
Abstract
The calcium-binding protein S100A9 has emerged as a pivotal biomolecular actor in oncology, implicated in numerous malignancies. This comprehensive bioinformatics study transcends traditional boundaries, investigating the prognostic and therapeutic potential of S100A9 across diverse neoplastic entities. Leveraging a wide array of bioinformatics tools and publicly available cancer genomics databases, such as TCGA, we systematically examined the S100A9 gene. Our approach included differential expression analysis, mutational burden assessment, protein interaction networks, and survival analysis. This robust computational framework provided a high-resolution view of S100A9’s role in cancer biology. The study meticulously explored S100A9’s oncogenic facets, incorporating comprehensive analyses of its relationship with prognosis, tumor mutational burden (TMB), microsatellite instability (MSI), DNA methylation, and immune cell infiltration across various tumor types. This study presents a panoramic view of S100A9 expression across a spectrum of human cancers, revealing a heterogeneous expression landscape. Elevated S100A9 expression was detected in malignancies such as BLCA (Bladder Urothelial Carcinoma), CESC (Cervical squamous cell carcinoma and endocervical adenocarcinoma), COAD (Colon adenocarcinoma), ESCA (Esophageal carcinoma), and GBM (Glioblastoma multiforme), while reduced expression was noted in BRCA (Breast invasive carcinoma), HNSC (Head and Neck squamous cell carcinoma), and KICH (Kidney Chromophobe). This disparate expression pattern suggests that S100A9’s role in cancer biology is multifaceted and context-dependent. Prognostically, S100A9 expression correlates variably with patient outcomes across different cancer types. Furthermore, its expression is intricately associated with TMB and MSI in nine cancer types. Detailed examination of six selected tumors—BRCA, CESC, KIRC (Kidney renal clear cell carcinoma), LUSC (Lung squamous cell carcinoma), SKCM (Skin Cutaneous Melanoma); STAD (Stomach adenocarcinoma)—revealed a negative correlation of S100A9 expression with the infiltration of most immune cells, but a positive correlation with neutrophils, M1 macrophages, and activated NK cells, highlighting the complex interplay between S100A9 and the tumor immune environment. This bioinformatics synthesis posits S100A9 as a significant player in cancer progression, offering valuable prognostic insights. The data underscore the utility of S100A9 as a prognostic biomarker and its potential as a therapeutic target. The therapeutic implications are profound, suggesting that modulation of S100A9 activity could significantly impact cancer management strategies.
Similar content being viewed by others


Introduction
The relentless onslaught of cancer continues to undermine human health, precipitating an acute degradation in the quality of life for affected individuals. Among contemporary therapeutic strategies, cancer immunotherapy—most notably, immune checkpoint blockade—has risen to prominence, revolutionizing the treatment paradigm for numerous malignancies1. This urgency is driven by the need to improve patient outcomes and extend survival rates, given the limited efficacy and significant side effects of traditional cancer treatments. Cancer immunotherapy, particularly immune checkpoint blockade, has transformed the therapeutic landscape by leveraging the body's immune system to combat cancer cells. The burgeoning field of cancer immunotherapy underscores the necessity for continuous exploration and validation of new targets. High-throughput screening techniques, combined with advanced bioinformatics tools, are instrumental in identifying and characterizing these targets2. The advent and maturation of public repositories such as The Cancer Genome Atlas (TCGA) and the Gene Expression Omnibus (GEO) have catalyzed a renaissance in the discovery of novel immunotherapeutic targets via pan-cancer gene expression analyses. By interrogating the nexus of gene targets, clinical prognostication, and signal transduction pathways, a rich tapestry of insights is being revealed. These databases provide extensive information on gene expression, genetic mutations, and epigenetic modifications, facilitating the identification of novel targets that could be exploited for cancer treatment. By linking these molecular targets to clinical outcomes, researchers can develop more precise prognostic models and tailor therapies to individual patients' genetic profiles.
Within the expansive landscape of calcium and zinc-binding proteins, the S100 protein family stands out, represented in vertebrates exclusively. S100A9, a protein of relatively low molecular weight within this family, binds calcium with specificity. However, despite a breadth of studies scrutinizing its role across a diversity of neoplasms, the precise mechanistic contributions of S100A9 to oncogenesis remain enigmatic and subject to debate. The S100A9 gene, a member of the S100 family of proteins, encodes a calcium-binding protein involved in the regulation of a variety of cellular processes, including cell cycle progression and differentiation. S100A9, often found in a complex with S100A8 (calprotectin), plays a crucial role in the inflammatory response, immune regulation, and leukocyte trafficking3. A growing corpus of evidence indicates that S100A9 may play a facilitative role in the cascade of tumor metastasis4. Observational studies have detected heightened S100A9 expression levels in the context of metastatic melanoma and prostate cancer5, positing a correlation with the mechanistic intricacies of carcinogenesis and the attenuation of cellular differentiation6. Recent studies have highlighted the significant role of S100A9 in tumor biology. S100A9 is frequently overexpressed in various malignancies, including breast, prostate, colorectal, and lung cancers. This overexpression is associated with poor prognosis and increased tumor aggressiveness7. The protein contributes to the tumor microenvironment by promoting chronic inflammation, which supports tumor growth and metastasis8. Mechanistically, S100A9 facilitates tumor progression through several pathways. It can interact with the receptor for advanced glycation end products and Toll-like receptor 4, activating downstream signaling cascades such as NF-κB and MAPK9. These pathways lead to the secretion of pro-inflammatory cytokines and chemokines, enhancing the inflammatory milieu and promoting angiogenesis, tumor cell proliferation, and invasion. Moreover, S100A9 has been implicated in the modulation of the immune response within the tumor microenvironment10. It can recruit myeloid-derived suppressor cells and regulatory T cells, which suppress anti-tumor immunity and facilitate immune evasion by cancer cells. This immunosuppressive environment enables tumor cells to thrive and evade detection by the immune system11. Given its multifaceted role in cancer progression, S100A9 is emerging as a potential biomarker for cancer diagnosis and prognosis. Targeting S100A9 and its signaling pathways holds promise for developing novel therapeutic strategies aimed at modulating the tumor microenvironment, enhancing anti-tumor immunity, and improving clinical outcomes for cancer patients.
Amid the landscape of oncologic intervention, where chemoradiotherapy, targeted therapy, and immunotherapy constitute the modern therapeutic armamentarium, there exists a disquieting realization: survival outcomes for an array of clandestine or recalcitrant malignancies languish as suboptimal12. This underscores an imperative for deeper mechanistic dissection of tumorigenesis to propel advances in early detection and intervention. The tumor microenvironment (TME) is a complex and dynamic milieu that surrounds and interacts with cancer cells, significantly influencing tumor development, progression, and response to therapy13. This intricate environment is composed of various cellular and non-cellular components, including immune cells, fibroblasts, endothelial cells, extracellular matrix, cytokines, and chemokines. A critical feature of the TME is its heterogeneity, reflecting the diverse cell types and the spatial and temporal variations within the tumor14. Immune cells within the TME, such as tumor-associated macrophages, myeloid-derived suppressor cells, and regulatory T cells, often adopt immunosuppressive phenotypes that facilitate immune evasion by cancer cells. These cells can secrete immunosuppressive cytokines like IL-10 and TGF-β, which inhibit the activity of cytotoxic T lymphocytes and natural killer cells, undermining the body's anti-tumor immune response15. Fibroblasts, particularly cancer-associated fibroblasts, are another vital component of the TME. They contribute to tumor progression by remodeling the ECM, promoting angiogenesis, and secreting growth factors and cytokines that support tumor cell proliferation and invasion16. The ECM itself, through its biochemical and mechanical properties, provides structural support and signaling cues that regulate tumor cell behavior. The TME is also characterized by an abnormal vascular network, resulting in hypoxia and nutrient deprivation within the tumor17. Hypoxia-inducible factors are stabilized under low oxygen conditions, driving the expression of genes that promote angiogenesis, metabolic adaptation, and survival of cancer cells under stress. The hypoxic environment further contributes to the immunosuppressive nature of the TME by attracting immunosuppressive cells and altering their function18. The strategic delineation of immunophenotypes and the validation of emerging immune-centric targets remain critical for the advancement of tumor therapeutics. Despite this, the role of S100A9 across the cancer spectrum is inadequately understood. Addressing this knowledge gap, our investigation rigorously examines S100A9 expression across thirty-three disparate malignancies, seeking to elucidate its influence within the immune-oriented TME. By placing an analytical lens over pivotal immunomodulators and evolving immune biomarkers, we aim to deepen the understanding of the complex and dynamic tumor-immune crosstalk, as presented in Fig. 1 of our study.

