
Abstract
Breast cancer is a prevalent malignancy affecting women globally, necessitating effective treatment strategies. This study explores the potential of ergosterol, a bioactive compound found in edible mushrooms, as a candidate for breast cancer treatment. Breast cancer cell lines (MCF-7 and MDA-MB-231) were treated with ergosterol, revealing its ability to inhibit cell viability, induce cell cycle arrest, and suppress spheroid formation. Mechanistically, ergosterol demonstrated significant inhibitory effects on the Wnt/beta-catenin signaling pathway, a critical regulator of cancer progression, by attenuating beta-catenin translocation in the nucleus. This suppression was attributed to the inhibition of AKT/GSK-3beta phosphorylation, leading to decreased beta-catenin stability and activity. Additionally, ergosterol treatment impacted protein synthesis and ubiquitination, potentially contributing to its anti-cancer effects. Moreover, the study revealed alterations in metabolic pathways upon ergosterol treatment, indicating its influence on metabolic processes critical for cancer development. This research sheds light on the multifaceted mechanisms through which ergosterol exerts anti-tumor effects, mainly focusing on Wnt/beta-catenin pathway modulation and metabolic pathway disruption. These findings provide valuable insights into the potential of ergosterol as a therapeutic candidate for breast cancer treatment, warranting further investigation and clinical application.
Similar content being viewed by others

Breast cancer is the most common cancer in women worldwide; in 2020, 2.3 million new cases were reported by GLOBALCAN1. There are several treatment options for breast cancer patients depending on the stage and type of breast cancer. Breast cancer cells that express hormone receptors, such as the estrogen receptor (ER +) or progesterone receptor (PR +), typically exhibit a slower progression and favorable prognosis following treatment involving hormone receptor blockade2. Furthermore, in the case of breast cancer patients with hormone receptor-negative tumors expressing the human epidermal growth factor receptor-related protein 2 (HER2), endocrine blockers may not be applicable. However, a promising prognosis was observed when employing targeted therapies and chemotherapeutic drugs designed to combat HER23. Twelve percent of breast cancer patients diagnosed with triple-negative breast cancer who do not express either ER + , PR + or HER2 tend to have the most aggressive behaviors4. Standard treatments for breast cancer are surgery, chemotherapy, and radiation. However, the side effects from chemotherapy often worsen the quality of a patient's life, causing drug resistance and eventually disease relapse5. Breast cancer metastasis refers to the dissemination of tumors from the primary tumor site to the secondary site, such as bone, lung, and liver6. Epithelial-Mesenchymal Transition (EMT) is involved in metastasis, altering the critical features of epithelial to mesenchymal characters and cancer stemness. Wnt/β-catenin plays a role in breast cancer metastasis and cancer stemness by upregulating regulatory proteins like c-Myc and cyclin D1, resulting in cellular transformation, invasion, and proliferation7. Generally, Wnt pathway is categorized into two major pathways, including non-canonical and canonical. Non-canonical Wnt signaling is related to the Ras homolog gene family A (RHOA), which activates the c-Jun N-terminal kinase (JNK) and involves cell polarity and migration. On the other hand, the canonical Wnt pathway is initiated by the binding of Wnt ligands to frizzled-related receptors. This binding event inhibits the destruction complex, which consists of casein kinase 1 (CK1), adenomatous polyposis coli (APC), Axin, and glycogen synthase kinase 3β (GSK-3β). Consequently, this leads to the dissociation of β-catenin, its translocation into the nucleus, and the subsequent regulation of downstream targets of the Wnt signaling pathway8,9. The β-catenin activity and breast cancer progression are reported to be correlated and marked as a novel therapeutic target for breast cancer10.
Natural products are essential for drug discovery and development, including plants, microbes, fungi, and living organisms11. This study focuses on the bioactive compound from edible mushrooms. Ergosterol, a phytosterol frequently present in mushrooms and serves as a precursor to vitamin D2. Ergosterol has been reported in its biological activities, including antioxidant12, anti-diabetic13, anti-microbial14, anti-viral15, and neuroprotection16. Additionally, ergosterol has demonstrated anti-tumor properties, as evidenced by studies conducted on diverse models, including mice with sarcoma 180 tumors17, cases of liver cancer18, lung cancer19, bladder cancer20 and breast cancer21,22. However, the underlying molecular mechanism of ergosterol in metastasis and cancer stemness is not clearly understood. The primary objective of this study was to investigate the molecular mechanisms through which ergosterol exerts its anti-tumor effects on breast cancer cells.
