
Abstract
Ichthyoplankton monitoring is crucial for stock assessments, offering insights into spawning grounds, stock size, seasons, recruitment, and changes in regional ichthyofauna. This study evaluates the efficiency of multi-marker DNA metabarcoding using mitochondrial cytochrome c oxidase subunit I (COI), 12S rRNA and 16S rRNA gene markers, in comparison to morphology-based methods for fish species identification in ichthyoplankton samples. Two transects with four coastal distance categories were sampled along the southern coast of Portugal, being each sample divided for molecular and morphological analyses. A total of 76 fish species were identified by both approaches, with DNA metabarcoding overperforming morphology—75 versus 11 species-level identifications. Linking species-level DNA identifications with higher taxonomic morphological identifications resolved several uncertainties associated with traditional methods. Multi-marker DNA metabarcoding improved fish species detection by 20–36% compared to using a single marker/amplicon, and identified 38 species in common, reinforcing the validity of our results. PERMANOVA analysis revealed significant differences in species communities based on the primer set employed, transect location, and distance from the coast. Our findings underscore the potential of DNA metabarcoding to assess ichthyoplankton diversity and suggest that its integration into routine surveys could enhance the accuracy and comprehensiveness of fish stock assessments.
Similar content being viewed by others


Introduction
Ichthyoplankton monitoring offers invaluable insights into the ecological conditions of marine ecosystems, providing essential data for fish stock assessments and informed fisheries management1. By studying these early life stages, researchers can gain a deeper understanding of local fish reproductive dynamics, including the identification of spawning sites and seasons, estimation of recruitment and spawning stock size, assessment of annual variations, identification of nursery areas, and detection of potential changes in regional ichthyofauna and species distribution2. This information serves as a foundation for decision-makers to develop and implement effective conservation strategies to ensure the sustainability of fisheries.
Until recently, ichthyoplankton studies primarily relied on morphological inspection to identify fish eggs and larvae. While this traditional approach has provided valuable contributions over the years, its limitations can impede the precision required for effective management purposes3. The process is highly time-consuming and requires specialists who dedicate years to refining their expertise. Even with considerable experience, precise identifications are not always guaranteed due to the underdeveloped form of fish larvae and the lack of distinctive characteristics during the egg stage4.
DNA metabarcoding emerges as a molecular complement to morphology, enabling higher sampling frequency, cost and time efficiency, and rapid generation of extensive data5,6. This technique can identify species at any life stage while resolving taxonomic uncertainties associated with morphological identification5. Combining DNA barcoding with High-Throughput Sequencing (HTS), eliminates the need for individual processing and sequencing of organisms, enabling simultaneous identifications of multiple species within bulk samples5,7. The application of this methodology in ichthyoplankton studies has demonstrated remarkable precision and reliability for fish species identification3,8–10, showcasing its ability to unveil greater diversity than the traditional approach in bulk samples of fish eggs and larvae11,12. However, several stages in the DNA metabarcoding workflow can affect its efficiency, ranging from the selected DNA extraction method13 to the bioinformatic pipelines14. Among these challenges, the selection of genetic markers and primer sets stands out as one of the most crucial aspects of this application15,16.
The mitochondrial cytochrome c oxidase subunit I (COI) gene has been shown to generally perform very well in DNA barcoding-based identification of fish species17, owing to its high interspecific variability and the availability of comprehensive and curated reference databases, which are crucial for taxonomic assignment18,19. However, challenges such as the recognition of conserved priming regions encompassing a broad range of taxonomic groups, and the propensity to amplify DNA from non-target organisms, (e.g., bacteria), particularly when using seawater or sediment eDNA samples20,21, have prompted researchers to explore alternative genes to explore alternative genes. The mitochondrial 12S rRNA and 16S rRNA have shown promising results in fish metabarcoding studies11,22,23,24,25. Furthermore, studies employing multiple genetic markers and primer sets have detected a greater diversity compared to those using only one marker26,27,28.
Despite the potential of DNA metabarcoding, its main limitation lies in the inability to directly quantify individuals and determine their subsequent abundances, due to the several factors contributing to amplification bias in the process29. However, some studies demonstrated that under certain conditions and proper parametrisation, relative species abundance can eventually be inferred from sequence data3,26.
This study aimed to assess the efficiency of a 3-marker DNA metabarcoding protocol for identifying fish species in ichthyoplankton samples and compare results to morphology-based identifications. To this end, we analysed the species composition of ichthyoplankton samples collected along two perpendicular transects on the south coast of Portugal.
Methods
Ichthyoplankton collection
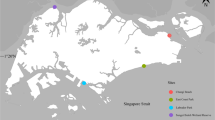
In June 2022, eight points along the southern coast of Portugal were sampled at varying distances from the coast, each corresponding to specific depths: inner shore (< 30 m), middle shore (30–100 m), outer shore (100–200 m), and offshore (> 500 m). The samples were distributed across two perpendicular transects along the coast in the adjacent areas of Arade River (A1–A4) and Ria Formosa (F1–F4) (Fig. 1). Ichthyoplankton samples were collected at night-time, starting immediately after sunset, to maximize the collection of ichthyoplankton due to their vertical migration near the surface at night30. Samples for each transect were collected on separate consecutive nights: the three closest points to the shore were sampled on the first nights (F1–F3 and A1–A3), and the offshore points were sampled on the following nights (F4 and A4). The trawls were performed using a Bongo net with a mesh size of 335 µm. The net was deployed obliquely, sampling the water column from a depth of 10–20 m below the sea surface for 10 min at a tow speed of 2 knots. To prevent contamination between sampling points, the net was rinsed and cleaned with seawater. Additionally, the Bongo collection buckets and all handling materials were thoroughly cleaned with detergent, followed by rinsing and cleaning with 96% ethanol and a 10% bleach solution. The equipment was deployed and retrieved from the sea with the assistance of a crane. Each collected sample was subdivided and randomly preserved using different methods for subsequent analysis. Specifically, 96% ethanol was used for samples for molecular analyses, while 4% formalin was employed to preserve samples for morphological identification. In total, 16 samples were processed in this manner. The samples designated for molecular analyses were stored at − 20 °C until DNA extraction could be performed. Every step of the experimental protocol was approved and adhered to the ethical standards established by the Animal Welfare and Protection guidelines of the former General Direction of Veterinary (DGV) under the former Ministry of Agriculture, Rural Development, and Fisheries in Lisbon, Portugal, for ecological research and the collection of ichthyoplankton samples.

Map with the eight sampling points. Points were distributed along two perpendicular transects to the southern coast of Portugal—the Arade River transect (A1–A4) and the Ria Formosa transect (F1–F4). Maps were created using Ocean Data View v.5.6.531 (https://odv.awi.de).
Morphological processing
Formalin-preserved samples were inspected with a Leica S8 APO stereoscope microscope for ichthyoplankton morphological identification. Samples were sorted to isolate fish eggs and larvae, and the respective total numbers were counted. The identification of eggs was feasible and reliable only for sardines and anchovies, as the morphology-based approach lacked sufficient resolution for the other taxa, which do not exhibit distinguishing characteristics. Fish larvae were identified to the lowest possible taxonomic level using morphological traits, following region-specific guides for the identification of planktonic eggs and larval stages30,32,33.