Framework. In this study, we extracted single gene expression data from TCGA database and performed differential analysis of S100A9. The differential expression data sets were obtained for clinical correlation analysis, survival prognosis analysis and immune correlation analysis. In addition, we also used the GEO dataset for validation and gene mutation analysis.
Materials and methods
Data acquisition
Data processing
Genomic alterations, including copy number amplification, severe loss, unknown missense mutations, and mRNA overexpression, were identified through analysis of the S100A9 gene using the cBioPortal. The TCGA dataset provided information on the differential expression of S100A9 between tumor and matched normal tissues. We obtained gene expression patterns and clinical information from The Cancer Genome Atlas (TCGA)19 for 33 cancers. ACC (Adrenocortical carcinoma); BLCA (Bladder Urothelial Carcinoma); BRCA (Breast invasive carcinoma); CESC (Cervical squamous cell carcinoma and endocervical adenocarcinoma); CHOL (Cholangiocarcinoma); COAD (Colon adenocarcinoma); DLBC (Lymphoid Neoplasm Diffuse Large B-cell Lymphoma); ESCA (Esophageal carcinoma); GBM (Glioblastoma multiforme); HNSC (Head and Neck squamous cell carcinoma); KICH (Kidney Chromophobe); KIRC (Kidney renal clear cell carcinoma); KIRP (Kidney renal papillary cell carcinoma); LAML (Acute Myeloid Leukemia); LGG (Brain Lower Grade Glioma); LIHC (Liver hepatocellular carcinoma); LUAD (Lung adenocarcinoma); LUSC (Lung squamous cell carcinoma); MESO (Mesothelioma); OV (Ovarian serous cystadenocarcinoma); PAAD (Pancreatic adenocarcinoma); PCPG (Pheochromocytoma and Paraganglioma); PRAD (Prostate adenocarcinoma); READ (Rectum adenocarcinoma); SARC (Sarcoma); SKCM (Skin Cutaneous Melanoma); STAD (Stomach adenocarcinoma); TGCT (Testicular Germ Cell Tumors); THCA (Thyroid carcinoma); THYM (Thymoma); UCEC (Uterine Corpus Endometrial Carcinoma); UCS (Uterine Carcinosarcoma); UVM (Uveal Melanoma) were included in 33 types. The S100A9 status change was discovered using the cBioPortal database20. The genomic changes include copy number amplification, severe loss, an unknown missense mutation, and mRNA overexpression. The TCGA provides data on S100A9 expression differences between tumor and matched normal tissue. After extracting the S100A9 data with the limma package, we used log2 (TPM + 1) transformed expression data to illustrate the difference analysis findings in parameter selection.
Statistical analysis
In an endeavor to elucidate the expression dynamics of S100A9 across a spectrum of tissue types and pathological states, we embarked on an exhaustive investigation encompassing 31 normal tissues and 33 discrete cancer specimens. The expression data underwent a meticulous log2 transformation, subsequently facilitating the implementation of a two-tiered comparative analysis designed to scrupulously assess the differential expression landscapes between tumor and normal tissues within each designated cancer typology. Differential expression analysis was meticulously conducted employing a criterion encapsulating an FDR < 0.05 and |log2FC| ≥ 1. The hazard ratio (HR) was judiciously utilized as a discriminant screening criterion, adopting values either exceeding 1 or, conversely, residing below 1 as pivotal thresholds. The statistical analyses were conducted using the R programming language, and box plots were generated using the “ggpubr” package. In our comprehensive analysis of gene activities, we employed sophisticated computational methods facilitated by renowned packages such as plyr, reshape2, and ggpubr within the R ecosystem. This rigorous approach enabled us to accurately discern differential gene activities. For enhanced clarity and effective communication of our findings, the activity patterns of each gene were elegantly represented as ranked boxplots. In contemplation of antecedent scholarly discourses encompassing pan-cancer and affiliated neoplastic entities, a pervasive adoption of a 65-year demarcation for age cutoff has been discerned. The rationale underpinning this demarcational choice predominantly resides in the substantive alteration observed in cancer-related mortality trajectories, which is conspicuously marked in patient cohorts upon attaining this particular chronological milestone. Additionally, the status changes in S100A9 were determined by examining the genomic alterations, including copy number amplification, severe loss, unknown missense mutations, and mRNA overexpression, observed in the S100A9 gene. This comprehensive analysis provides a nuanced understanding of S100A9's role across various cancers and its potential as a biomarker and therapeutic target.
Relationship between S100A9 and survival and clinical stage
Overall survival (OS), disease-specific survival (DSS), disease-free survival (DFS), and progression-free survival (PFS) were employed to assess the impact of S100A9 on cancer survival. The log-rank test and univariate Cox proportional hazards models were utilized for the analyses. Clinical factors such as age, sex, and stage were taken into consideration for multivariate Cox regression analysis. The stage survival plot module was utilized to investigate the association between S100A9 expression and clinical stage. Specifically, two clinical phenotypes, tumor stage and patient age, were selected to explore their correlation with S100A9 expression. The patients were divided into two groups based on an age cutoff value of 65 years. Correlation analyses between clinical phenotypes and S100A9 expression were conducted using the R-packages “limma” and “ggpubr.” Statistical significance was defined as P < 0.05. The Kaplan–Meier method and log-rank tests were applied to perform survival analyses for each cancer type, with significance defined as P < 0.05. Survival curves were plotted using the R packages “survival” and “survminer.” Furthermore, Cox analysis was conducted using the R packages “survival” and “forestplot” to examine the pan-cancer relationship between S100A9 expression and survival.
Relationship between S100A9 and the TME