The research encompassed a comprehensive screening of its impact on cell viability, cell cycle regulation, spheroid formation, protein synthesis, and the modulation of signal cascading proteins within the Wnt/β-catenin pathway. Notably, this study is the first to highlight the influence of ergosterol on breast cancer stemness, particularly concerning the Wnt/β-catenin pathway. Our findings also contribute to a more comprehensive understanding of the pharmacological effects of ergosterol. By unraveling the specific mechanisms through which ergosterol combats breast cancer, this study contributes to the broader understanding and advancement of cancer treatment strategies.
Materials and methods
Chemicals and reagents
Ergosterol (PHR1512, purity > 99%) and cycloheximide (C6255, purity > 98%) were purchased from Sigma Aldrich (St. Louis, MO, USA). Dimethyl sulfoxide (DMSO) (DR1022, purity > 99.5%) was purchased from Biosesang (Gyeonggi-do, Korea).
Cell culture maintenance
Human breast cancer cell lines (MCF-7 and MDA-MB-231) were purchased from American Type Culture Collection (ATCC, Rockville, MD, USA). Cells were maintained in Dulbecco’s Modified Eagle Medium (DMEM) medium (Welgene, Gyeongsan, Korea) supplemented with 10% Fetal bovine serum (FBS) (Gibco, Grand Island, NY, USA), 1% penicillin–streptomycin (Gibco) and 1% sodium pyruvate (Gibco). Cells were cultured at 37 °C in a 5% CO2 humidified incubator.
Cell viability
Cells were plated at a density of 1 × 103 cells/well in a 96-well culture plate and allowed to incubate overnight. Ergosterol (ER) in the range of 2.5–40 μM was treated into cells, and 0.1% DMSO was used as vehicle control. After 24, 48 and 96 h of incubation time, CellTiter 96Ⓡ AQueous One Solution Cell Proliferation Assay (Promega, Madison, WI, USA) was added and measured the absorbance by a microplate reader at 492 nm (Thermo Fisher Scientific, Waltham, MA, USA).
Cell cycle analysis
Treated cells were collected by trypsinization with 0.25% trypsin (Gibco) and centrifugation at 3000 rpm, 5 min, and 4 °C. Then, cells were washed with cold PBS twice and fixed with 70% ethanol by dropwise adding and stored at − 20 °C until analysis. Fixed cells were washed with PBS twice and stained with 400 μL of propidium iodine solution (100 μg/mL PI, 10 μg/mL RNAse A, 0.1% triton X-100 in PBS) for 30 min incubation on ice and stored in the dark until analysis. The cell phases (G0/G1, S, and G2/M phases) were analyzed by SH800S Cell sorter (Sony, Japan) with 10,000 events.
Spheroid formation
To investigate the spheroid formation with and without Matrigel, cells were plated at a density of 2,000 cells/well into an ultra-low attachment (ULA) 96-well round-bottom plate (Corning, Kennebunk, ME, USA). For the Matrigel-embedded spheroids, the 50 μL of 2.5% Matrigel (Corning, NY, USA) was added into wells and incubated at 37 °C for 1 h before the cell plating. spheroids were cultured for 3 days to generate spheroids. Cells were treated with 0.1% DMSO or ER 20 μM and replaced with fresh treatments every two days. Five spheroids were conducted per conditions with three replicates. Spheroids were captured on Days 0, 1, 3, 5 and 7 at 40X magnification. Data were analyzed by measuring spheroid diameter in spheroid formation without Matrigel. For Matrigel-embedded spheroid, mass intensity was analyzed from integrated density after threshold adjustment by ImageJ software.