Molecular processing
DNA extraction
DNA extraction was conducted following an adapted protocol for these ichthyoplankton bulk samples described by Steinke et al.34 and based on Ivanova et al.35. This method involved an overnight incubation step with agitation, using a buffer solution (100 mM NaCl, 50 mM Tris–HCl pH 8.0, 10 mM EDTA pH 8.0, and 0.5% SDS) designed to promote cell lysis while preserving the main characteristics of each specimen. From each sample lysate, two 1 ml aliquots were transferred to microtubes and used for DNA extraction. These aliquots were subsequently pooled together at the end of the procedure, resulting in a final volume of approximately 60 µl (30 µl + 30 µl). As a negative control for the DNA extraction step, 1 ml of the solution buffer was processed alongside the ichthyoplankton samples to check for contamination of the solutions and labware materials used. These negative controls were used as templates in subsequent PCR amplification reactions. To ensure the recovery of the target DNA before PCR amplification, DNA concentration levels were measured using the Thermo Scientific™ NanoDrop™ One/OneC at the end of the protocol.
PCR amplification and high-throughput sequencing
DNA amplification was conducted for three different mitochondrial gene regions using specific primers (Table 1). For all primer sets, two PCR reaction replicates were performed for each sample using the KAPA HiFi HotStart PCR Kit (Kapabiosystems, Cape Town, South Africa) according to manufacturer instructions. Regarding COI, two primer sets were used: one generalist primer pair targeting marine metazoans—mICOIintF/LoboR136,37—and a fish-specific cocktail—FishATL_Cocktail238 (Table 1). For mICOIintF/LoboR1, the PCR reaction contained 0.3 μM of each PCR primer: forward primer mICOIintF and reverse primer LoboR1 and 5 μl of template DNA in a total volume of 25 μl. The PCR conditions involved a 3 min denaturation at 95 °C, followed by 35 cycles of 98 °C for 20 s, 60 °C for 30 s, 72 °C for 30 s, and a final extension at 72 °C for 5 min. For the FishATL_Cocktail 2, the PCR reaction contained 3 μM of each PCR primer: a pool of forward primers 219F, 217F, and 214F and a pool of reverse primers FishR1, FishR2, and FishR7 and 1 μl of template DNA in a total volume of 25 μl. The PCR conditions involved a 3 min denaturation at 95 °C, followed by 5 cycles of 95 °C for 30 s, 56 °C for 60 s and 72 °C for 60 s, 30 cycles of 95 °C for 30 s, 60 °C for 60 s, 72 °C for 60 s and a final extension at 72 °C for 10 min. For the 12S rRNA gene, a primer set combining the primer pairs miFISH-U and miFISH-E was used39. The PCR reaction contained 0.6 μM of each PCR primer: a pool of forward primers miFISH-U_F and miFISH-E_F and reverse primers miFISH-U_R and miFISH-E_R and 2.5 μl of template DNA in a total volume of 25 μl. The PCR conditions involved a 3 min denaturation at 95 °C, followed by 38 cycles of 98 °C for 20 s, 60 °C for 30 s, 72 °C for 30 s, and a final extension at 72 °C for 5 min. For the 16S mitochondrial gene, the primer pair Fish 16S used by Berry et al.22 was chosen. The PCR reaction contained 0.3 μM of each PCR primer: forward primer 16SF/D and reverse primer 16s2R-deg and 5 μl of template DNA in a total volume of 25 μl. The PCR conditions involved a 3 min denaturation at 95 °C, followed by 45 cycles of 98 °C for 20 s, 54 °C for 30 s, 72 °C for 30 s, and a final extension at 72 °C for 5 min. PCR-negative controls were included in each amplification. The 2 PCR replicates of each region for all samples were pooled before the second PCR reaction where Nextera XT indexes and sequencing adapters were added to both ends of the amplified target region according to the manufacturer’s recommendations40. Finally, PCR products were purified and normalised using SequalPrep 96-well plate kit (ThermoFisher Scientific, Waltham, USA)41, pooled and pair-end sequenced in the Illumina MiSeq® sequencer with the MiSeq Reagent Kit v3 (600 cycles), according to manufacturer’s instructions (Illumina, San Diego, CA, USA) at Genoinseq (Cantanhede, Portugal).
Bioinformatic processing
Raw data from high-throughput sequencing, contained in FASTQ files were processed using PMiFish version 2.4.1 (available at https://github.com/rogotoh/PMiFish.git)23. PMiFish allowed the analysis of all information obtained from the different molecular markers using a uniform pipeline. This approach improved the standardisation of the analysis process, facilitating comparisons among primer sets. The data preprocessing and subsequent analysis involved several steps based on USEARCH version 10.0.24045: (1) paired-end reads (Forward and Reverse) were merged, with reads containing more than 5 differences within the aligned region or lacking a minimum size (< 100 bp) being removed; (2) primers were then removed from the merged reads and (3) submitted to a quality filtering step; (4) the remaining sequences were dereplicated and (5) denoising was performed, creating amplicon sequence variants (ASVs) that excluded chimeras and sequences with errors; (6) finally, the ASVs were taxonomically assigned to species level, with a required identity threshold of > 97%. This resulted in the creation of molecular operational taxonomic units (MOTUs), where each MOTU was represented with a representative sequence corresponding to the ASV with the highest number of reads within that group of sequences. The COI reference database was compiled using the BAGS software (https://github.com/tadeu95/BAGS)46. The 12S and 16S reference databases were generated by mining available sequences from NCBI GenBank47. For the 12S and 16S reference data sets, unverified records were removed (i.e., submitted records that the GenBank staff could not confirm in terms of accuracy), as well as records without species-level identifications. All records were scrutinised for synonyms or unaccepted names, and any taxonomic discrepancies were rectified to align with the accepted nomenclature based on WoRMS48. Furthermore, for COI datasets, each record was assigned a Barcode Index Number (BIN)49, which was cross-checked on BOLD50 after taxonomic assignment to ensure unambiguous identifications and minimise misidentifications. Following taxonomic assignment, conservative criteria were implemented to prevent overestimations of abundance and ambiguous assignments. Specifically, for a species record to be validated, at least 5 reads for that specific species were required to be detected in the respective sample. This threshold value was determined for this study using the ratio described by Macé et al.14, which is based on the total read counts of true haplotypes relative to the total number of reads in certain samples. Additionally, to avoid false negatives, some records with less than 5 reads were retained under the following conditions: (a) if the species was identified by three different sets of primers within the same sample; (b) if the species was identified with more than 5 reads by another set of primers within the same sample; (c) if the species was identified morphologically in the same sample.
Decision tree for species inference
To gain further insights into the diversity that may not have been identified through morphology inspections, we devised a classification system inspired by Hoffman et al.51 that was tailored to our specific results. We hypothesised that several species-level identifications (IDs) made through DNA metabarcoding might have been hidden within higher taxonomic-level IDs made through morphological inspection. This classification system enabled us to associate discrepant IDs within a sample by assessing the agreement between both methodologies.