In this study, we aimed to investigate and quantify the association between S100A9 expression and the gene markers of tumor-infiltrating immune cells (TIICs) in malignant tumors, as well as identify immune cell infiltration patterns. The tumor mutation burden (TMB) of each tumor sample was calculated as the total count of somatic mutations (excluding silent mutations) detected in the tumor genome. To assess the TME, we computed the stromal score, immune score, and ESTIMATE score using the ESTIMATE algorithm. Tumor purity was determined inversely relative to these scores. Subsequently, the limma package was employed to examine the differences in the TME among multiple cancer samples based on immunological, ESTIMATE, and stromal scores. Scatterplots were generated to evaluate tumor cell purity. Higher predicted immune or stromal scores indicated a greater proportion of immune or stromal components in the TME. Consequently, a higher associated score reflected the increased significance of the relevant component within the TME. The ESTIMATE score was calculated as the sum of these two components, representing their respective proportions within the TME. The TMB has gained recognition as a distinct and reliable biomarker for predicting the response to immunotherapy. It was calculated as the total number of mutations per DNA megabase, categorized into nucleotide insertions, base substitutions, or deletions21. Microsatellite instability (MSI), characterized by spontaneous nucleotide loss or gain in short tandem repeat DNA sequences, was also examined to explore its relationship with TMB22. The fmsb package was utilized for this analysis. By integrating these comprehensive methodologies, we aimed to elucidate the complex interplay between S100A9 expression, immune cell infiltration, and tumor mutational characteristics within the TME. This study provides valuable insights into the potential of S100A9 as a biomarker and therapeutic target in cancer immunotherapy.
Immunotherapeutic response
The assessment of immunotherapeutic response was conducted by compiling and evaluating datasets GSE78220, GSE67501, and IMvigor210. Inclusion criteria for the analysis involved patients who exhibited either a complete or partial response. To compare the levels of S100A9 between the respondent and non-respondent groups, the Wilcoxon test was employed.
Analysis of GSEA and comprehensive gene regulatory networks
To investigate the underlying biological signaling pathways, Gene Set Enrichment Analysis (GSEA) was performed by segregating the high and low-expression groups based on the median level of S100A9 expression. GO and KEGG gene sets were obtained from the official GSEA website, while the gene set from the MSigDB was utilized for Gene Set Variation Analysis (GSVA). GSVA scores were calculated for all tumors, and subsequently, samples within each tumor were categorized into high and low expression groups using the median of differentially expressed genes with the R-package “limma.” Enrichment analysis identified gene sets with significant findings indicated by |NES| > 1, NOM p < 0.05, and false discovery rate q-value < 0.0523. In addition, to unravel the fundamental mechanisms underlying S100A9, we constructed a comprehensive Gene regulatory networks from GeneMANIA.
Mendelian randomization analysis
To ensure the independence of exposure and outcome variables in our genome-wide association study (GWAS) summary data, we engaged in an association analysis via the TwoSampleMR package in R. Designating S100A9-related expression as the some cancers (BLCA, CESC, COAD, etc.) as the outcome, we aimed to explore potential causal relationships. The analysis entailed: (1) Instrumental Variables (IVs) Configuration: S100A9-related expressions were screened with a P-value threshold of < 5 × 10−8 to identify strongly associated exposures. (2) Independence Configuration: Linkage disequilibrium (LD) between SNPs was calculated using the PLINK clustering method, excluding SNPs with LD coefficient r2 > 0.001 and within 10,000 kb to ensure SNP independence and reduce pleiotropic biases. (3) Statistical Strength Configuration: The robustness of instrumental variables was assessed using the F-statistic (F = β2/SE2), with variables having F < 10 deemed inadequate to mitigate confounding effects.
Leveraging GWAS data, SNPs associated with the instrumental variables were identified, and through the “harmonise_data” function within TwoSampleMR, we aligned allelic directions of exposure and outcome, excluding incompatible SNPs. The inverse variance-weighted (IVW) method served as the cornerstone for causal inference, employing the variance of instrumental variables as weights to determine causal dynamics, thereby advancing our understanding of the genetic architecture underlying disease states.
Ethics approval and consent to participation
This manuscript is a Bioinformatics analysis, hence the ethics approval and consent to participation is not applicable.
Results
Clinical landscape of S100A9 expression
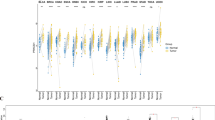
Given its remarkable sensitivity, S100A9 holds significant promise as a critical target and biomarker for cancer diagnosis. To evaluate S100A9 expression in tumors and adjacent normal tissues, we conducted a comprehensive analysis of S100A9 mRNA expression levels. Our results revealed significantly elevated S100A9 mRNA levels in cancer samples from BLCA, BRCA, CESC, COAD, ESCA, GBM, HNSC, KICH, KIRC, KIRP, LIHC, LUSC, PAAD, PRAD, THCA, and UCEC. These findings suggest a potential oncogenic role for S100A9 in the progression of these diverse cancers (Fig. 2a). Notably, S100A9 expression was particularly elevated in HNSC, CESC, LUSC, and ESCA, as highlighted in Fig. 2b. S100A9 activity was markedly enhanced in tumor categories including BLCA, CESC, CHOL, COAD, ESCA, GBM, KIRC, LUAD, LUSC, PAAD, READ, STAD, THCA, and UCEC, while it exhibited a significant reduction in BRCA, KICH, PCPG, and PRAD, as depicted in Fig. 2c. Furthermore, significantly higher activity levels were observed in HNSC, CESC, LUSC, and ESCA, as shown in Fig. 2d. Figure 3a illustrates distinct expression patterns of S100A9 in older patients with GBM, while lower expression levels were observed in BRCA, ESCA, LAML, SARC, STAD, and THYM. Gender disparities in S100A9 expression were significant in BRCA, HNSC, SARC, and SKCM, as depicted in Fig. 3b. Additionally, associations between S100A9 expression and cancer grade were observed in several cancer types, including ESCA, HNSC, and LGG, as illustrated in Fig. 3c. Moreover, S100A9 expression was linked to tumor stage in multiple cancers, including HNSC, KICH, LIHC, LUAD, PAAD, TGCT, and THCA, as shown in Fig. 3d. These findings underscore the multifaceted role of S100A9 in cancer biology, emphasizing its potential utility as a diagnostic biomarker and therapeutic target.