Immunofluorescence
Immunofluorescence staining was performed by seeding cells onto coverslips in a 6-well culture plate at a density of 3 × 105 cells/well, and the cells were left to incubate overnight before treatments. To examine the expression of β-catenin, cells were treated with 0.1% DMSO or ER 20 μM in a serum-free culture medium for 24 h. After incubation, cells were washed with PBS and fixed with 4% paraformaldehyde for 20 min at RT. Cells were permeabilized with 0.1% triton X-100 for 15 min and blocked with 2% BSA for 1 h. Cells were washed with 1X PBS, and the anti-β-catenin antibody (1:200, Cat. #610154) (BD Biosciences, Franklin Lakes, USA) was added overnight at 4 °C. After the incubation, cells were washed and replaced with the FITC-anti-Mouse secondary antibody (1:1000, Cat. #F2765) (Invitrogen, Waltham, USA) for 1 h. Then, cells were washed and counter-stained with Hoechst 33342 (1 μg/mL, Sigma-Aldrich) for 30 min at RT, respectively. Slides were mounted with mounting medium (Agilent, CA, USA) and sealed with nail polish. Samples were analyzed by a confocal microscope at 200X magnification. The nuclear translocation of β-catenin was examined using Image J software, and Pearson's correlation coefficient was used for data analysis.
Nuclear and cytoplasmic subfraction
To further confirm the nuclear translocation of β-catenin from cytoplasm to nucleus, the cytoplasmic and nuclear subfraction was carried out by the NE-PER™ Nuclear and Cytoplasmic Extraction Reagents (Thermo Scientific, UT, USA) according to the manufacturer's instructions.
Transient transfection
To confirm the role of β-catenin with the underlying mechanism, the transient overexpression of β-catenin was conducted. The pCDNA3.1 empty vector (EV) and pCDNA3.1 β-catenin plasmid construct23 (1 μg) were transfected by PolyJet™ reagent (SignaGen Laboratories, Frederick, MD, USA) into a 6-well culture plate according to the manufacturer's instruction. After transfection, the transfected cells were replaced with a fresh culture medium and left to incubate for an additional 6 h. Following this incubation period, the cells were subjected to various treatments in a serum-free medium for 24 h. Cells were collected and analyzed by using Western blot analysis.
Dual luciferase activity
The luciferase activity was carried out using a dual luciferase reporter assay kit (Promega). The wild-type binding site of TCF/LEF (TOP) and mutant binding site (FOP) plasmids were a generous gift from Dr. Eek-Hoon Jho (University of Seoul, Seoul, South Korea). The TOP/FOP plasmid constructs (500 ng) and pRL-null control construct (50 ng) were transfected using PolyJet™ reagent (Signagen) into a 24-well plate. Then, cells were treated for 24 h and detected the luminescence activity as described in the manufacturer's instructions. The dual luciferase activity was detected using a BioTek Synergy HTX Plate reader (Biotex) normalized to the Renilla luciferase activity. Data were expressed as the relative TOP/FOP activity and compared to the DMSO control.
Protein synthesis assay
To investigate the protein synthesis rate, cells were plated at a density of 1 × 104 cells/well into a 96-well culture plate and allowed to incubate overnight. Subsequently, the cells were treated using a completed medium containing either 0.1% DMSO, ER 20 μM, or 10 μM cycloheximide (CHX) (Sigma Aldrich, St. Louis, MO, USA). The latter served as the positive control for the protein synthesis assay. After 24 h of incubation, Click-iT™ HPG Alexa Fluor™ 594 Protein Synthesis Assay Kit (Cat. #C10429, Life Technologies) was used to measure the protein synthesis data according to the manufacturer's protocol. Imaging and analysis were performed by the CellInsight™ CX7 High Content Screening (HCS) Platform (Thermo Fisher Scientific, Waltham, MA, USA). The mean HPG fluorescence intensity (MFI) was analyzed and compared to the vehicle control.