Five different categories were established to classify our records: (a) Match—when both methodologies identified a species in a sample; (b) DNA No-ID—when a specimen was identified to species-level solely by morphology and was not detected by metabarcoding; (c) Morph No-ID—when metabarcoding identified certain species while morphology did not. Morphological identifications at the Family/Genus level were reviewed, and if they were non-existent or could not be associated with the DNA ID, the record was included in this category; (d) Possible Match—if morphologic inspection identified organisms only to the Family/Genus level and DNA identified multiple species belonging to these taxonomic groups, the record was classified as a possible match. This category was used because we had several DNA matches to the species-level that could explain and resolve those morphological IDs, and (e) Reliable Match—a similar scenario to the previous classification, but this category was applied when the morphological identification at the Family/Genus level could only be matched to a single ID made by DNA metabarcoding. Our confidence in assuming those IDs are related was higher in this scenario (Fig. 2).

Decision tree for species inference. Each record was classified based on the identification (ID) agreement between morphology and DNA metabarcoding, allowing the association of higher taxonomic-level IDs obtained from morphology with species-level IDs derived from DNA metabarcoding.
Diversity analysis
The proportion of species with overlapping or exclusive detections by morphology versus metabarcoding-based identification, and the metabarcoding dataset among molecular markers and primer sets, was assessed using Venn Diagrams through the web-based tool InteractiVenn (https://interactivenn.net/)52 and edited using Inkscape 1.253. Jaccard dissimilarity index was computed between each sampled point, considering the primer set used, distance to the coast, and transect allocation using “vegdist” (vegan 2.6-4 R package)54. To investigate significant differences within these variables, we conducted a Permutational Multivariate Analysis of Variance (PERMANOVA) to assess dissimilarities in our fish taxa. The analysis was performed using the “adonis2” command from the aforementioned R package, employing 999 permutations by default. For visualisation, we conducted a principal coordinate analysis (PCoA) through Primer v6.1.1655 software to identify differences in species recovery among primer sets and potential clustering among points within the same transect or distance from the coast.
Ethical standards
This study was conducted in accordance with the ethical standards set by the Animal Welfare and Protection of the former General Direction of Veterinary (DGV) of the former Ministry of Agriculture, Rural Development and Fisheries (Lisbon, Portugal) for ecological research and collection of ichthyoplankton samples, complying with the European Union Directive 2010/63/EU and Portuguese Law (DL 113/2013). Necessary authorisations for sample collection were obtained from the Portuguese Institute for the Sea and the Atmosphere (IPMA). The methods reported align with the ARRIVE guidelines for ethical and laboratory quality standards, ensuring the integrity and reproducibility of the results.
Results
Morphology identifications
In total, 10,805 fish eggs and larvae were counted across the eight sampled locations with 7750 eggs and 3055 larvae observed. Among the sampled locations, F1 displayed the highest number of ichthyoplankton collected (2825), while F4 had the lowest number recorded (68) (Table 2). Egg identification was conducted only for Sardina pilchardus and Engraulis encrasicolus, accounting for 42.25% and 38.48% of the total eggs inspected, respectively. The remaining 1494 eggs were not identified due to the lack of distinctive morphological characteristics. Of the 3055 fish larvae counted, 43.37% were identified at the species level, 7.56% at the genus level, and 43.01% at the family level, with only 6.06% remaining unidentified. In total, 11 different species were identified, with Engraulis encrasicolus being the most common larval species, accounting for 38.66% of the total larvae observed, followed by larvae from the families Sparidae, Labridae, and Gobiidae.
DNA metabarcoding identifications
High-throughput sequencing generated a total of 2.4 million reads for the three markers and four primer sets used. Among these, 83% passed the quality control and filtration steps and were submitted to taxonomic assignment (Supplementary Table S1). After filtering, the average number of reads for each library was 63,107 ± 3536 (SEM).
Upon taxonomic assignment, the miFISH U-E primer set for 12S obtained the highest number of reads assigned to fish species, accounting for 46.35% of the total reads. This was closely followed by the Fish16S primer pair, which accounted for 31.57% of the reads. In the case of COI, while the mICOIintF/LoboR1 primer pair preserved a higher number of reads for taxonomic assignment post-processing, its effectiveness in retrieving fish reads was comparatively lower with 10.22% compared to 22.02%, achieved with the FishATL_Cocktail2 primer set.
In total, 75 fish species were identified by DNA metabarcoding when accounting for all primer sets (Fig. 3, Supplementary Tables S2–S5). The efficiency in recovering the desired taxonomic group varied among markers, with 16S exhibiting the highest number of identified species (60). This was followed by both COI primer sets, which identified 56 species using FishATL_Cocktail2 and 51 species with mICOIintF/LoboR1. The 12S primer combination identified a slightly lower number of fish species (48).

Partitioning of the total fish species identified by each primer pair/set. The numbers in parentheses indicate the total number of fish species identified by each primer combination.
All primer sets retrieved in common 29 fish species in our samples, representing 38.67% of the total species identified with DNA metabarcoding. Regarding shared identifications across the three molecular markers, 38 species (50.67%) were detected (Fig. 3). Among the primer pairs, the mICOIintF/LoboR1 demonstrated the highest capacity for detecting unique species (6), closely followed by miFISH U-E (5), while Fish16S identified 3 unique species, and FishATL_Cocktail2 detected 1 unique species.
Morphology vs DNA metabarcoding
Figure 4 illustrates a disparity in species-level identifications between morphology and DNA metabarcoding. The traditional method identified 11 species, while the molecular analysis with the 4 primer sets revealed 75 species. Only one species—Pomatoschistus pictus—was not identified by DNA metabarcoding. Ten species were identified simultaneously by both approaches: Arnoglossus laterna, Engraulis encrasicolus, Gadiculus argenteus, Mullus barbatus, Mullus surmuletus, Parablennius gattorugine, Parablennius pilicornis, Pomatoschistus microps, Sardina pilchardus and Trachurus trachurus.

Partitioning of the total fish species identified by both methodologies—morphology versus DNA metabarcoding. The numbers in parentheses indicate the total number of fish species identified by each approach.
We applied our decision tree classification to the records to understand the connection between species-level DNA identifications and higher taxonomic-level morphology identifications. Of the 235 species-level records made across our total samples, 38 exhibited agreement between both methodologies and were classified as Match. Ninety-four records showed varying degrees of association between DNA and the traditional method: 7 directly corresponded to a single species classified as Reliable Match, while 87 were linked to multiple species within the same genus or family, classified as Possible Match. Additionally, 96 DNA species records could not be correlated with morphological records categorised as Morph No-ID, and 7 morphological records could not be linked to DNA, categorised as DNA No-ID.
When considering the 76 fish species identified by both methodologies, it became apparent that at least 31 taxa identified at the species level by DNA metabarcoding were likely classified to a higher taxonomic level by morphology. These species were categorised in our classification tree as either Reliable or Possible Match.
Fish community structure
Analysing the outcomes from a community perspective, the 76 Teleostei fish species identified in this study were distributed across 20 orders, 33 families, and 60 genera (Supplementary Table S6). The most prominent orders were Eupercaria incertae sedis (27.63%), Gobiiformes (14.47%), and Perciformes (11.84%), with a substantial portion of the orders represented by a single species (9). Regarding fish families, Sparidae, with 15 species, stood out as the most represented (19.74%), followed by Gobiidae with 11 species (14.47%). The remaining families were represented by three or fewer species.