S100A9 activity. (a) Differential analysis of Tumor and normal. (b) Mean expression. (c) Activity analysis. (d) Mean activity.

Clinical information. (a) Age. (b) Gender. (c) Grade. (d) Stage.
Prognostic expression of S100A9 in cancers
Forest plots were constructed to evaluate the prognostic significance of S100A9 across various cancer types. The analysis revealed a favorable association between S100A9 expression and overall survival (OS) in LAML, LGG, LIHC, THYM, and UVM, as depicted in Fig. 4. Notably, a clear positive correlation between S100A9 expression and disease-free survival (DFS) was observed in LIHC and READ, whereas a negative correlation was evident in lung squamous cell carcinoma (LUSC). Regarding disease-specific survival (DSS), elevated S100A9 expression emerged as a risk factor in BLCA, COAD, KIRC, LGG, LIHC, and UVM. The forest plot for progression-free survival (PFS) further substantiated the risk associated with S100A9 expression in BLCA, COAD, KIRC, LGG, LIHC, and UVM. Moreover, the graphical representation facilitated the identification of additional malignancies where S100A9 expression was determined to be a concomitant risk factor, notably in THYM and UVM. While not directly interfacing with clinical attributes, S100A9 expression demonstrated a robust association with survival outcomes across diverse neoplastic entities, particularly in LAML, LGG, and LIHC. These findings underscore the critical role of S100A9 as a prognostic biomarker, highlighting its potential impact on the management and therapeutic stratification of various cancers.

Univariate Cox regression analyses.
S100A9 expression and immune infiltration levels in cancer
To evaluate the correlation between S100A9 expression and immune infiltration levels across various malignancies, we calculated the coefficients of S100A9 expression and immune infiltration. Figure 5 provides an overview of the stromal and immunological scores. S100A9 expression demonstrated significant associations with stromal scores in DLBC, GBM, KICH, LAML, LGG, PCPG, SARC, TGCT, THCA, and UVM. Additionally, S100A9 expression showed correlations with immune scores in COAD, GBM, KICH, KIRP, LAML, LGG, LIHC, PAAD, PCPG, PRAD, READ, SARC, THCA, and UVM (Table 1). Moreover, the analysis of immune cell infiltration revealed significant associations between S100A9 expression and specific immune cell subsets. Notably, S100A9 expression was negatively associated with CD4 memory resting T cells in ACC, monocytes in LAML, M1 macrophages in DLBC, activated natural killer (NK) cells in DLBC and KICH, naive B cells in LAML and TGCT, and neutrophils in ACC, CHOL, COAD, GBM, KICH, KIRC, PCPG, READ, and STAD, as depicted in Fig. 6. These findings highlight the intricate relationship between S100A9 expression and the tumor microenvironment, suggesting its potential role in modulating immune infiltration and influencing cancer progression.

ESTIMATE analyses. (a) StromalScore. (b) ImmuneScore.

Immune infiltration analyses.
Analysis of S100A9 expression and immune modulators
To investigate the intricate relationship between S100A9 expression and immune modulators, a comprehensive analysis was conducted. As shown in Fig. 7, a meticulous examination was carried out on 24 distinct types of immune inhibitors. Notably, S100A9 exhibited significant associations with specific immune modulators in particular cancer types. Specifically, S100A9 demonstrated a correlation with IL10 in GBM, HAVCR2 in THCA, and LGALS9 in SARC, while no significant association was observed with CD160 in CHOL. Moving forward, a thorough exploration was undertaken to assess the relationship between S100A9 expression and 45 immune stimulators, as illustrated in Fig. 8. The findings revealed intriguing patterns of association. S100A9 expression displayed a positive correlation with IL6 in GBM, CD86 in THCA, and IL2RA in SARC. Conversely, a negative association was observed with TNFSF13 in ESCA, suggesting a potential regulatory role of S100A9 in modulating immune responses. Furthermore, it is noteworthy that S100A9 expression exhibited distinct associations with HLA genes. Figure 9 highlights these associations, where S100A9 showed a positive correlation with HLA-DPA1 in KICH, HLA-DPB1 in THCA, and HLA-DRA in THCA. Conversely, a negative association was identified with HLA-A in CHOL, indicating potential intricate mechanisms underlying the interplay between S100A9 expression and HLA genes. These findings shed light on the complex interrelationships between S100A9 expression and immune modulators, providing valuable insights into the underlying mechanisms of immune regulation in various cancer types.

Immune inhibitors.

Immune stimulators.

MHC molecules.
Immunotherapeutic markers and response
The present study aimed to investigate the correlation between S100A9 and two novel dynamic markers associated with immune checkpoint blockade, namely TMB and MSI. The analysis revealed that S100A9 expression exhibited a positive association with TMB in BRCA, CESC, KIRC, and LGG. However, this association was not observed in ESCA, LAML, PAAD, PCPG, and PRAD. Conversely, MSI demonstrated a positive relationship with S100A9 expression in BRCA, CESC, KIRC, and LGG, while exhibiting a negative relationship in ESCA, LAML, PAAD, PCPG, and PRAD. The immune checkpoint pathway plays a pivotal role in cancer therapy, particularly the programmed cell death protein 1 (PD-1) pathway. Previous literature has reported the utilization of three commonly employed microarray datasets, namely GSE67501 (n = 11), GSE78220 (n = 28), and IMvigor210, to investigate the expression of individual genes in relation to PD-1. Accordingly, we investigated the association between S100A9 and immune checkpoints, specifically focusing on these three datasets. However, the analysis revealed no statistically significant differences in S100A9 expression between responder and non-responder groups in any of the three cohorts, implying that S100A9 may not be influenced by the microarray sets commonly employed in these immune checkpoint studies. Notably, patients exhibiting lower S100A9 expression displayed greater susceptibility to immunotherapy in the analyzed cohorts (Fig. 10). It is important to acknowledge that although the three GEO datasets used in this study are widely recognized and frequently employed in classic GEO microarray studies, they do possess certain limitations such as small sample sizes, which may introduce some inherent biases into the results. Consequently, future investigations should endeavor to employ larger datasets to validate these findings.

