pathogenicity” (106), together these represented 84.27% of all proteins predicted with PHI-base. The functional annotation results in the GO database showed that 5,053 genes (58.46% of all the genes) could be classified into three types, including genes related to cell composition (26.73%), molecular function (29.05%) and biological process (44.22%).
In the biological process function, the metabolic process-related gene is at most 2720, followed by the cellular process (2668). The GO terms with the highest numbers of genes in the corresponding function genes classified as related to molecular function were for catalytic activity (2725) and binding (2682). Cell part had the most genes related to cell composition function (1851) (Fig. 6). Cytochrome P450 (CYP) is a superfamily of hemoproteins that use heme as a cofactor. CYPs have various substrates in different enzymatic reactions and are present in all kingdoms. Eighty-three putative CYPs genes were identified in P. longispora through a BLAST search that was classified into 8 families. The E-class P450, group I family had the highest number of enriched genes (39), followed by Undeterminded (34), and E-class P450, group IV (16) (Fig. S5).

GO functional annotation of Pseudodiploöspora longispora GUCCX-091.
Analysis of homologous gene families in Hypocreales fungi
Orthologs are genes that exist in different species, usually play the same function in different species, and diverge with the formation of species66. Here, we identified 15,705 gene families encoding 138,392 genes in these species, in addition to 14,315 unclustered genes (Fig. 7A). Among them, P. longispora GUCCX-091 has 7381 protein-coding genes including 2723 single-copy orthologs, 21 unique paralogs, 312 unclustered genes, 3556 common families genes and 769 other paralogs. In addition, we also obtained the number of coding gene families of P. longispora (6606), Z. penicillatus (7805) and C. protrusum (8481), respectively. The Venn diagram analysis showed that P. longispora shared 5603 genes with the other two pathogenic fungi. P. longispora and Z. penicillatus had 739 homologous genes, significantly more than 185 with C. protrusum. C. protrusum had more unique genes (1519), P. longispora had the lowest number of unique genes (79), and Z. penicillatus had 289 unique genes (Fig. 7B).

Evolutionary and functional analysis of the of P. longispor GUCCX-091 genome. (A) Ortholog clustering analysis of the protein-coding genes in the P. longispora genome; (B) Venn diagram showing shared and distinct orthologous gene families in Hypocreales fungi. The proteomes of P. longispora, Z. penicillatus and C. protrusum were clustered using orthoMCL. The numbers of genes in each species are shown in parenthesis; (C) Whole-genome collinearity analysis based on protein-coding genes; (D) Phylogenetic tree and number of expanded and contracted gene families among 15 fungal genomes. Calibrated nodes are indicated by red dots, and the overall timeline is shown below the phylogenetic tree.
Synteny analysis and phylogenetic analysis
The collinearity of P. longispora, Z. penicillatus and C. protrusum was analyzed by collinear arrangement of genome-wide protein-coding genes. Only sequences with a sequence length of more than 1 M were shown in the figure. Compared with the genome of P. longispora, the results of genome collinearity analysis showed that there were 13 collinearity regions between the genome of Z. penicillatus and the genome of P. longispora. There were 11 collinearity regions with a collinearity sequence transfer length of more than 1000 kp, and the total was 38,488,948 bp, accounting for 94.23 of the whole genome, which was highly conserved collinearity. However, the collinearity of P. longispora to C. protrusum was 22,652,435 bp, which was significantly lower than that of P. longispora and Z. penicillatus (Fig. 7C).
Based on the phylogenetic relationship of Hypocreales fungi, we used 2,723 single-copy homologous proteins to construct a phylogenetic tree according to the results of gene family clustering, so as to provide a basis for clarifying their taxonomic status (Fig. 7D). The differentiation time of N. crassa 222 (202.3–240.2) MYA was used as a reference for time calibration. Phylogenomic analysis revealed that P. longispora, Z. penicillatus and C. protrusum differentiated into two branches before 80.3 (72.9–84.3) MYA, which was greater than that of 54.7 (51.1–57.1) MYA C. protrusum and E. weberi. It is earlier than 50 (44.3–61.4) MYA of P. longispora and Z. penicillatus. We identified 6488 gene families across those species, the expansion and contraction of their gene families were compared. Except for A. muscularius, T. breve and F. oxysporum, the contraction of the gene families of 13 fungi in Hypocreales exceeded the expansion along each lineage. Among them, P. longispora has 46 gene family expansion, 890 contraction, Z. penicillatus expansion 42, contraction 177, C. protrusum contraction 135, expansion 203.