The inner shore points (F1 and A1) emerged as the locations where the highest number of species were identified by each primer set, except for the COI fish cocktail in the Arade River transect. In contrast, the offshore locations (F4 and A4) were characterised by lower diversity (Table 3). The same trend was observed in the total richness of fish species identified by all primer sets: F1 and A1 exhibited the highest number of different species identified (38 and 32), while F4 and A4 displayed lower richness in each transect (12 and 25) (Table 3). If the morphologic identifications are included in this analysis, the results remain very similar, with an additional species identified at four sampled locations (F2, A1, A2, and A3) (Table 3, Supplementary Fig. S1).
Both transects shared the presence of 31 fish species, with the Ria Formosa transect exhibiting 26 exclusive species and the Arade River transect 19 unique species (Supplementary Fig. S2). Within the Ria Formosa transect, three species were identified across all points. Excluding the offshore point (F4), 16 fish species were distributed among the area of the remaining points (F1, F2, and F3). In the case of the Arade River transect, 14 species were observed at all four locations. The endpoints, representing the inner shore and offshore locations (A1 and A4), exhibited the highest numbers of exclusive species, with 9 species limited to the inner shore and 5 to the offshore location.
The PERMANOVA analysis revealed significant differences in fish taxa composition based on the primer set used (pseudo-F = 3.8645, df = 3, p = 0.001), the distance to the coast of each sampling location (pseudo-F = 6.3268, df = 3, p = 0.001), and the respective transects (pseudo-F = 8.7065, df = 1, p = 0.001) (Supplementary Table S7). This statistical differentiation is visually depicted in the PCoA plot (Fig. 5), where distinct clusters emerge. A clear separation was found between transects—with fish compositions showing greater similarity within locations from the same transect compared to those from the other transect (filled versus normal symbols). Regarding the distance to the coast, a greater dissimilarity was found among points of the Ria Formosa transect than those of the Arade River transect. In particular, the Ria Formosa F4 offshore point stands out with a markedly distinct composition, forming an isolated group in the PCoA plot, for all primer sets used. Generally, within each transect, samples sequenced using the same primer set tended to cluster together, in particular for 12S (blue symbols) and some 16S samples (red symbols).

Principal Coordinates Analysis (PCoA) plot based on the Jaccard dissimilarity index. The plot displays the spatial distribution of each sequenced sample using different primer sets, highlighting the differences in community composition retrieved by each primer set. Each point is categorised by its distance to the coast and corresponding transect (see detailed legend in the figure). Closer points indicate more similar communities, while points further apart indicate greater dissimilarity. The dashed clusters were defined without statistical basis for the purpose of better interpreting the image.
Discussion
Our primary objective was to evaluate the efficacy of a multi-marker DNA metabarcoding protocol for identifying fish species in ichthyoplankton bulk samples, using morphology-based identifications as a comparison method. While this molecular technique is increasingly used for aquatic species assessments and monitoring3,9,10,22,56,57,58,59, there is a paucity of ichthyoplankton studies in the literature comparing multi-marker DNA metabarcoding with morphology-based identifications, particularly in the Northeast Atlantic. Notably, employing 3 markers and 4 target DNA regions enhanced the detection of fish species, identifying between 20 and 37.33% more species than using a single marker/amplicon.
Globally, our findings confirm that DNA metabarcoding is a valuable complement to traditional morphological inspection in ichthyoplankton studies. It demonstrates superior species-level discrimination and higher throughput, allowing for the recovery of a much higher spatial–temporal density of information on the occurrence of eggs and larvae for virtually all fish species present in a given area and time. In addition to these findings, our study provides updated records for historically documented fish species in this region, including both economically important and discarded species in commercial fisheries. It also highlights non-abundant taxa and potentially new species records for the region and country.
The DNA metabarcoding approach was able to detect a much larger number of fish species compared to morphology. This result was not unexpected, given that DNA metabarcoding sensitivity and discrimination power have been reported to outperform morphological approaches in numerous aquatic monitoring studies12,59,60,61. In the case of this study, we identified combined justifications for the very large difference observed (an extra 65 species through DNA metabarcoding). First, except for Sardina pilchardus and Engraulis encrasicolus eggs, the remaining eggs present in the samples could not be reliably identified through morphological methods. Considering the substantial number of remaining fish eggs counted in this study (Table 2), it is plausible that several species identified through molecular means were “hidden” within these unidentified organisms. It is also noteworthy that the multi-marker/primer approach employed in DNA metabarcoding further bolstered the total number of species retrieved. Second, the inherent challenge of identifying and distinguishing specimens in ichthyoplankton inspection also contributed to the disparity. The similarity of characteristics among these early-life individuals, combined with their variable state of preservation, imposed a more conservative approach to identification using morphology. Therefore, several identifications had to be assigned at ranks above the species (genus or family), rather than the species-level. These limitations were particularly evident in certain orders or families of fish. Morphological analyses were not able to recognise 11 orders including Acanthuriformes, Beloniformes, Callionymiformes, Carangaria incertae sedis, Lophiiformes, Mugiliformes, Myctophiformes, Ophidiiformes, Scombriformes, Stomiiformes, and Syngnathiformes, encompassing 15 distinct species, as revealed through metabarcoding. Moreover, although our taxonomists identified Eupercaria incertae sedis and Perciformes through morphology, these orders were not classified down to species level. In turn, metabarcoding detected 30 species within these two orders. This trend extends to the family level, with morphology being unable to identify 25 fish families recorded with DNA.
The creation of the decision tree depicted in Fig. 2 aimed to elucidate the discrepancy observed in fish species identified through the different approaches. Our objective was to explore whether we could establish connections between higher-level identifications made via morphology and the species-level identifications obtained through molecular assessment. For instance, within certain family and genus morphology-based identifications it was possible to distinguish multiple larvae belonging to different fish species. However, the absence of distinctive features hindered precise species-level identifications. We uncovered potential associations between metabarcoding and morphology across seven of the eight sampled locations by subjecting each identification obtained from both approaches to our decision tree. As previously mentioned, some orders and families contain species with identical morphological traits, particularly during early-life stages, posing challenges to taxonomists in achieving accurate identifications. This was particularly evident for the Eupercaria incertae sedis order, where no species-level identifications were achieved through morphological inspection. Nonetheless, upon comparison with metabarcoding results, we inferred that at least 18 species from the Sparidae and Labridae families likely correspond to these higher classifications across different samples. A similar pattern emerged for the Gobiiformes order, where 8 species were identified within the conservative identifications made via morphology. We inferred 31 species from 6 different orders that could be linked to higher-rank morphological identifications through the decision tree. However, as many as 14 orders detected by DNA metabarcoding remained unlinked with morphology. This could be due to morphologically unidentified eggs and some larvae, but the possible occurrence of some false positives cannot be discarded. That is if the DNA from fish species that were not truly present in the ichthyoplankton was somehow transported with eggs and larvae from other species. The likelihood of false positives arising from contamination in our laboratory procedures is exceedingly low, given that the negative controls showed no evidence of amplification. The only missed detection of Pomatoschistus pictus by DNA metabarcoding can be primarily attributed to the low number of larvae present in the samples (2 larvae at two points and 1 larva at one point), leading to insufficient biomass for successful DNA extraction and amplification for this species. Additionally, primer bias or mismatches during the sequencing process could have contributed to the non-detection by metabarcoding.