Immunotherapeutic markers and response.
The association between changes in fatty acid metabolism regulating genes (CNV, SNP, and mutation) and clinicopathological characteristics in patients was investigated. Correlation study of S100A9 expression in the prognostic model and SNP revealed 6 SNP-driven cancers, including BLCA,STAD, BRCA,COAD, CSEC, BRCA (Fig. 11a–f). A correlation analysis of S100A9 expression in the prognostic model and CNV found numerous cancers driven by CNV. The expression of 6 cancers were upregulated in the single mutations group versus that of the non-mutations group. (P < 0.05), indicating that dysregulation of key genes might be driven by SNP in S100A9.

SNP and mutation analysis. (a–f) Prognostic signatures and SNP. (a) BLCA, (b) STAD, (c) BRCA, (d) COAD, (e) CSEC, (f) BRCA, (g) CNV analysis.
Comprehensive gene regulatory networks and GSEA
To unravel the fundamental mechanisms underlying S100A9, we constructed a comprehensive Comprehensive Gene Regulatory Networks (Fig. 12). Within this network, S100A9 exhibited a strong interaction with S100A8, S100A12, CD33, CSTA, MMP9, S100A7, which has been implicated in cancer metastasis (Table 2). These proteins exhibit diverse roles in tumor progression, inflammation, and immune response modulation, with their expression levels often being dysregulated in various malignancies. S100A8 and S100A9, members of the S100 calcium-binding protein family, are known to form heterodimers and are markedly upregulated in several types of cancer. They function as damage-associated molecular patterns, promoting pro-inflammatory responses and enhancing the recruitment of myeloid-derived suppressor cells, which are known to facilitate tumor progression by suppressing anti-tumor immunity24. These proteins also contribute to the remodeling of the extracellular matrix, thus aiding in metastasis. S100A12, another member of the S100 family, shares functional similarities with S100A8 and S100A925. It is often co-expressed with these proteins and plays a role in inflammatory processes and cancer. Its interaction with the receptor for advanced glycation end products is particularly notable, as it activates key signaling pathways involved in tumor growth and metastasis. CD33, a transmembrane receptor expressed on myeloid cells, plays a crucial role in modulating immune responses. In the context of cancer, CD33 is expressed on MDSCs and contributes to their immunosuppressive functions, thereby facilitating tumor immune evasion26. Targeting CD33-positive MDSCs has emerged as a potential therapeutic strategy to enhance anti-tumor immunity. CSTA, a cysteine protease inhibitor, is implicated in cancer progression through its regulatory effects on proteolytic activity. CSTA can influence tumor invasion and metastasis by modulating the activity of cathepsins, a family of proteases involved in extracellular matrix degradation27. Its expression levels have been correlated with tumor aggressiveness in various cancers. Collectively, these investigations provide compelling support for the credibility and plausibility of our findings, suggesting that S100A9 could serve as a novel diagnostic and prognostic biomarker in human cancers.

Comprehensive Gene Regulatory Networks of S100A9.
Subsequently, we employed GSEA to identify functional enrichments related to S100A9 (Fig. 13). The analysis revealed that elevated levels of S100A9 were significantly associated with metabolic-related activities, including olfactory transduction, autophagy regulation, the rig I like receptor signaling pathway, systemic lupus erythematosus, and taste transduction, as indicated by the KEGG pathway database. Moreover, based on the GO annotations, high levels of S100A9 were primarily associated with epidermis development, sensory perception of chemical stimulus, and sensory perception of smell.

GSEA. (a + c) Low expression. (b + d) High expression sample. (a + b) KEGG. (c + d): GO.
Mendelian randomization analysis
In our exploration of the intrinsic connection between BLCA, CESC, COAD, ESCA, GBM, BRCA, HNSC, and S100A9, forest plots were meticulously employed to visually articulate the associations. Further dissecting the heterogeneity inherent in our analysis, the funnel plot tailored to KIRC revealed a deviation from the expected symmetrical distribution, albeit maintaining a general symmetry. This nuanced observation was further scrutinized through sensitivity analysis, employing a “leave-one-out” approach. Remarkably, the omission of any individual SNP from the analysis had a negligible effect on the results of the Inverse Variance Weighted (IVW) analysis, indicating that the remaining SNPs consistently mirrored the outcomes of the aggregate dataset. Substantiating the validity of our findings, the MR-Egger regression analysis was invoked, providing a solid foundation that bolsters both the robustness and authenticity of our results and the methodologies applied. This Mendelian randomization analysis unequivocally confirms the intimate association of BLCA, CESC, COAD, ESCA, GBM, BRCA, HNSC with S100A9. Hence, it delineates a potential pathway to modulate the incidence, evolution, and progression of S100A9 by intervening in the functions of BLCA, CESC, COAD, ESCA, GBM, BRCA, HNSC, presenting a promising avenue for therapeutic intervention and a deeper understanding of the disease mechanism (Table 3, Fig. 14).