Western blot analysis
Cells (3 × 105 cells) were plated into a 6-well culture plate overnight. Then, cells were washed with PBS once and treated with DMSO or various concentrations of ER (0, 2, and 20 μM) in a serum-free medium and incubated for different time points (1, 3, 6, 12, and 24 h). RIPA buffer (BIOMAX, Seoul, South Korea) containing protease inhibitors (1:200, Cat. # BPI0001) (BIOMAX) was added into cells for cell lysis and collected homogenized proteins at 13,000 rpm for 20 min at 4 °C. Whole-cell lysates were transferred and stored at − 80 °C until analysis. Pierce BCA Protein Assay Kit (Thermo Fisher Scientific, Waltham, MA, USA) measured the protein concentration before loading 30–50 μg of protein into the sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS-PAGE). The SDS-PAGE was transferred to a polyvinylidene fluoride (PVDF) membrane. The membrane was blocked with 1% BSA for 1 h at RT then primary antibodies were incubated overnight at 4 °C. The primary antibodies used in this study include anti-β-catenin (1:1,000, Cat. #610154) (BD Biosciences, Franklin Lakes, USA), anti-c-Myc (1:1000, Cat. #5605P), anti-p-Akt (S473) (1:1,000, Cat. #9271), anti-Akt (1:1,000, Cat. #9272), p-GSK-3α/β (Ser21/9) (1:1,000, Cat. #9331), anti-p-β-catenin (Ser33/Ser37/T41) (1:1,000, Cat. #9561P), anti-p-β-catenin (Thr41/Ser45) (1:1,000, Cat. #9565P), anti-p-β-catenin (S552) (1:1000, Cat. #9566P), anti-USP9x (1:1,000, Cat. #5751), anti-p-4EBP1 (Thr37/46) (1:1,000, Cat. #2855) (Cell Signaling Technology (Danvers, MA, USA). Anti-cyclin D1 (1:1000, Cat. #sc-8396), anti-α-tubulin (1:200, Cat. #sc-8035), anti-lamin B1 (:200, Cat. #sc-374015), anti-USP47 (1:1,000, Cat. #SC-100633), and anti-β-actin-HRP (C4) (1:5,000, Cat. #sc-47778) (Santa Cruz Biotechnology, Dallas, TX, USA). The membrane was washed with Tris-buffered saline containing 0.1% Tween® 20 detergent (TBST) buffer three times and followed by adding a Goat anti-rabbit secondary antibody HRP conjugated (1:5000, Cat. #31460) or Rabbit anti-Mouse secondary antibody HRP conjugated (1:5000, Cat. #31450) (Invitrogen, Rockford, USA). The membrane was probed for 1 h at RT. The chemiluminescence signal was detected under the Alliance Q9 mini (Cambridge, UK) using SuperSignal™ ECL substrate reagents (Thermo Fisher Scientific, Rockford, IL, USA). Image J software analyzed Imaging and data to calculate the relative protein expression.
Antibody array
The metabolic antibody array was performed to examine the effect of ergosterol on breast cancer metabolism. MDA-MB-231 cells were treated with 0.1% DMSO or ER 20 μM in a serum-free medium for 24 h. Then, cells were collected, and the 200 μg of protein lysates were loaded to the RayBio C-Series Human Metabolic Pathways Antibody Array 2 (RayBiotech, Peachtree Corners, GA, USA). The sample preparation and detection followed the manufacturer's protocol. Data were analyzed by Image J software and clustered the metabolic pathway with the downregulated proteins by using the STRING database (https://string-db.org/).
Statistical analysis
Data were shown as mean ± standard derivation (S.D.) of three independent experiments. The statistical analysis was analyzed by GraphPad Prism 9.2.0 using unpaired t-test or One-way ANOVA with Dunnett's post-hoc analysis. The p-value (p < 0.05*; p < 0.01**) was considered statistically significant.