An interesting case arose in our study concerning species from the genus Arnoglossus. Our results revealed a discrepancy between the species identified through DNA metabarcoding and those identified via morphology in several samples. Morphological inspections consistently identified the species as Arnoglossus laterna in six samples, but in four of them, none of the primer sets detected this species, detecting instead A. imperialis and A. thori. The morphological identification keys we used for the Portuguese region only provided detailed information about A. laterna, potentially biasing the identification process. On the other hand, genetically these three species exhibit a significant distance from their sister species, making them more distinguishable at the molecular level. Furthermore, A. imperialis was exclusively identified in five locations using the 12S primer set, while A. thori was identified by the remaining primer sets. A review of each marker’s database revealed an underrepresentation of A. thori in the 12S database, with only one record present. This absence of information may have contributed to inaccurate taxonomic assignments. For instance, with limited information available, the discrimination power of the 12S marker for these species remains unassessed. Therefore, we cannot discard the possibility that the bioinformatic pipeline might have matched some sample reads to the closest species with sufficient representation in the database (in this case, A. imperialis) rather than the correct one (A. thori). Cross-verifying morphological and molecular species identifications can significantly enhance the accuracy of the identification process. The multi-marker approach (further explored in the subsequent section) is crucial as it provides “internal” molecular confirmation through redundant detections across various primers and markers. This approach also facilitates molecular-based identifications exclusively detected by a single primer set. Employing this methodology would enable a thorough review of specimens and facilitate subsequent identifications based on a more robust foundation, regardless of the similar morphological features exhibited by each individual.
The employment of multiple genetic markers in DNA metabarcoding has demonstrated considerable success in expanding the breadth of diversity captured, including in ichthyoplankton studies12,16,26. In our approach, we carefully selected a set of primer pairs from different genetic markers taking into consideration their documented efficacy in fish species identification22,36–39, while acknowledging their differences in taxonomic coverage of reference sequences, PCR amplification and species discrimination abilities16,20,62. The COI gene, characterised by its high interspecific variability63 and comprehensive database coverage for fish species of the northern Atlantic Ocean19,64, was included in our analysis. However, the use of COI primers of a broad taxonomic spectrum may favour non-specific amplification and primer bias21,65. To address these challenges we tested a primer cocktail for COI metabarcoding specifically designed for fish species—FishATL_Cocktail238—alongside a generalist primer pair that has exhibited successful results in amplifying DNA from zooplankton and benthic communities—mlCOIintF/LoboR160,66,67,68,69.
The FishATL_Cocktail2 primer set exhibited higher efficacy in species identification, detecting 56 species, compared to the mICOIintF/LoboR1 set, which identified 51 species. This slight difference can be attributed to the fish-tailored design of FishATL_Cocktail2, whereas mICOIintF/LoboR1 is a generalist primer pair. Furthermore, the extra length of the cocktail amplicon compared to mICOIintF/LoboR1 (~ 100 bp) increases the likelihood of including additional informative sites, thereby enhancing the discrimination and identification of more species70. Nevertheless, when considering the exclusive species detected, results suggest that mICOIintF/LoboR1 may complement the 12S and 16S primer pairs more effectively than the FishATL_Cocktail2.
In addition to the COI gene, we used primer pairs targeting the 12S gene (miFISH-U + E)39 and the 16S gene (Fish16S)22. Both primer sets have been extensively used in fish environmental DNA (eDNA) and DNA metabarcoding studies16,21,23,24. Like the COI markers, both deal with specific concerns such as the incompleteness of reference databases and the potential limitations in discriminating genetically close species due to conserved regions22,39,70. Among the 76 species identified in our study, we found 8 species missing in the 12S gene database (10.53%) and 6 missing species in the 16S gene database (7.90%), which could have been the primary reason for some of the failed detections with those primers. Despite these incomplete databases, and limited species discrimination in some genera, our results indicate the suitability of these primer sets for fish species identification in ichthyoplankton bulk samples. Notably, Fish16S displayed the highest diversity of fish species recovered (60), and 12 species (15.79%) were exclusively identified by either the 12S or the 16S gene. Specifically, these two genetic markers identified in common 4 species—Capros aper, Lesueurigobius suerii, Lophius budegassa, and Sarda sarda; 5 species were only identified by the 12S gene—Diplodus cervinus, Diplodus puntazzo, Diplodus sargus, Macroramphosus scolopax, and Nezumia sclerorhynchus; and 3 species only by the 16S gene—Cyclothone atraria, Cynoscion regalis, and Micromesistius poutassou. It is worth mentioning that most of these species are represented by a relatively low number of reads compared to other species in the respective sample, highlighting the detection power of these markers. These findings are similar to previous observations regarding the 12S gene performance, as described by Teixeira et al.16.
In the context of multi-marker/primer studies, our findings align with previous reports emphasising the enhanced diversity recovery achieved using multiple primer sets16,26,27,28. Conducting our study with only one primer set would have resulted in numerous species going undetected, thereby constituting false negatives in our samples. The most substantial deficit in species identification would have occurred with solely the 12S primer set, leading to the omission of 27 species, constituting a 36% loss relative to the total number of fish species identified. Following this, mICOIintF/LoboR1 missed 24 species, while the FishATL_Cocktail2 failed to identify 19 species. Interestingly, Fish16S exhibited the most favourable outcome, missing only 15 species (a 20% loss), six of which were absent from the corresponding reference library. These differences in the number of species identified by each marker/primer pair can be attributed to several factors. Firstly, the completeness of each reference database plays a crucial role19, as does the discriminatory power of each marker to differentiate closely related species24. Furthermore, each molecular marker exhibits distinct sensitivity during the sequencing process, potentially amplifying information related to non-target organisms. This phenomenon has been reported particularly for some COI primer sets in other studies, which may amplify mostly non-metazoan DNA21. However, that occurs mainly with eDNA studies where bacteria retained in the filter may be co-amplified with metazoan DNA21. In the case of the present study, the non-metazoa amplification was residual, and most of the non-target amplification was due to other metazoans present in the sample, namely marine invertebrates. Such variability may introduce a level of bias that can significantly influence the final results, including the number of fish species recovered in our study. Therefore, the utilization of multiple primer sets not only enhances species detection but also helps mitigate the impact of such biases, ultimately leading to a more comprehensive and accurate assessment of biodiversity.
These findings are significant for future research, as they indicate the potential for using a combination of primer sets to maximise species detection while minimising redundancy. By employing a multi-primer approach, the ability to uncover a broader diversity of species is enhanced, providing valuable insights into optimising primer selection for such endeavours. Additionally, it is important to emphasise that internal cross-validation within metabarcoding that was previously mentioned, which involves the simultaneous detection of multiple species using different markers (38 fish species representing 50.67% of the total fish diversity identified in this study by DNA metabarcoding, shown in Fig. 3), significantly bolsters confidence in our molecular results. This is particularly crucial when existing databases are incomplete or prone to errors19. Moreover, employing a higher-resolution marker can assist in refining species identities inferred from lower-resolution markers—for instance, a genus-level identification obtained in a sample for one genetic marker, can be upgraded to a species-level identification if another marker as the discriminatory power to differentiate species within that specific genus. Based on this study, for fish and ichthyoplankton-specific research, we recommend using the FishATL_Cocktail2 combined with the 12S and 16S primer sets to obtain a comprehensive profile of the ichthyofauna. Conversely, if the study aims to gather additional information about the zooplankton community, employing the mICOIintF/LoboR1 primer set would yield better results. If only two primer sets are to be utilized, we recommend selecting one COI primer set along with either the 12S primer set, which is extensively used in the literature, or the 16S primer set, which demonstrated excellent results in this study and appears to be underutilized for fish metabarcoding despite its significant potential.