Mendelian randomization analysis. (a) BLCA, (b) CESC, (c) COAD, (d) ESCA, (e) GBM, (f) BRCA, (g) HNSC.
Discussion
Cancer remains a formidable adversary in the pursuit of sustaining optimal quality of life and reducing mortality. Despite significant advances in cancer treatment, its meticulous management remains an intricate clinical challenge. The advent of cancer immunotherapy (CIT) has ushered in a transformative era in therapeutics, effectively leveraging the interactions between the immune system and malignant cells28. However, to fully realize the potential of CIT, there is a critical need to identify predictive biomarkers that can accurately evaluate responses to various immunotherapeutic modalities and determine early therapeutic efficacy for patients. While previous studies have highlighted several factors influencing CIT response, the precise and reliable identification of biomarkers remains an elusive goal29. S100A9, a pivotal transcription factor intricately woven into the immunological fabric of TME, emerges as a promising candidate for immunotherapeutic targeting. It is essential to meticulously explore the interplay between S100A9 and various facets of the TME, including immune cellular components, immunomodulators, and responses to immunotherapy. This study aimed to conduct a comprehensive examination of S100A9 expression across 33 malignancies, focusing on its impact on the immunological TME. Emphasis was placed on vital immunomodulators and dynamic immunological biomarkers. Our initial analysis of the relationship between S100A9 expression and clinical variables revealed no significant differences in age, gender, or tumor stage across most cancer types, consistent with previous findings. Although the prognostic value of S100A9 expression was only marginally significant in several cancers, including breast cancer, S100A9 has been identified as a proto-oncogene in various malignancies, such as bladder, colorectal, and pancreatic cancers30. In pancreatic cancer, S100A8 and S100A9 proteins create a paracrine feedback loop between cancer cells and monocytes31. Tumor-infiltrating monocytes/macrophages enhance tumor invasion and migration, driven by upregulated S100A8 and S100A9 expression within cancer cells32. Based on the compelling evidence presented in this study, we propose that therapeutic modulation of S100A9 activity across diverse tumor types may represent a viable clinical strategy.
Moreover, a comparison between the S100A9 activity score and transcriptional levels revealed partial concordance in the overall activation of S100A9 across various malignancies, including BLCA, CESC, CHOL. However, discrepancies between S100A9 expression and activity were observed in certain cancers, such as HNSC, KIRP, LIHC, CHOL, LUAD, READ, STAD, and PCPG. These perturbations in S100A9 functional activity might be attributed to post-transcriptional modifications or shifts in protein metabolism, revealing inconsistencies that necessitate deeper exploration. Strikingly, in LIHC and READ, S100A9 demonstrated a positive association with DFS, while presenting a negative association in LUSC. Additionally, concomitant elevations in the blood levels of both S100A8/A9 and CA15-3 were observed in breast cancer patients, with these augmentations positively correlating with tumor size. This suggests the S100A8/A9 heterodimer as a potential dual-purpose biomarker, offering both diagnostic and prognostic utility for BRCA. Transcriptome profiling of KIRP, which exhibited significantly elevated S100A9 expression levels, unveiled an association with disease progression33. Given its pivotal role in activating inflammatory pathways that fuel cancer progression, S100A9 has emerged as a promising target for anticancer therapeutic research34. Thus, S100A9 may be implicated in the oncogenic processes of GBM, KIRP, BRCA, and LIHC, further underscoring its potential as a critical biomarker and therapeutic target in these malignancies.
Additionally, a comparison between transcriptional levels and S100A9 activity scores revealed partial concordance, suggesting that transcriptional levels partially reflect the overall activation of S100A9 in various tumors, including BLCA, CESC, CHOL, COAD, ESCA, GBM, KIRC, LUAD, LUSC, PAAD, READ, STAD, THCA, and UCEC. The transcriptional level serves as a paradigmatic indicator of S100A9 activation within these tumor categories. However, discrepancies between S100A9 expression and activity have been observed in several cancers, including HNSC, KIRP, LIHC, CHOL, LUAD, READ, STAD, and PCPG. These inconsistencies may stem from post-transcriptional protein modifications or perturbations in protein metabolism, influencing S100A9 expression. Aurora De Ponti et al. highlighted the pivotal role of S100A8 in the progression of non-inflammatory liver tumors, suggesting S100A8 as a promising therapeutic target in managing LIHC35. Concurrently, Paul R. Gielen et al. documented a notable elevation of MDSCs in the blood of glioma patients, correlating with tumor grade. The presence and activation status of MDSCs were assessed via the quantification of S100A8/A9 and arginase levels36. Furthermore, elevated levels of both S100A8/A9 and CA15-3 were identified in the serum of breast cancer patients, with these levels positively correlating with tumor size. This observation suggests that the S100A8/A9 heterodimer could serve as a biomarker for the diagnosis and prognosis of BRCA37. In the context of KIRC, transcriptome profiles revealing markedly elevated S100A9 expression levels have been implicated in the progression of this cancer subtype. S100A9 has emerged as a significant target for anticancer therapeutic research due to its integral role in initiating inflammatory pathways that promote cancer metastasis38. The evidence from these studies collectively supports our findings, indicating a potential association between S100A9 and oncogenic processes in patients with GBM, KIRC, BRCA, and LIHC. Thus, our findings suggest that S100A9 may function as an oncogene in these specific cancer populations.