Carbohydrate-active enzymes in Pseudodiploöspora longispora
The fungal cell wall is generally composed of 90% polysaccharides (glucan and chitin), which are the main polymers of carbohydrates, such as α-1,3-glucan, α-1,4-glucan and so on. P. longispora requires carbohydrate enzymes capable of degrading and modifying polysaccharides in the process of infecting morel. Therefore, we compared the assembled genome sequence of P. longispora with the CAZyme database, and 247 CAZyme-encoding genes were annotated in 93 CAZyme gene families. Compared with Z. penicillatus encoding 268 CAZyme proteins from 101 families, P. longispora has fewer carbohydrate enzyme genes. Compared with Z. penicillatus encoding 268 CAZyme proteins from 101 families and C. protrusum annotating 298 genes in 100 families of CAZymes. The carbohydrate enzymes of P. longispora include AAs (20), GTs (55), CEs (5), PLs (1), GHs (149) and CBMs (17) (Table 3).
Comparing the CAZyme types of the three fungi, it was found that GH, GT and AA accounted for the largest proportion. Therefore, we clustered and compared the encoded 108 CAZyme types, and found that the number of GHs was significantly higher than that of other types (Fig. 8). GHs belongs to plant cell wall degrading enzyme (PCWDEs), an enzyme that hydrolyzes glycosidic bonds and plays an important role in the hydrolysis and synthesis of sugars and glycoconjugates in organisms67. Among them, chitinases include GH18, GH19 and GH20, which can hydrolyze the β-1, 4-glycosidic bond between C1 and C4 of two GlcNAc in chitin, and play a very important role in the successful penetration and infection of pathogens68. As predicted, GH18 content was the highest in all three fungi, and GH18 content in C. protrusum (27) was significantly higher than that in P. longispora (18) and Z. penicillatus (17). In addition, more GH16 family genes P. longispora (14), C. protrusum (14) and Z. penicillatus (16) were annotated. GH16 contains a number of glucanases that are widely present in many organisms, including bacteria, fungi, plants and animals69. Previous studies have shown that Z. penicillatus can secrete glucanase, mannanase and protease to degrade the cell wall of morel during the infection of morel. GH18 and GH16 were the two most annotated carbohydrate enzyme families in the three species, accounting for P. longispora (12.96%), C. protrusum (13.76%) and Z. penicillatus (12.31%) of the total carbohydrate enzymes, respectively. It shows that the three pathogenic fungi have strong chitinase and glucanase synthesis abilities, which is helpful for them to adapt and infect morels.

Cluster analysis of carbohydrate-active enzymes of three pathogenic fungi.
Secondary metabolite gene clusters
One of the key weapons possessed by necrotic pathogens is the production of toxic secondary metabolites, including polyketides, non-ribosomal peptide synthase (NRPS), terpenes and alkaloids, to kill host cells70. P. longispora, Z. penicillatus and C. protrusum can lead to the collapse and necrosis of fruiting bodies in the late stage of infection. Therefore, the secondary metabolic gene clusters of the three fungi were predicted. The results showed that P. longispora had 27 non-ribosomal peptide synthase (NRPS) and type I polyketide synthase (T1PKS) gene clusters, encoding 29 NRPS and 31 T1PKS core genes, respectively, and 11 terpene gene clusters. The abundance was lower than C. protrusum NRPS (30), T1PKS (51) and Terpene (18). However, Z. penicillatus has the least nonribosomal peptide synthetase (NRPS) and type I polyketide synthase (T1PKS) gene clusters (23) except Terpene (18). In general, the secondary metabolic gene cluster encoded by C. protrusum was significantly more than that of P. longispora and Z. penicillatus (Table 4).
In addition, the genomes of P. longispora and Z. penicillatus were predicted to have genes for the synthesis of peptaibols. peptaibols are linear antimicrobial peptides synthesized by different NRPS, which are resistant to a variety of bacteria and fungi. They were highly similar to the known gene cluster BGC0001358 (NCBI GenBank: LSBH01000002.1) encoding leucinostatin A and leucinostatin B of Purpureocillium lilacinum. The antiSMASH 7.0.1 and BLAST analyses showed that the whole genomes of P. longispora and C. protrusum contained gene clusters that were highly similar to those responsible for the Ochratoxin A (OTA) biosynthetic gene cluster from Aspergillus steynii (GenBank: MG701897.1), which was not predicted in Z. penicillatus. Ochratoxins are toxic secondary metabolites produced by Penicillium and Aspergillus species, which widely contaminate food and feed, among them, Ochratoxin A is the most toxic71,72.