Regarding species richness at each sampling location, there was a tendency for a gradual increase in the number of fish species detected as the distance to the coast decreased for each transect (Supplementary Fig. S1). This trend was also evident when combining the fish species identified across both transects, with 49 inner shore identifications compared to 34 in offshore areas. This is commonly observed in marine ecosystems due to a multitude of ecological factors that can influence the distribution and abundance of different fish species, as previously highlighted for Portuguese estuaries and adjacent areas by Nicolas et al.71. Inner and middle shore areas are characterised by a multiplicity of habitats, sustained by ample food resources and critical breeding grounds that are vital for the survival of various fish species' life stages. In particular, on the Portuguese coast, this is enhanced by the seasonal events of upwelling that generate highly productive zones extremely suitable for fish72. Our results also revealed a distinct fish community at F4 (offshore point) compared to the other points within the same transect (Fig. 5). This discrepancy may be attributed to the more homogeneous environment subject to the stressful hydrodynamic conditions that typify these areas73. Moreover, considering that our sampled points encompassed different depths, it is important to note that some fish communities, particularly benthonic species, and different life stages associated with greater depths, may be underrepresented. This is because our sampling focused primarily on the water column with depths ranging from 10 to 20 m.
In terms of fish distribution, two species were found in all sampled locations—Arnoglossus thori and Pagellus acarne—being the majority of the taxa present in three or more points. Additionally, 29 species were exclusively identified in a single location, while 14 species were found in two locations. It should be noted that 20 species exhibited a broad distribution, being identified from the nearest points to the coast (inner shore) to the most distant points (offshore). Interestingly, these depth categories had the highest number of unique species (10 and 9, respectively), revealing a community composition specific to each area, likely associated with their environmental characteristics.
Three species detected in our study—Chromogobius zebratus, Cyclothone atraria, and Vanneaugobius dollfusi—are not listed in the recent compilation of Portuguese ichthyofauna by Carneiro et al.74. Notably, all these species belong to genera that have reported members in Portugal. Although this may indicate some of the first documented occurrences of these species in the area, conclusions must be drawn cautiously. Chromogobius zebratus and V. dollfusi are known to inhabit the Mediterranean Sea, making plausible an introduction or migration from those waters. However, the case of C. atraria is more debatable, given that its known records are associated with the North Pacific Ocean. Furthermore, the species recorded in Portugal for these genera are underrepresented in the databases for the different markers, as highlighted before in the Arnoglossus case. While this study employed conservative criteria to minimise false positives, it is crucial to note that C. atraria was identified with just 5 reads in F4 using Fish16S, and V. dollfusi was detected with fewer than 15 reads in two locations (A2 and A3), by FishATL_Cocktail2. In contrast, C. zebratus presented a different scenario, being identified in three points (F1, F2, and A1) by multiple markers (COI and 16S), and even emerging as one of the top records in A1.
This brings to consideration the importance of establishing a suitable threshold to confirm the presence of a taxon in a study, a challenge for which there is no consensus in the field, as noted by Breitbart et al.75. In our study, we adopted a ratio proposed by Macé et al.14 that aligned well with our research objectives. However, it became evident that adjusting the minimum number of reads required to validate a species record, whether increased or decreased, could lead to an increase in false negatives or false positives, respectively. Considering this information, future investigations in the sampled region will contribute to clarifying the status of these possible new records and exploring the potential ecological implications of these species in the area.
Our findings underscored the superior efficacy of DNA metabarcoding in identifying species from ichthyoplankton samples, compared to traditional morphological methods. By linking DNA species-level identifications with higher taxonomic-level morphological identifications, DNA metabarcoding provided potential solutions for some genera and family identifications that traditional methods struggled with. Furthermore, our research reaffirmed the importance of using multiple molecular markers and respective amplicons in metabarcoding studies. This comprehensive approach revealed a greater extent of diversity than using a single marker/primer pair, leading to more robust analyses and greater certainty in the results. In conclusion, our results align with our objectives, demonstrating that DNA metabarcoding is a powerful tool for uncovering fish diversity in ichthyoplankton samples. This technology has immense potential for supporting future fish stock assessments and aiding in the management of marine ecosystems.
Data availability
The data supporting the findings of this study are available within the main body of the manuscript and the supplementary material.
References
Botsford, L. W. et al. Connectivity, sustainability, and yield: Bridging the gap between conventional fisheries management and marine protected areas. Rev. Fish Biol. Fish. 19, 69–95 (2009).
Santos, A. M. P. et al. Sardine (Sardina pilchardus) larval dispersal in the Iberian upwelling system, using coupled biophysical techniques. Prog. Oceanogr. 162, 83–97 (2018).
Nobile, A. B. et al. DNA metabarcoding of Neotropical ichthyoplankton: Enabling high accuracy with lower cost. Metabarcoding Metagenom. 3, 69–76 (2019).
Ko, H. L. et al. Evaluating the accuracy of morphological identification of larval fishes by applying DNA barcoding. PLoS One 8, e53451 (2013).
Taberlet, P., Coissac, E., Hajibabaei, M. & Rieseberg, L. H. Environmental DNA. Mol. Ecol. 21, 1789–1793 (2012).
Grant, D. M. et al. The future of DNA barcoding: Reflections from early career researchers. Diversity 13, 313 (2021).
Yu, D. W. et al. Biodiversity soup: Metabarcoding of arthropods for rapid biodiversity assessment and biomonitoring. Methods Ecol. Evol. 3, 613–623 (2012).
Kimmerling, N. et al. Quantitative species-level ecology of reef fish larvae via metabarcoding. Nat. Ecol. Evol. 2, 306–316 (2018).
Mariac, C. et al. Metabarcoding by capture using a single COI probe (MCSP) to identify and quantify fish species in ichthyoplankton swarms. PLoS One 13, 1–15 (2018).
Maggia, M. E. et al. DNA metabarcoding of Amazonian ichthyoplankton swarms. PLoS One 12, 1–14 (2017).
Ratcliffe, F. C. et al. Quantitative assessment of fish larvae community composition in spawning areas using metabarcoding of bulk samples. Ecol. Appl. 31, 1–10 (2021).
Jiang, R., Lusana, J. L. & Chen, Y. High-throughput DNA metabarcoding as an approach for ichthyoplankton survey in Oujiang River Estuary, China. Diversity 14, 1–12 (2022).
Lamia, D. et al. Comparison and validation of ichthyoplankton DNA extraction methods. Methods Protoc. 4, 1–8 (2021).
Macé, B. et al. Evaluating bioinformatics pipelines for population-level inference using environmental DNA. Environ. DNA 4, 674–686 (2022).
Polanco, F. A. et al. Comparing the performance of 12S mitochondrial primers for fish environmental DNA across ecosystems. Environ. DNA 3, 1113–1127 (2021).