This study aimed to explore the potential utility of S100A9 by investigating its relationship with immune cell infiltration. In ACC, CHOL, and COAD, S100A9 exhibited a significant correlation with neutrophils. Previous research suggests that S100A9 influences tumor growth and immune responses through its interaction with TME-associated macrophages39. Specifically, S100A9 may modulate neutrophils and subsequent activation of an immunosuppressive response40. Notably, in CHOL, S100A9 displayed a significant negative association with CD160. Apart from ESCA, most immune stimulants and MHC molecules exhibited a positive relationship with S100A9. This intriguing finding may uncover a novel regulatory mechanism in the context of ESCA immunotherapy. Enrichment analysis revealed that elevated levels of S100A9 were predominantly associated with metabolic-related activities. Metabolic inflammation is characterized by dysregulated expression of cytokines and adipocytokines in adipose tissue41. S100A9 has the potential to promote phagocytosis, induce neutrophil chemotaxis and adhesion, and enhance neutrophil bactericidal activity through the activation of SYK, PI3K/AKT, and ERK1/2 signaling pathways42. Furthermore, its extracellular proinflammatory activity involves leukocyte recruitment, promotion of cytokine and chemokine production, and regulation of leukocyte adhesion and migration43. Increased S100A9 levels in certain malignancies may disrupt innate immunity by activating metabolic-related pathways. These findings highlight the multifaceted role of S100A9 in cancer progression and immune modulation, suggesting its potential as a biomarker and therapeutic target in various malignancies.
In general, tumors with a higher number of somatic mutations tend to exhibit increased neoantigen production, making TMB a reasonable estimate of the tumor-neoantigen burden44. MSI, characterized by defective DNA mismatch repair, is considered a potent mutator phenotype and a potential prognostic indicator for immunotherapy45. Within a spectrum of neoplasias, including ESCA, LAML, PAAD, PCPG, and PRAD, S100A9 delineated a negative correlation with both TMB and MSI. In stark contrast, in BRCA, CESC, KIRC, and LGG, a positive relationship with both biomarkers was discerned. These observations suggest that S100A9 might indirectly modulate immunotherapeutic responses within these cancers. While the exploration into the relationship between S100A9 and immunotherapeutic responses was meticulously undertaken, discernible disparities were not evident among the populations under scrutiny. Nonetheless, the present study unveils pivotal insights into the nexus between S100A9 and tumor immunology, highlighting its potential as a biomarker in oncology. However, the investigation into immunotherapeutic responses was constrained to a limited number of cohorts, inhibiting the derivation of unequivocal conclusions regarding the specific immunotherapeutic responses associated with S100A9. Future research should focus on probing larger cohorts within immunotherapeutic contexts to further unravel its clinical relevance.
This investigation enhances the existing understanding of S100A9’s role in cancer immunotherapy, providing an initial framework elucidating potential correlations between S100A9 and key immunological markers. However, the present study has limitations that warrant discussion. Primarily, the derivation of a validated predictive signature for S100A9 was based solely on public data repositories, such as TCGA, limiting the extent of external validation. Secondly, although bioinformatic analyses provide valuable insights into S100A9’s role in oncology, these findings should be considered preliminary. The complexity of protein expression, often divergent from RNA expression, necessitates further validation using larger, more comprehensive datasets. To corroborate and enhance the therapeutic applicability of our findings, biological evaluations through in vitro or in vivo studies are imperative46. Additionally, considering the pivotal role of post-translational modifications in modulating intracellular signaling and regulatory factor functionality, further meticulously designed investigations are required to fully understand these aspects. Overall, while this study establishes a foundational understanding of S100A9's involvement in cancer immunotherapy, extensive validation and additional research are essential to confirm and expand upon these findings.
Conclusions
S100A9 emerges as a molecule of profound prognostic and therapeutic significance, demonstrating substantive associations across numerous malignancies and positioning itself as a promising therapeutic target. This study elucidates the pivotal role of S100A9 in oncogenesis, while also shedding light on potential mechanisms through which it influences tumor immunology and metabolic activity. Notably, we have developed a predictive model based on the distinctive S100A9 signature, which holds potential for forecasting the clinical trajectory of specific malignancies. This model underscores the critical importance of S100A9 as both a biomarker and a therapeutic target in cancer treatment.
Data availability
Patients who granted informed consent to use their data have been uploaded to the public-accessible TCGA databases. The datasets generated and/or analysed during the current study are available in the [TCGA] repository [https://portal.gdc.cancer.gov/].
Abbreviations
- S100A9:
-
S100 calcium binding protein A9
- GO:
-
Gene ontology
- KEGG:
-
Kyoto Encyclopedia of Genes and Genomes
- TME:
-
Tumor microenvironment
- TMB:
-
Tumor mutational burden
- DSS:
-
Disease-specific survival
- HR:
-
Hazard ratios
- PPI:
-
Protein–protein interaction
- CR:
-
Complete response
- PD:
-
Progressing disease
- TCGA:
-
The Cancer Genome Atlas
- GEO:
-
Gene Expression Omnibus
- GSEA:
-
Gene set enrichment analysis
- MSI:
-
Microsatellite instability
- OS:
-
Overall survival
- CI:
-
Confidence intervals
- TIICs:
-
Tumor-infiltrating immune cells
- PFs:
-
Progression-free survival
- PR:
-
Partial response
- SD:
-
Stable illness
References
Abdelgawad, I. Y. et al. Molecular mechanisms and cardiovascular implications of cancer therapy-induced senescence. Pharmacol. Ther. 221, 107751 (2021).
Chen, G. et al. GPC2 is a potential diagnostic, immunological, and prognostic biomarker in pan-cancer. Front. Immunol. 13, 857308 (2022).
Liu, Y. et al. Pan-cancer analysis on the role of PIK3R1 and PIK3R2 in human tumors. Sci. Rep. 12(1), 5924 (2022).
Tomonobu, N., Kinoshita, R. & Sakaguchi, M. S100 soil sensor receptors and molecular targeting therapy against them in cancer metastasis. Transl. Oncol. 13(4), 100753 (2020).
Wagner, N. B. et al. Tumor microenvironment-derived S100A8/A9 is a novel prognostic biomarker for advanced melanoma patients and during immunotherapy with anti-PD-1 antibodies. J. Immunother. Cancer 7(1), 343 (2019).
Liu, L. et al. Targeting the IRAK1-S100A9 axis overcomes resistance to paclitaxel in nasopharyngeal carcinoma. Cancer Res. 81(5), 1413–1425 (2021).
Inciarte-Mundo, J., Frade-Sosa, B. & Sanmarti, R. From bench to bedside: Calprotectin (S100A8/S100A9) as a biomarker in rheumatoid arthritis. Front. Immunol. 13, 1001025 (2022).
Bao, X. et al. An immunometabolism subtyping system identifies S100A9(+) macrophage as an immune therapeutic target in colorectal cancer based on multiomics analysis. Cell Rep. Med. 4(4), 100987 (2023).
Chen, Y., Ouyang, Y., Li, Z., Wang, X. & Ma, J. S100A8 and S100A9 in cancer. Biochim. Biophys. Acta Rev. Cancer 1878(3), 188891 (2023).
Zhong, C. et al. S100A9 derived from chemoembolization-induced hypoxia governs mitochondrial function in hepatocellular carcinoma progression. Adv. Sci. (Weinh) 9(30), e2202206 (2022).