Discussion
White mold is the most common disease of Morchella, which occurs in a large area in China. The pathogen mainly infects and destroys the cap of the fruiting body, and spreads rapidly during the harvest period, causing serious losses to the yield and quality of Morchella. In this study, we collected the samples of white mold in the cultivation area of Morchella in Dafang county, Guizhou province. The pathogen was identified as P. longispora by pathogenicity test, molecular biology and morphology73. In the investigation and isolation of white mold in the main cultivation area of Morchella in Guizhou, the pathogen was P. longispora, and other pathogens were not isolated, which may be related to different geographical and climatic environments. The main fungal pathogens of Morchella have been reported to be Z. penicillatus, Aspergillus sp., C. mycophilum, Lecanicillium aphanocladii and T. atroviride. Among them, the symptoms of white rot disease caused by Aspergillus sp., rot disease caused by T. atroviride are similar to white mold in the field, which brings some difficulties to the prevention and control of the disease17,21. Therefore, the identification of pathogens is of great significance in the prevention and control of Morchella diseases.
The genome size assembled by P. longispora in the study was 40.846 Mb, which was larger than C. protrusum (39.1 Mb) and Z. penicillatus (39.8 Mb), however, P. longispora annotated significantly fewer genes than them. This may be related to the expansion and contraction of their genes during evolution, because we found that the number of contraction genes of P. longispora (890) was much larger than that of Z. penicillatus (177) and C. protrusum (135). Genome and gene evolution of fungi enable them to exist in diverse environments, patterns of gene family expansion or contraction can reflect particular selection pressure that a species has been subjected to over evolutionary time scales74. Whether this is related to their parasitic adaptability to Morchella needs further study. Annotation results of KEGG, GO, TCDB and KOG databases showed that the coding gvenes were mainly enriched in functional areas including protein transport, metabolism and biological process, and exercised molecular functions such as catalytic activity, binding and transport, which was conducive to adapting to the environment. From the perspective of metabolic pathways, P. longispora still has many secondary metabolites to be explored, which has important research value.
Cluster analysis of the genome-encoded proteins of the three Morchella pathogens showed that there were 739 homologous genes in P. longispora and Z. penicillatus, of which significantly more than 185 with C. protrusum. Moreover, the genomes of P. longispora and Z. penicillatus were highly similar in synteny. Most of the shared genes may indicate a high degree of genetic similarity between them, which may be due to the common genetic background and common ancestors. Based on genome-wide phylogenetic tree analysis, we found that P. longispora and Z. penicillatus differentiated into two branches before 50 (44.3–61.4) MYA. However, we noticed that a previous study reported divergence time for P. longispora and Z. penicillatus based on (SSU, ITS, LSU, RPB2 and TEF) phylogenetic tree analysis as about 14 MYA. Based on the differences between the two methods, genome-wide data can provide a more comprehensive and accurate estimation of differentiation time, helping us to better understand the evolutionary history and genetic relationship of fungi.
The destruction of host cell wall by CAZymes is the key to the success of early infection of pathogenic fungi75. The number of CAZymes predicted by the genome of P. longispora is 247, lower than that of Z. penicillatus (268) and C. protrusum (298), the ability of P. longispora to infect Morchella may be weaker than that of Z. penicillatus and C. protrusum. Notably, the number of GHs is significantly higher than other types, GHs is a cell wall degrading enzyme that exists in almost all organisms and plays a very important role in the successful penetration and infection of hosts. Further analysis showed that the content of GH18 content in C. protrusum (27) was significantly higher than that in P. longispora (18) and Z. penicillatus (17). GH18 chitinase has been shown to be involved in many physiological processes, including tissue degradation, developmental regulation, pathogenicity and immune defense27. In addition, in the process of Z. penicillatus infecting morel, it can secrete cell wall degrading enzymes, mannanase and so on to degrade the cell wall. Therefore, we speculate that the three pathogens may have the same infection mechanism for the infection of morel fruiting bodies, that is, in the early stage of pathogen infection, the host cell wall is destroyed mainly by secreting a large number of GHs.