Results
Ergosterol inhibited the cell viability, cell cycle progression, and spheroid formation of breast cancer cells
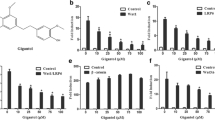
Human breast cancer cells (MCF-7 and MDA-MB-231) were treated with various concentrations of ergosterol (ER 2.5–40 μM) for 1, 2, and 4 days. The cell viability was investigated and found that ER could significantly affect cell survival rate in both cell lines in a dose-dependent manner (Fig. 1a). The cell survival rate of MCF-7 cells decreased, with a half-maximal inhibitory concentration (IC50) of 6.70 ± 1.51 μM and 9.52 ± 5.77 μM observed after 1 and 2 days of incubation periods, respectively. In the case of the MDA-MB-231 cell line, cell viability was significantly reduced to 71.12 ± 4.18% after 1 day of treatment with 20 μM ER. The IC50 value was determined to be 15.55 ± 6.84 μM after 2 days. To explore the further biological effects of ER on cell growth arrest, we opted to use aconcentration of 20 μM for 24 h. As shown in Fig. 1b, the cell cycle analysis revealed that MCF-7 and MDA-MB-231 cells treated with 20 µM of ER demonstrated a substantial cell cycle arrest at the G0/G1 phase after 24 h treatment. The cell cycle regulation is the crucial circuit for cell proliferation and sustains the cancer progression. Hence, we investigated the ability of ER to inhibit spheroid formation in the presence and absence of Matrigel. Cells were allowed to form spheroids for three days before being exposed to the treatment. In spheroids without Matrigel, both MCF-7 and MDA-MB-231 spheroids exhibited limited proliferation in terms of size when treated with 20 μM of ER, as observed for 7 days (Fig. 1c). In the context of the spheroids with 2.5% Matrigel, the typical outcome involved the formation of spherical scaffolds. Our result indicated that treatment with 20 µM of ER resulted in a significant reduction in the volume of the tumor spheres after 7 days, compared to the control group treated with DMSO (Fig. 1d). This suggests that ER has a suppressive effect on the growth of tumor masses in both cell types.

The inhibitory effect of Ergosterol (ER) against MCF-7 and MDA-MB-231 breast cancer cells. (a) The cell viability of MCF-7 and MDA-MB-231 was screened with various concentrations of ER (2.5–40 μM) by MTS assay for 24, 48 and 96 h. (b) The cell cycle assay was analyzed by flow cytometer with propidium iodide staining. The representative histogram of cell phase distribution was displayed as G0/G1, S, and G2/M phases after DMSO control and ER 20 μM treatment for 24 h. (c) The spheroids without Matrigel with DMSO or ER 20 μM treatment after 1, 3, 5 and 7 days were photographed and analyzed for the spheroid diameter (scale bar: 2500 μm). (d) The spheroids were embedded in 2.5% Matrigel and treated with DMSO or ER 20 μM. Spheroids were performed five wells per condition and photographed after 1, 3, 5, and 7 days of incubation (scale bar: 2500 μm), and the mass intensity was analyzed by Image J software. Data were expressed as mean ± S.D. of three independent experiments. The statistical significance was analyzed and compared to the DMSO control by One-way ANOVA, and Dunnett’s post-hoc analysis with P-value (P < 0.05 *; P < 0.01 **) was considered a significance.
Ergosterol suppresses the nuclear translocation of β-catenin and its downstream targets
The Wnt/β-catenin pathway is involved in breast cancer proliferation, stemness, and metastasis7. To investigate the effect of ER on β-catenin signaling, β-catenin and its downstream targets, such as c-Myc and Cyclin D1, were examined. Western blot analysis showed that ER (2 and 20 μM) treatments after 24 h significantly inhibited c-Myc and cyclin D1 protein expression in both MCF-7 and MDA-MB-231 (Fig. 2a). Although the β-catenin seems to be marginally downregulated by ER, the β-catenin accumulation in the nucleus was dramatically decreased in the ER treated samples as assessed by the immunofluorescence staining of β-catenin in MCF-7 and MDA-MB-231(Fig. 2b). The effect of ER on nuclear translocation was confirmed the β-catenin expression between cytoplasm and nucleus by Western blot (Fig. 2c). These data indicated that ER had the ability to diminish the translocation of β-catenin and its subsequent downstream targets.

Ergosterol inhibited the β-catenin signaling pathway. (a) The effects of ER treatment (2 and 20 μM) were assessed by conducting Western blot analysis on β-catenin, c-Myc, and cyclin D1 after a 24 h period. (b) The translocation of β-catenin to the nucleus was evaluated using immunofluorescence staining after subjecting cells to 24 h treatments with DMSO or ER 20 at μM. Co-localization of β-catenin and the nucleus was visualized using confocal microscopy at a magnification of 200X, aided by the application of Hoechst 33,342 for staining the nuclei. A scale bar representing 50 μm was included. The nuclear translocation was quantified using Pearson’s correlation coefficient and analyzed using Image J software with the JACoP plugin. (c) The verification of β-catenin translocation was accomplished through the separation of cytoplasmic and nuclear fractions. Western blot analysis of β-catenin was conducted on these fractions, employing Lamin B1 and α-tubulin as internal controls for nuclear and cytoplasmic compartments. The presented data represents the mean ± S.D. from three independent experiments. Statistical significance was determined by unpaired t-tests or One-way ANOVA, followed by Dunnett’s post-hoc analysis. Significance levels were denoted by asterisks (* for P < 0.05, ** for P < 0.01). These analyses were carried out to compare the results with the DMSO control. The original images for western blot analysis were shown in “Supplementary information”.