Marinkovic, G. et al. S100A9 links inflammation and repair in myocardial infarction. Circ. Res. 127(5), 664–676 (2020).
Abbott, M. & Ustoyev, Y. Cancer and the immune system: The history and background of immunotherapy. Semin. Oncol. Nurs. 35(5), 150923 (2019).
Giese, M. A., Hind, L. E. & Huttenlocher, A. Neutrophil plasticity in the tumor microenvironment. Blood 133(20), 2159–2167 (2019).
Anderson, N. M. & Simon, M. C. The tumor microenvironment. Curr. Biol. 30(16), R921–R925 (2020).
Elhanani, O., Ben-Uri, R. & Keren, L. Spatial profiling technologies illuminate the tumor microenvironment. Cancer Cell 41(3), 404–420 (2023).
Hinshaw, D. C. & Shevde, L. A. The tumor microenvironment innately modulates cancer progression. Cancer Res. 79(18), 4557–4566 (2019).
Vitale, I., Manic, G., Coussens, L. M., Kroemer, G. & Galluzzi, L. Macrophages and metabolism in the tumor microenvironment. Cell Metab. 30(1), 36–50 (2019).
Bejarano, L., Jordao, M. & Joyce, J. A. Therapeutic targeting of the tumor microenvironment. Cancer Discov. 11(4), 933–959 (2021).
Wang, Z., Jensen, M. A. & Zenklusen, J. C. A practical guide to the cancer genome atlas (TCGA). Methods Mol. Biol. 1418, 111–141 (2016).
Cerami, E. et al. The cBio cancer genomics portal: An open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2(5), 401–404 (2012).
Greillier, L., Tomasini, P. & Barlesi, F. The clinical utility of tumor mutational burden in non-small cell lung cancer. Transl. Lung Cancer Res. 7(6), 639–646 (2018).
Hause, R. J., Pritchard, C. C., Shendure, J. & Salipante, S. J. Classification and characterization of microsatellite instability across 18 cancer types. Nat. Med. 22(11), 1342–1350 (2016).
Subramanian, A. et al. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA 102(43), 15545–15550 (2005).
Chung, Y. H. et al. Viral nanoparticle vaccines against S100A9 reduce lung tumor seeding and metastasis. Proc. Natl. Acad. Sci. USA 120(43), e2073108176 (2023).
Wang, S. et al. S100A8/A9 in inflammation. Front. Immunol. 9, 1298 (2018).
Albinger, N. et al. Primary CD33-targeting CAR-NK cells for the treatment of acute myeloid leukemia. Blood Cancer J. 12(4), 61 (2022).
Wei, R., Zhang, L., Hu, W., Wu, J. & Zhang, W. CSTA plays a role in osteoclast formation and bone resorption by mediating the DAP12/TREM2 pathway. Biochem. Biophys. Res. Commun. 627, 12–20 (2022).
Silva, D. M. B. et al. S100A9 drives the chronification of psoriasiform inflammation by inducing IL-23/type 3 immunity. J. Investig. Dermatol. 143(9), 1678–1688 (2023).
Wang, L. et al. CELF2 is a candidate prognostic and immunotherapy biomarker in triple-negative breast cancer and lung squamous cell carcinoma: A pan-cancer analysis. J. Cell. Mol. Med. 25(15), 7559–7574 (2021).
Gunaldi, M. et al. Diagnostic importance of S100A9 and S100A12 in breast cancer. Biomed. Pharmacother. 76, 52–56. https://doi.org/10.1016/j.biopha.2015.10.029 (2015).
Nedjadi, T. et al. S100A8 and S100A9 proteins form part of a paracrine feedback loop between pancreatic cancer cells and monocytes. BMC Cancer 18, 1255. https://doi.org/10.1186/s12885-018-5161-4 (2018).
Lim, S. Y., Yuzhalin, A. E., Gordon-Weeks, A. N. & Muschel, R. J. Tumor-infiltrating monocytes/macrophages promote tumor invasion and migration by upregulating S100A8 and S100A9 expression in cancer cells. Oncogene 35, 5735–5745. https://doi.org/10.1038/onc.2016.107 (2016).
Khorrami, S., Tavakoli, M. & Safari, E. Clinical value of serum S100A8/A9 and CA15-3 in the diagnosis of breast cancer. Iran. J. Pathol. 14(2), 104–112 (2019).
Mirza, Z. et al. Molecular interaction of a kinase inhibitor midostaurin with anticancer drug targets, S100A8 and EGFR: Transcriptional profiling and molecular docking study for kidney cancer therapeutics. PLoS One 10(3), e119765 (2015).
De Ponti, A. et al. A pro-tumorigenic function of S100A8/A9 in carcinogen-induced hepatocellular carcinoma. Cancer Lett. 369(2), 396–404 (2015).
Gielen, P. R. et al. Elevated levels of polymorphonuclear myeloid-derived suppressor cells in patients with glioblastoma highly express S100A8/9 and arginase and suppress T cell function. Neuro Oncol. 18(9), 1253–1264 (2016).
Lin, C. et al. S100A8/S100A9 promote progression of multiple myeloma via expansion of megakaryocytes. Cancer Res. Commun. 3(3), 420–430 (2023).
Razmkhah, F., Kim, S., Lim, S., Dania, A. J. & Choi, J. S100A8 and S100A9 in hematologic malignancies: From development to therapy. Int. J. Mol. Sci. 24(17), 13382 (2023).
Willers, M. et al. S100A8 and S100A9 are important for postnatal development of gut microbiota and immune system in mice and infants. Gastroenterology 159(6), 2130–2145 (2020).
Yoshioka, Y. et al. Neutrophils and the S100A9 protein critically regulate granuloma formation. Blood Adv. 1(3), 184–192 (2016).
Wu, H. & Ballantyne, C. M. Skeletal muscle inflammation and insulin resistance in obesity. J. Clin. Investig. 127(1), 43–54 (2017).
Vogl, T. et al. MRP8 and MRP14 control microtubule reorganization during transendothelial migration of phagocytes. Blood 104(13), 4260–4268 (2004).
Koike, A. et al. Dynamic mobility of immunological cells expressing S100A8 and S100A9 in vivo: A variety of functional roles of the two proteins as regulators in acute inflammatory reaction. Inflammation 35(2), 409–419 (2012).
Chan, T. A. et al. Development of tumor mutation burden as an immunotherapy biomarker: Utility for the oncology clinic. Ann. Oncol. 30(1), 44–56 (2019).
Yamamoto, H. & Imai, K. An updated review of microsatellite instability in the era of next-generation sequencing and precision medicine. Semin. Oncol. 46(3), 261–270 (2019).
Berg, V. et al. miRs-138 and -424 control palmitoylation-dependent CD95-mediated cell death by targeting acyl protein thioesterases 1 and 2 in CLL. Blood 125(19), 2948–2957 (2015).
Acknowledgements
Thank reviewers and editors for their sincere comments.
Funding
2022 State Administration of Traditional Chinese Medicine, Chinese Traditional Medicine Personnel Teaching Letter [2022] 76; Project of Hunan Provincial Administration of Traditional Chinese Medicine (No. B2024049).
Author information
Authors and Affiliations
Contributions
The manuscript was written and corrected by Yingying Chen and Zixuan Wu. Data gathering was overseen by Xingxing Yi and Zixuan Wu. The final manuscript version has been read and approved by all of the writers. Xingxing Yi and Yingying Chen conceived and designed this article, in charge of syntax modification and revised of the manuscript. All the authors have read and agreed to the final version manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Chen, Y., Wu, Z. & Yi, X. Elucidating the pan-oncologic landscape of S100A9: prognostic and therapeutic corollaries from an integrative bioinformatics and Mendelian randomization analysis. Sci Rep 14, 19071 (2024). https://doi.org/10.1038/s41598-024-70223-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-024-70223-x
- Springer Nature Limited