SMs is an important determinant of the pathogenicity of pathogenic fungi76. SMs gene cluster were identified in C. protrusum than in P. longispora and Z. penicillatus (Table 4). It shows that the potential of C. protrusum to produce secondary metabolites is stronger than that of P. longispora and Z. penicillatus (Table S2). Importantly, Peptaibols (including alamethicin F-50, polysporin B, septocylindrin B (1 − 3), and longisporin A) were confirmed to be the virulence factors of P. longispora infected Morchella. Although this study did not predict the gene encoding this type of peptaibols substance, but the SMs gene clusters of P. longispora and Z. penicillatus were highly similar to BGC0001358 (NCBI GenBank: LSBH01000002.1), which encodes peptaibols leucinostatin A and leucinostatin B of P. lilacinum. Whether leucinostatin A and leucinostatin B are one of the virulence factors of pathogen infection needs to be verified by further research. In addition, we also predicted the protein sequence encoding OTA in P. longispora and C. protrusum. OTA is a secondary metabolite of fungi such as Aspergillus and Penicillium, causing serious economic losses to agricultural products and feed71. The hepatorenal toxicity and three-induced effects (teratogenic, carcinogenic, mutagenic) of OTA have been supported by more and more data, confirming that it poses a great threat to human health77. The physical and chemical properties of OTA and its derivatives have been studied comprehensively, but the synthesis process and regulation mechanism are still unclear78. This study speculates that P. longispora and C. protrusum have the potential to synthesize the toxic substance OTA, which can provide materials for the synthesis and regulation mechanism of OTA. After the disease infection of edible fungi, the pathogen may produce toxic substances, which pose a potential threat to the quality and safety of agricultural products.
In general, we identified the pathogen causing white mold of Morchella in Dafang County, Guizhou Province, and revealed the genomic characteristics of the pathogen P. longispora, including its genome and repeats, and predicted its coding function and evolutionary relationship. Through the comparative genomic analysis of Morchella pathogens, some carbohydrate enzymes and secondary metabolites such as chitinase, leucinostatin A, leucinostatin B and Ochratoxin A may play a key role in pathogenicity. However, only by comparing their omics analysis, the molecular mechanism of pathogen selection on hosts cannot be fully revealed. These findings will help us to better understand the pathogenic mechanism of this fungus and provide new candidate targets for the development of antifungal drugs in the future.
Data availability
The sequences generated in this study can be found in GenBank. The whole genome accession number of GUCCX-091 is: SAMN38441546; GUCCX-090 (ITS: OR889858; LSU: OR889860; SSU: OR889862; EF-1α: OR908784); GUCCX-091 (ITS: OR889859; LSU: OR889861; SSU: OR889863; EF-1α: OR908785).
References
Liu, Q., Ma, H. S., Zhang, Y. & Dong, C. H. Artificial cultivation of true morels: Current state, issues and perspectives. Crit. Rev. Biotechnol. 38, 259–271. https://doi.org/10.1080/07388551.2017.1333082 (2018).
Pan, Z. H., Lan, Y. & Zhang, S. Study on the Antioxidative and antiaging effects of the exopolysaccharide extract from Morchella conica. J. South China Normal Univ. 02, 124–128 (2011).
Bao, M., Zeng, Y., Zhang, L. Y. & Peng, S. J. Study on antioxidant effect of extracellular polysaccharide from Morchella crassipes in vitro. Edible Fungi. 36, 63–64 (2014).
Nitha, B., Meera, C. R. & Janardhanan, K. K. Anti-inflammatory and antitumour activities of cultured mycelium of morel mushroom Morchella esculenta. Curr. Sci. 92, 235–239. https://doi.org/10.1126/science.1135926 (2007).
Liu, C. et al. Characteristics and antitumor activity of Morchella esculenta polysaccharide extracted by pulsed electric field. Int. J. Mol. Sci. 17, 986. https://doi.org/10.3390/ijms17060986 (2016).
Liu, B. et al. Nutrient analysis of morel in northwest Yunnan Province. Sci. Technol. Food Industry. 33, 363–365. https://doi.org/10.13386/j.issn1002-0306 (2012).
He, P. et al. Involvement of autophagy and apoptosis and lipid accumulation in sclerotial morphogenesis of Morchella importuna. Micron. 109, 34–40. https://doi.org/10.1016/j.micron.2018.03.005 (2018).
Cai, Y. L., Ma, X. L. & Liu, W. Review of nutrition value and health efficacy of Morel. Edible Med. Mushrooms. 29, 20–27 (2021).
Shi, X., Liu, D., He, X., Liu, W. & Yu, F. Epidemic identification of fungal diseases in Morchella cultivation across China. J. Fungi. 8, 1107. https://doi.org/10.3390/jof8101107 (2022).
Kuo, M. Morels; University of Michigan Press: Ann Arbor (MI, 2005).
Wang, Q. et al. Current situation and development thinking of Morchella industry in Guizhou Province. Edible Fungi 44(02), 67–69 (2022).
He, X. L. et al. White mold on cultivated morels caused by Paecilomyces penicillatus. FEMS Microbiol. Lett. 364(5), 1. https://doi.org/10.1093/femsle/fnx037 (2017).