Teixeira, D. F., Hilário, H. O., Santos, G. B. & Carvalho, D. C. DNA metabarcoding assessment of Neotropical ichthyoplankton communities is marker-dependent. Ecol. Evol. 13, 1–9 (2023).
Ward, R. D. DNA barcode divergence among species and genera of birds and fishes. Mol. Ecol. Resour. 9, 1077–1085 (2009).
Costa, F. O. et al. A ranking system for reference libraries of DNA barcodes: Application to marine fish species from Portugal. PLoS One 7, 1–9 (2012).
Weigand, H. et al. DNA barcode reference libraries for the monitoring of aquatic biota in Europe: Gap-analysis and recommendations for future work. Sci. Total Environ. 678, 499–524 (2019).
Deagle, B. E., Jarman, S. N., Coissac, E., Pompanon, F. & Taberlet, P. DNA metabarcoding and the cytochrome c oxidase subunit I marker: Not a perfect match. Biol. Lett. 10, 2–5 (2014).
Collins, R. A. et al. Non-specific amplification compromises environmental DNA metabarcoding with COI. Methods Ecol. Evol. 10, 1985–2001 (2019).
Berry, T. E. et al. DNA metabarcoding for diet analysis and biodiversity: A case study using the endangered Australian sea lion (Neophoca cinerea). Ecol. Evol. 7, 5435–5453 (2017).
Miya, M., Gotoh, R. O. & Sado, T. MiFish metabarcoding: A high-throughput approach for simultaneous detection of multiple fish species from environmental DNA and other samples. Fish. Sci. 86, 939–970 (2020).
Zhang, S., Zhao, J. & Yao, M. A comprehensive and comparative evaluation of primers for metabarcoding eDNA from fish. Methods Ecol. Evol. 11, 1609–1625 (2020).
Shaw, J. L. A. et al. Comparison of environmental DNA metabarcoding and conventional fish survey methods in a river system. Biol. Conserv. 197, 131–138 (2016).
Duke, E. M. & Burton, R. S. Efficacy of metabarcoding for identification of fish eggs evaluated with mock communities. Ecol. Evol. 10, 3463–3476 (2020).
Stefanni, S. et al. Multi-marker metabarcoding approach to study mesozooplankton at basin scale. Sci. Rep. 8, 1–13 (2018).
Zhang, G. K., Chain, F. J. J., Abbott, C. L. & Cristescu, M. E. Metabarcoding using multiplexed markers increases species detection in complex zooplankton communities. Evol. Appl. 11, 1901–1914 (2018).
Carvalho, D. C. Ichthyoplankton DNA metabarcoding: Challenges and perspectives. Mol. Ecol. 31, 1612–1614 (2022).
Ré, P. Ictioplâncton Estuarino Da Península Ibérica-Guia de Identificação Dos Ovos e Estados Larvares Planctónicos. Câmara Municipal de Cascais (1999).
Schlitzer, R. Ocean Data View, version 5.6.5. https://odv.awi.de/ (2022).
Ré, P. & Meneses, I. Early Stages of Marine Fishes Occurring in the Iberian Peninsula. INIAP/IPIMAR-IMAR (2008).
Rodriguez, J. M., Alemany, F. & Garcia, A. A Guide to the Eggs and Larvae of 100 Common Western Mediterranean Sea Bony Fish Species. Food and Agricultural Organization of the United Nations (2017).
Steinke, D. et al. Message in a Bottle—Metabarcoding enables biodiversity comparisons across ecoregions. Gigascience 11, 1–11 (2022).
Ivanova, N. V., Dewaard, J. R. & Hebert, P. D. N. An inexpensive, automation-friendly protocol for recovering high-quality DNA. Mol. Ecol. Notes 6, 998–1002 (2006).
Leray, M. et al. A new versatile primer set targeting a short fragment of the mitochondrial COI region for metabarcoding metazoan diversity: Application for characterizing coral reef fish gut contents. Front. Zool. 10, 1–14 (2013).
Lobo, J. et al. Enhanced primers for amplification of DNA barcodes from a broad range of marine metazoans. BMC Ecol. 13, 1 (2013).
Costa, G. O. da. Aprimoramento do protocolo de DNA metabarcoding para aplicação na identificação de áreas prioritárias à conservação da ictiofauna do rio Mogi-Guaçu. (Instituto de Biociências de Botucatu, Universidade Estadual Paulista “Júlio de Mesquita Filho”, 2021).
Miya, M. et al. MiFish, a set of universal PCR primers for metabarcoding environmental DNA from fishes: Detection of more than 230 subtropical marine species. R. Soc. Open Sci. 2, 150088 (2015).
Illumina. 16S Metagenomic Sequencing Library Preparation Preparing 16S Ribosomal RNA Gene Amplicons for the Illumina MiSeq System. Illumina Tech. Doc. (Part. No. 15044223 Rev. B.) (2013).
Comeau, A. M., Douglas, G. M. & Langille, M. G. I. Microbiome helper: A Custom and streamlined workflow for microbiome research. mSystems 2, e00127–16 (2017).
Ward, R. D., Zemlak, T. S., Innes, B. H., Last, P. R. & Hebert, P. D. N. DNA barcoding Australia’s fish species. Philos. Trans. R. Soc. B Biol. Sci. 360, 1847–1857 (2005).
Jennings, W. B. et al. Barcoding the Neotropical freshwater fish fauna using a new pair of universal COI primers with a discussion of primer dimers and M13 primer tails. Genome 62, 77–83 (2019).
Deagle, B. E. et al. Studying seabird diet through genetic analysis of faeces: A case study on macaroni penguins (Eudyptes chrysolophus). PLoS One 2, e831 (2007).
Edgar, R. C. Search and clustering orders of magnitude faster than BLAST. Bioinformatics 26, 2460–2461 (2010).
Fontes, J. T., Vieira, P. E., Ekrem, T., Soares, P. & Costa, F. O. BAGS: An automated Barcode, Audit & Grade System for DNA barcode reference libraries. Mol. Ecol. Resour. 21, 573–583 (2021).
Sayers, E. W. et al. GenBank. Nucleic Acids Res. 47, D23–D28 (2019).
WoRMS Editorial Board. World Register of Marine Species. https://www.marinespecies.org (2024) https://doi.org/10.14284/170.
Ratnasingham, S. & Hebert, P. D. N. A DNA-based registry for all animal species: The Barcode Index Number (BIN) System. PLoS One 8, e66213 (2013).
Ratnasingham, S. & Hebert, P. D. N. BOLD: The barcode of life data system: Barcoding. Mol. Ecol. Notes 7, 355–364 (2007).
Hoffman, J. C. et al. Comparison of larval fish detections using morphology-based taxonomy versus high-throughput sequencing for invasive species early detection. Can. J. Fish. Aquat. Sci. 78, 752–764 (2021).
Heberle, H., Meirelles, V. G., da Silva, F. R., Telles, G. P. & Minghim, R. InteractiVenn: A web-based tool for the analysis of sets through Venn diagrams. BMC Bioinform. 16, 1–7 (2015).
Inkscape Project. Inkscape. https://inkscape.org (2020).
Oksanen, J. et al. Vegan: Community ecology package. R package version 2.6-4. Community Ecol. Packag. 2.6-4, 1–297 (2022).
Clarke, K. R. & Gorley, R. N. PRIMER v6: Primer V6: User Manual/Tutorial at (2006).