Ergosterol affected the β-catenin signaling pathway by inhibiting AKT/GSK-3β/β-catenin axis
The activation of β-catenin by AKT causes β-catenin accumulation and nuclear translocation24. To explore the AKT signaling pathway regulated by ER, a range of time points (3–24 h) were examined following ER treatment (20 µM) to elucidate the signaling cascade in greater detail. The results demonstrated that ER significantly suppressed the phosphorylation of Akt (S473) from 6 to 24 h in both MCF-7 and MDA-MB-231 cell lines (Fig. 3a). Consequently, the six-hour time point was selected for examining GSK-3 β/β-catenin phosphorylation. The Wnt/β-catenin activation depends on the homeostasis of β-catenin phosphorylation and ubiquitination-dependent proteolysis consisting of adnomatous polyposis coli (APC), Axin, and glycogen synthase kinase-325. The phosphorylation at S33, S37, T41, and S45 of β-catenin was recognized by the GSK-3 complex and promoted β-catenin degradation. In contrast, the phosphorylation at residue S552 responds to β-catenin stabilization. Our data demonstrated that ER (2 and 20 μM) could inhibit the phosphorylated Akt (S473), phosphorylated β-catenin (S33/S37/T41 and T41/S45), and phosphorylated GSK-3β (S9) in a dose-dependent manner on MCF-7 cells. In contrast, MDA-MB-231 cells exhibited significant inhibition in Akt phosphorylation at S473 and phosphorylated GSK-3α/β at S21/S9. Additionally, there was a slight induction in phosphorylated β-catenin at T41/S45, although this change was not statistically significant (Fig. 3b,c). These results indicate that ER could promote β-catenin degradation by inhibiting AKT/GSK-3β phosphorylation.

Ergosterol inhibited the phosphorylation of AKT/GSK-3 β/β-catenin. (a) The effect of ER 20 μM on AKT phosphorylation in a time-dependent manner after 3, 6, 12, and 24 h. The western blot analysis of phospho-Akt (S473), phospho-β-catenin (T41/S45), (S33/S37/T41), (S552) and phospho-GSK-3α,β (S21/S9) was performed and analyzed after treated with ER (2 and 20 μM) for 6 h. Total Akt and β-actin were used to normalize the protein expression on (b) MCF-7 and (c) MDA-MB-231, respectively. Data were expressed as mean ± S.D. of three independent experiments. The statistical significance was analyzed and compared to the DMSO control by unpaired t-test or One-way ANOVA. Dunnett’s post-hoc analysis with P-value (P < 0.05 *; P < 0.01 **) was considered significant. The original images for western blot analysis were shown in “Supplementary information”.
Ergosterol downregulated β-catenin activity
To gain deeper insights into the impact of ER on β-catenin, the overexpression experiment with wild-type β-catenin construct was conducted. The results revealed that the overexpression of wild-type β-catenin significantly induced the expression of c-Myc. However, ER treatment was unable to suppress c-Myc expression back to control levels in both MCF-7 and MDA-MB-231 cell lines (Fig. 4a,b). Immunofluorescence data also demonstrated the overexpression of β-catenin did not alter the ratio of nuclear/cytoplasm β-catenin after ER treatment (Fig. 4c). Finally, the transcriptional activity of β-catenin was investigated by TOP/FOP flash reporter assay. ER 20 μM treatment in breast cancer cells showed that ER diminished the transcription activity of the promoter containing the TCF/LEF site (Fig. 4d). This suggests that ER modulates β-catenin activity, resulting in the suppression of gene regulation at the TCF binding sites.