He, P. X. et al. First report of pileus rot disease on cultivated Morchella importuna caused by Diploöspora longispora in China. J. Gen. Plant Pathol. 84, 65–69. https://doi.org/10.1007/s10327-017-0754-3 (2018).
Lan, Y. F. et al. First report of Cladobotryum protrusum causing cobweb disease on cultivated Morchella importuna. Plant Dis. 104, 977–977. https://doi.org/10.1094/PDIS-08-19-1611-PDN (2020).
Liu, Z. H. et al. Characterization and genome analysis of Cladobotryum mycophilum, the causal agent of cobweb disease of Morchella sextelata in China. J. Fungi. 9, 411. https://doi.org/10.3390/jof9040411 (2023).
Guo, M. P., Chen, K., Wang, G. Z. & Bian, Y. B. First report of stipe rot disease on morchella importuna caused by Fusarium incarnatum—F. equiseti species complex in China. Plant Dis. 100, 2530–2530. https://doi.org/10.1094/PDIS-05-16-0633-PDN (2016).
Yu, M., Yin, Q. & He, P. X. lsolation and identification of pathogen of morel white rot. Northern Hortic. 7, 142–145. https://doi.org/10.11937/bfyy.20193757 (2020).
Liu, T. H. et al. A new stipe rot disease of the cultivated Morchella sextelata. Mycosystema 40, 2229–2243. https://doi.org/10.13346/j.mycosystema.210055 (2021).
Lv, B. B., Sun, Y., Chen, Y. F., Yu, H. L. & Mo, Q. First report of Lecanicillium aphanocladii causing rot of Morchella sextelata in China. Plant Dis. https://doi.org/10.1094/PDIS-12-21-2656-PDN (2022).
Fu, Y. W. et al. First Report of Clonostachys rosea causing rot of Morchella sextelata in Anhui Province. China. Plant Dis. https://doi.org/10.1094/PDIS-08-22-1794-PDN (2022).
Fu, Y. W. et al. First report of Trichoderma atroviride causing rot of Morchella sextelata in Anhui Province. China. Crop Protect. 1, 1. https://doi.org/10.1016/j.cropro.2023.106206 (2023).
Fu, B. et al. Isolation, identification and biological characteristics of causal pathogen causing pileus rot of cultivated Morchella importuna. Acta Agricult. Boreali Occid. Sin. 31, 640–647. https://doi.org/10.7606/j.issn.1004-1389.2022.05.013 (2022).
Xu, R. et al. Genomic features of Cladobotryum dendroides, which causes cobweb disease in edible mushrooms, and identification of genes related to pathogenicity and mycoparasitism. Pathogens. 20(9), 232. https://doi.org/10.3390/pathogens9030232 (2020).
Dai, Y. T. et al. Genomic analyses provide insights into the evolutionary history and genetic diversity of Auricularia species. Front. Microbiol. 1(10), 2255. https://doi.org/10.3389/fmicb.2019.02255 (2019).
Dong, W. et al. Identification and characterization of peptaibols as the causing agents of Pseudodiploöspora longispora infecting the edible mushroom Morchella. J. Agric. Food Chem. 29(71), 18385–18394. https://doi.org/10.1021/acs.jafc.3c05783 (2023).
Liu, Q. et al. Infection process and genome assembly provide insights into the pathogenic mechanism of destructive mycoparasite Calcarisporium cordycipiticola with host specificity. J. Fungi (Basel). 7(11), 918. https://doi.org/10.3390/jof7110918 (2021).
Chen, W., Jiang, X. & Yang, Q. Glycoside hydrolase family 18 chitinases: The known and the unknown. Biotechnol. Adv. 1(43), 107553. https://doi.org/10.1016/j.biotechadv.2020.107553 (2020).
Wang, X. et al. Genome Sequencing of Paecilomyces Penicillatus Provides Insights into Its Phylogenetic Placement and Mycoparasitism Mechanisms on Morel Mushrooms. Pathogens. 9(10), 834. https://doi.org/10.3390/pathogens9100834 (2020).
Chen, C. et al. Genome sequence and transcriptome profiles of pathogenic fungus Paecilomyces penicillatus reveal its interactions with edible fungus Morchella importuna. Comput. Struct. Biotechnol. J. 19, 2607–2617. https://doi.org/10.1016/j.csbj.2021.04.065 (2021).