Fais, M. et al. Meiofauna metabarcoding in Lima estuary (Portugal) suggests high taxon replacement within a background of network stability. Reg. Stud. Mar. Sci. 38, 101341 (2020).
Garcia-Vazquez, E., Georges, O., Fernandez, S. & Ardura, A. eDNA metabarcoding of small plankton samples to detect fish larvae and their preys from Atlantic and Pacific waters. Sci. Rep. 11, 1–14 (2021).
Leite, B. R., Vieira, P. E., Troncoso, J. S. & Costa, F. O. Comparing species detection success between molecular markers in DNA metabarcoding of coastal macroinvertebrates. Metabarcoding Metagenomics 5, 249–260 (2021).
Lobo, J., Shokralla, S., Costa, M. H., Hajibabaei, M. & Costa, F. O. DNA metabarcoding for high-throughput monitoring of estuarine macrobenthic communities. Sci. Rep. 7, 1–13 (2017).
Duarte, S. et al. Macrozoobenthos monitoring in Portuguese transitional waters in the scope of the water framework directive using morphology and DNA metabarcoding. Estuar. Coast. Shelf Sci. 281, 108207 (2023).
Stepien, C. A., Schultz, H. K., McAllister, S. M., Norton, E. L. & Keister, J. E. Evaluating metabarcoding markers for identifying zooplankton and ichthyoplankton communities to species in the Salish Sea: Morphological comparisons and rare, threatened or invasive species. DNA 4, 1–33 (2023).
Claver, C., Canals, O., de Amézaga, L. G., Mendibil, I. & Rodriguez-Ezpeleta, N. An automated workflow to assess completeness and curate GenBank for environmental DNA metabarcoding: The marine fish assemblage as case study. Environ. DNA 5, 634–647 (2023).
Hebert, P. D. N., Cywinska, A., Ball, S. L. & DeWaard, J. R. Biological identifications through DNA barcodes. Proc. R. Soc. B Biol. Sci. 270, 313–321 (2003).
Oliveira, L. M. et al. Assembling and auditing a comprehensive DNA barcode reference library for European marine fishes. J. Fish Biol. 89, 2741–2754 (2016).
Elbrecht, V. & Leese, F. Can DNA-based ecosystem assessments quantify species abundance? Testing primer bias and biomass-sequence relationships with an innovative metabarcoding protocol. PLoS One 10, 1–16 (2015).
Leite, B. R. et al. Evaluation of DNA metabarcoding for identifying fish eggs: A case study on the West Florida Shelf. Front. Mar. Sci. 5, e76 (2021).
Lavrador, A. S., Fontes, J. T., Vieira, P. E., Costa, F. O. & Duarte, S. Compilation, revision, and annotation of DNA barcodes of marine invertebrate non-indigenous species (NIS) occurring in European Coastal Regions. Diversity 15, 174 (2023).
Moutinho, J., Carreira-Flores, D., Gomes, P. T., Costa, F. O. & Duarte, S. Assessing the seasonal and spatial dynamics of zooplankton through DNA metabarcoding in a temperate estuary. Animals 13, 1–24 (2023).
Leite, B. R., Duarte, S., Troncoso, J. S. & Costa, F. O. Artificial seaweed substrates complement ARMS in DNA metabarcoding-based monitoring of temperate coastal macrozoobenthos. Diversity 15, 657 (2023).
Fontes, J. T., Katoh, K., Pires, R., Soares, P. & Costa, F. O. Benchmarking the discrimination power of commonly used markers and amplicons in marine fish (e)DNA. ARPHA Prepr. https://doi.org/10.3897/arphapreprints.e128777 (2024).
Nicolas, D. et al. Fish under influence: A macroecological analysis of relations between fish species richness and environmental gradients among European tidal estuaries. Estuar. Coast. Shelf Sci. 86, 137–147 (2010).
Santos, A. M. P., Kazmin, A. S. & Peliz, Á. Decadal changes in the Canary upwelling system as revealed by satellite observations: Their impact on productivity. J. Mar. Res. 63, 359–379 (2005).
Martins, R., Quintino, V. & Rodrigues, A. M. Diversity and spatial distribution patterns of the soft-bottom macrofauna communities on the Portuguese continental shelf. J. Sea Res. 83, 173–186 (2013).
Carneiro, M., Martins, R., Reiner, F. & Batista, I. Ichthyofauna of Portugal: Taxonomic Diversity, Common and Scientific Names of Marine Fishes - Bibliography and Electronic References. IPMA, I.P. vol. 2 (2019).
Breitbart, M. et al. Evaluation of DNA metabarcoding for identifying fish eggs: a case study on the West Florida Shelf. PeerJ 11, e15016 (2023).
Acknowledgements
We are grateful to the Mário Ruivo crew for their help in the collection of the samples during the field campaign.
Funding
This research was funded by the project “A-Fish-DNA-Scan: Cutting-edge DNA-based approaches for improved monitoring and management of fisheries resources along Magellan-Elcano´s Atlantic route” (CIRCNA/BRB/0156/2019; https://doi.org/https://doi.org/10.54499/CIRCNA/BRB/0156/2019) financed by the Foundation for Science and Technology (FCT, I.P.), by the CBMA “Contrato-Programa” (UIDB/04050/2020; https://doi.org/https://doi.org/10.54499/UIDB/04050/2020) and by the ARNET “Contrato-Programa” (LA/P/0069/2020; https://doi.org/https://doi.org/10.54499/LA/P/0069/2020) funded by national funds through FCT, I.P. A.O.F. (DFA/BD/6653/2020; https://doi.org/https://doi.org/10.54499/2020.06653.BD) and S.D. (https://doi.org/https://doi.org/10.54499/CEECIND/00667/2017/CP1458/CT0001) were financially supported by FCT I.P. J.T.F. (UI/BD/150910/2021; https://doi.org/https://doi.org/10.54499/UI/BD/150910/2021) was supported by the Collaboration Protocol for Financing the Multiannual Research Grants Plan for Doctoral Students with financial support from FCT I.P. and the European Social Fund under the Northern Regional Operational Program—Norte2020. This work was also supported by the European Regional Development Fund (ERDF) through the Operational Programme for Competitiveness and Internationalisation—COMPETE 2020 and Portuguese national funds by FCT, I.P. under the strategic project UIDB/04539/2020, UIDP/04539/2020 and LA/P/0058/2020. It also contributes to the Strategic Research Plan of CCMAR under the contracts UIDB/04326/2020, UIDP/04326/2020 and LA/P/0101/2020 funded by FCT. I.P.
Author information
Authors and Affiliations
Contributions
A.O.F., S.D., A.M.P.S. and F.O.C. conception and design; A.O.F and A.M.P.S. sampling; O.M.A. and P.R. morphological identification; A.O.F. filtration and DNA extraction; C.E and C.B. amplification and sequencing; J.T.F. databases compilation; A.O.F bioinformatic and statistical analysis; A.O.F, S.D. and F.O.C. writing the article. All authors revised the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Ferreira, A.O., Azevedo, O.M., Barroso, C. et al. Multi-marker DNA metabarcoding for precise species identification in ichthyoplankton samples. Sci Rep 14, 19772 (2024). https://doi.org/10.1038/s41598-024-69963-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-024-69963-7
- Springer Nature Limited