Yu, Y. et al. Dual RNA-Seq analysis of the interaction between edible fungus Morchella sextelata and its pathogenic fungus Paecilomyces penicillatus uncovers the candidate defense and pathogenic factors. Front. Microbiol. 12, 760444. https://doi.org/10.3389/fmicb.2021.760444 (2021).
Sossah, F. L. et al. Genome sequencing of Cladobotryum protrusum provides insights into the evolution and pathogenic mechanisms of the cobweb disease pathogen on cultivated mushroom. Genes (Basel). 10(2), 124. https://doi.org/10.3390/genes10020124 (2019).
Zeng, X. Y. et al. Taxonomy and control of Trichoderma hymenopellicola sp. nov. responsible for the first green mould disease on Hymenopellis raphanipes. Front. Microbiol. 13, 991987. https://doi.org/10.3389/fmicb.2022.991987 (2022).
Yuan, X. X. et al. Complete genomic characterization and identification of Saccharomycopsis phalluae sp. Nov., a novel pathogen causes yellow rot disease on Phallus rubrovolvatus. J. Fungi. 7, 707. https://doi.org/10.3390/jof7090707 (2021).
Mu, J. Y. & Sang, W. J. ldentification of aniseed leaf spot pathogen with toxicity evaluation of five fungicides. J. Mount. Agric. Biol. 29, 360–363. https://doi.org/10.15958/j.cnki.sdnyswxb.2010.04.006 (2010).
Wang, J. S., Jiang, Y. L., Wang, D. F., Yang, Y. X. & Cheng, D. L. Pathogen identification of grape anthracnose in Guiyang area and fungicides screening in laboratory. J. Mount. Agric. Biol. 33, 59–61. https://doi.org/10.15958/j.cnki.sdnyswxb.2014.06.011 (2014).
Huang, H. et al. Dentification and cultural characterization of Diplospora longispora associated with pileus rot disease on cultivated morel. Plant Protect. 48, 66–72. https://doi.org/10.16688/j.zwbh.2020537 (2022).
Lim, H. J., Lee, E. H., Yoon, Y., Chua, B. & Son, A. Portable lysis apparatus for rapid single-step DNA extraction of Bacillus subtilis. J. Appl. Microbiol. 120, 379–387. https://doi.org/10.1111/jam.13011 (2016).
White, T. J., Bruns, S., Lee, S. & Taylor, J. Amplification and direct sequencing of fungal ribosomal RNA genes for phylogenetics. PCR Protocols, A guide to methods and application 1, 315–322 (1990).
Wu, S. et al. Identification and growth characteristics of tea leaf spot pathogenic. J. Mount. Agric. Biol. 42, 10–17. https://doi.org/10.15958/j.cnki.sdnyswxb.2023.04.002 (2023).
Qiao, L. J., Wen, T. C., Kang, J. C., Kang, C. & Wu, X. L. Morphology and phylogeny of seven Russula species from Mayang River National Nature Reserve of Guizhou. J. Mount. Agric. Biol. 35, 87–92. https://doi.org/10.15958/j.cnki.sdnyswxb.2016.06.015 (2016).
Zeng, X. Y., Tan, T. J., Tian, F. H., Wang, Y. & Wen, T. C. OFPT: a one-stop software for fungal phylogeny. Mycosphere. 14, 1730–1741. https://doi.org/10.5943/mycosphere/14/1/20 (2023).
Tian, F. H., Li, C. T. & Li, Y. Genomic analysis of Sarcomyxa edulis reveals the basis of its medicinal properties and evolutionary relationships. Front. Microbiol. 12, 652324. https://doi.org/10.3389/fmicb.2021.652324 (2021).
LaCava, M. E. F. et al. Accuracy of de novo assembly of DNA sequences from double-digest libraries varies substantially among software. Mol. Ecol. Resour. 20, 360–370. https://doi.org/10.1111/1755-0998.13108 (2020).
Nyström-Persson, J., Keeble-Gagnère, G. & Zawad, N. Compact and evenly distributed k-mer binning for genomic sequences. Bioinformatics. 37, 2563–2569. https://doi.org/10.1093/bioinformatics/btab156 (2021).
Saha, S., Bridges, S., Magbanua, Z. V. & Peterson, D. G. Empirical comparison of ab initio repeat finding programs. Nucleic Acids Res. 36, 2284–2294. https://doi.org/10.1093/nar/gkn064 (2008).
Benson, G. Tandem repeats finder: A program to analyze DNA sequences. Nucleic Acids Res. 27, 573–580. https://doi.org/10.1093/nar/27.2.573 (1999).
Haas, B. J. et al. Improving the Arabidopsis genome annotation using maximal transcript alignment assemblies. Nucleic Acids Res. 31, 5654–5666. https://doi.org/10.1093/nar/gkg770 (2003).
Haas, B. J. et al. Automated eukaryotic gene structure annotation using EVidenceModeler and the program to assemble spliced alignments. Genome Biol. 9, R7. https://doi.org/10.1186/gb-2008-9-1-r7 (2008).
Ashburner, M. et al. Gene ontology: Tool for the unification of biology The Gene Ontology Consortium. Nat. Genet. 25, 25–29. https://doi.org/10.1038/75556 (2000).
Kanehisa, M., Goto, S., Kawashima, S., Okuno, Y. & Hattori, M. The KEGG resource for deciphering the genome. Nucleic Acids Res. 32, D277–D280. https://doi.org/10.1093/nar/gkh063 (2004).
Galperin, M. Y., Makarova, K. S., Wolf, Y. I. & Koonin, E. V. Expanded microbial genome coverage and improved protein family annotation in the COG database. Nucleic Acids Res. 43, D261–D269. https://doi.org/10.1093/nar/gku1223 (2015).
Li, W., Jaroszewski, L. & Godzik, A. Tolerating some redundancy significantly speeds up clustering of large protein databases. Bioinformatics. 18, 77–82. https://doi.org/10.1093/bioinformatics/18.1.77 (2002).
Milton, H. S., Vamsee, S. R., Dorjee, G. T. & Ake, V. The transporter classification database. Nucleic Acids Res. 42, D251–D258. https://doi.org/10.1093/nar/gkt1097 (2014).
Bairoch, A. & Apweiler, R. The SWISS-PROT protein sequence database and its supplement TrEMBL in 2000. Nucleic Acids Res. 28, 45–48. https://doi.org/10.1093/nar/28.1.45 (2000).
Altschul, S. F., Gish, W., Miller, W., Myers, E. W. & Lipman, D. J. Basic local alignment search tool. J. Mol. Biol. 215, 403–410. https://doi.org/10.1016/S0022-2836(05)80360-2 (1990).
Tatusov, R. L., Galperin, M. Y., Natale, D. A. & Koonin, E. V. The COG database: A tool for genome-scale analysis of protein functions and evolution. Nucleic Acids Res. 28, 33–36. https://doi.org/10.1093/nar/28.1.33 (2000).
Petersen, T. N., Brunak, S., von Heijne, G. & Nielsen, H. SignalP 4.0: Discriminating signal peptides from transmembrane regions. Nat. Methods. 8, 785–786. https://doi.org/10.1038/nmeth.1701 (2011).
Urban, M. et al. The Pathogen-Host Interactions database (PHI-base): Additions and future developments. Nucleic Acids Res. 43, D645–D655. https://doi.org/10.1093/nar/gku1165 (2015).
Li, L., Stoeckert, C. J. Jr. & Roos, D. S. OrthoMCL: Identification of ortholog groupsfor eukaryotic genomes. Genome Res. 13, 2178–2189. https://doi.org/10.1101/gr.1224503 (2003).
Guindon, S. et al. New algorithms and methods to estimate maximum-likelihood phylogenies: assessing the performance of PhyML 3.0. Syst. Biol. 59, 307–321. https://doi.org/10.1093/sysbio/syq010 (2010).
Kurtz, S. et al. Versatile and open software for comparing large genomes. Genome Biol. 5, R12. https://doi.org/10.1186/gb-2004-5-2-r12 (2004).
Zheng, J. et al. dbCAN3: Automated carbohydrate-active enzyme and substrate annotation. Nucleic Acids Res. 51, W115–W121. https://doi.org/10.1093/nar/gkad328 (2023).
Lombard, V., Golaconda Ramulu, H., Drula, E., Coutinho, P. M. & Henrissat, B. The carbohydrate-active enzymes database (CAZy) in 2013. Nucleic Acids Res. 42, D490–D495. https://doi.org/10.1093/nar/gkt1178 (2014).
Blin, K. et al. antiSMASH 7.0: New and improved predictions for detection, regulation, chemical structures and visualisation. Nucleic Acids Res. 51, W46–W50. https://doi.org/10.1093/nar/gkad344 (2023).
Hittalmani, S., Mahesh, H. B., Mahadevaiah, C. & Prasannakumar, M. K. De novo genome assembly and annotation of rice sheath rot fungus Sarocladium oryzae reveals genes involved in Helvolic acid and Cerulenin biosynthesis pathways. BMC Genomics. 17, 271. https://doi.org/10.1186/s12864-016-2599-0 (2016).
Emms, D. M. & Kelly, S. SHOOT: Phylogenetic gene search and ortholog inference. Genome Biol. 23, 85. https://doi.org/10.1186/s13059-022-02652-8 (2022).
Graebin, N. G. et al. Immobilization of glycoside hydrolase families GH1, GH13, and GH70: state of the art and perspectives. Molecules. 21, 1074. https://doi.org/10.3390/molecules21081074 (2016).
Zhou, W. X., Tian, C. M. & You, C. M. Genomic sequencing analysis of Cytospora piceae associated with spruce canker disease andcomparative genomic analysis of Cytospora species. Acta Microbiol. Sin. 61, 3128–3148. https://doi.org/10.13343/j.cnki.wsxb.20200754 (2021).
Suriani Ribeiro, M. et al. Endo-β-1,3-glucanase (GH16 Family) from Trichoderma harzianum participates in cell wall biogenesis but is not essential for antagonism against plant pathogens. Biomolecules. 9, 781. https://doi.org/10.3390/biom9120781 (2019).
Brakhage, A. A. Regulation of fungal secondary metabolism. Nat. Rev. Microbiol. 11, 21–32. https://doi.org/10.1038/nrmicro2916 (2013).
Ding, L., Han, M., Wang, X. & Guo, Y. Ochratoxin A: Overview of prevention, removal, and detoxification methods. Toxins (Basel). 15, 565. https://doi.org/10.3390/toxins15090565 (2023).
Wang, Y. et al. A Consensus Ochratoxin A Biosynthetic Pathway: Insights from the Genome Sequence of Aspergillus ochraceus and a Comparative Genomic Analysis. Appl. Environ. Microbiol. 84, e01009–e1018. https://doi.org/10.1128/AEM.01009-18 (2018).
Castañeda, R. F. Fungi cubenses II. Instituto de Investigaciones Fundamentales en Agricultura Tropical “Alejandro Humboldt”. Academia de Ciencas de Cuba, La Havana, Cuba (1987).
Nygaard, S. et al. The genome of the leaf-cutting ant Acromyrmex echinatior suggests key adaptations to advanced social life and fungus farming. Genome Res. 21, 1339–1348. https://doi.org/10.1101/gr.121392.111 (2011).
Bashyal, B. M. et al. Whole genome sequencing of Fusarium fujikuroi provides insight into the role of secretory proteins and cell wall degrading enzymes in causing bakanae disease of rice. Front. Plant Sci. 8, 2013. https://doi.org/10.3389/fpls.2017.02013 (2017).
Garajova, S. et al. Single-domain flavoenzymes trigger lytic polysaccharide monooxygenases for oxidative degradation of cellulose. Sci. Rep. 6, 28276. https://doi.org/10.1038/srep28276 (2016).
Macheleidt, J. et al. Regulation and role of fungal secondary metabolites. Annu. Rev. Genet. 50, 371–392. https://doi.org/10.1146/annurev-genet-120215-035203 (2016).
Gao, J., Liu, H. Q., Zhang, Z. Z. & Liang, Z. H. Production, toxicity and biosynthesis of Ochratoxin A: A review. Microbiol. China. 50, 1265–1280. https://doi.org/10.13344/j.microbiol.china.220641 (2023).
Acknowledgements
We would like to thank all people listed for their substantial, direct, and intellectual contributions to the work.
Funding
This research was funded by the Guizhou Provincial Science and Technology Projects [ZK[2023] general 087]; Guizhou Provincial Support Fund of Science and Technology, grant number Support of QKH [2021] General 199; The National Natural Science Foundation of China (NSFC: 32260044); GZMARSEdible Fungi, grant number GZMARS-SYJ-2021-2025, and Guizhou Education Department (grant no. Qianjiaohe KY 2021] 054).; Tianjin Synthetic Biotechnology Innovation Capacity Improvement Project (TSBICIP-CXRC-006).
Author information
Authors and Affiliations
Contributions
Conceptualization, FT. Data Curation, JX and XL. Funding acquisition, Resources, Supervision and Visualization, FT. Investigation, JX and ZQ Methodology, JX and SM. Software, CL and Entaj. Validation, XZ and FT. Writing-original draft, JX. Writing-review and editing, XZ and FT. All authors have read and agreed to the published version of the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Xie, J., Liu, X., Qin, Z. et al. Evolution and related pathogenic genes of Pseudodiploöspora longispora on Morchella based on genomic characterization and comparative genomic analysis. Sci Rep 14, 18588 (2024). https://doi.org/10.1038/s41598-024-69421-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-024-69421-4
- Springer Nature Limited