Ergosterol inhibited β-catenin movement and transcriptional activity. The overexpression of empty vector (EV) and wild-type β-catenin were transfected into (a) MCF-7 and (b) MDA-MB-231 cells. After 24 h of DMSO or ER 20 μM treatments, the β-catenin and c-Myc protein expression were detected by Western blots. (c) The β-catenin expression of transfected cells with β-catenin expression vector was visualized under confocal microscopy at 200X magnifications. The scale bar indicates 50 µM. The nuclear translocation of β-catenin was measured using Image J software. (d) The transcriptional activity of the β-catenin was performed by TOP/FOP flash luciferase reporter system with DMSO or ER 20 μM treatments after 24 h of incubation period. Data were expressed as mean ± S.D. of three independent experiments. The statistical significance was analyzed and compared to the DMSO control by unpaired t-test or One-way ANOVA. Dunnett’s post-hoc analysis with P-value (P < 0.05 *; P < 0.01 **) was considered significant. The original images for western blot analysis were shown in “Supplementary information”.
Ergosterol affects protein synthesis and ubiquitination
Protein synthesis is the critical step for cancer cell growth and progression. The protein synthesis assay kit was employed to examine the correlation between protein synthesis and ubiquitination influenced by ER. Cycloheximide (CHX) was used as the positive control for the protein synthesis assay. Results showed that ER 20 μM and CHX 10 μM treatment after 24 h inhibited the overall protein in both breast cell lines (Fig. 5a). Furthermore, phosphorylated-4EBP1, a marker of protein synthesis from AKT/mTOR pathway, was reduced after ER treatment (Fig. 5b). The stabilization of β-catenin engages with proteins within the ubiquitin–proteasome system, notably USP47 and USP9x proteins. These proteins have been reported to play a role in β-catenin stabilization and are associated with poor cancer prognosis. Results from the study showed that ER could inhibit the USP47 and USP9x expression in both cell lines (Fig. 5c). These data supported that ER suppressed protein synthesis and downregulated the key proteins in β-catenin stabilization in the cytoplasm.

The inhibitory effect of Ergosterol on protein synthesis and proteasome degradation of breast cancer cells. (a) The protein synthesis assay using Click-iT™ HPG Alexa Fluor™ 594 Protein Synthesis Assay Kit was measured by the CellInsight™ CX7 High Content Screening (HCS) Platform (Thermo Fisher Scientific, Waltham, MA, USA). The mean fluorescence signal was analyzed after being treated with DMSO, ER 20 μM, or cycloheximide 10 μM into MCF-7 and MDA-MB-231 for 24 h. (b) Western blot analysis of the phosphorylation form of 4E-BP1, an initiator of protein synthesis, and (c) the ubiquitin–proteasome system proteins (USP47 and USP9x) after being treated with ER (2 and 20 μM) for 24 h. Data were expressed as mean ± S.D. of three independent experiments. The statistical significance was analyzed and compared to the DMSO control by One-way ANOVA, and Dunnett’s post-hoc analysis with P-value (P < 0.05 *; P < 0.01 **) was considered significant. The original images for western blot analysis were shown in “Supplementary information”.
Ergosterol altered the metabolic pathway in breast cancer cells
Emerging evidence suggests a strong link between aberrant metabolic pathways and breast cancer development26. The metabolic array analysis was conducted on MDA-MB-231 cells following treatments with DMSO or ER 20 μM for 24 h. The result obtained from the array indicated that 34 out of 41 protein targets were downregulated (Fig. 6a,b). The KEGG enrichment pathway analysis was performed utilizing the STRING database (https://string-db.org/), revealing five primary clusters within the downregulated proteins. These clusters encompassed pathways such as steroid hormone biosynthesis, nitrogen metabolism, sphingolipid metabolism, pyrimidine metabolism, and glycosaminoglycan biosynthesis (Fig. 6c). It has been known that these metabolic pathways are linked to the anti-cancer activity of ER on breast cancer27,28,29,30.

The metabolic antibody array with Ergosterol treatment on MDA-MB-231 cells. (a) After treating MDA-MB-231 cells with DMSO or ER 20 μM for 24 h, whole cell lysates (200 μg) were collected and loaded into the metabolic antibody array, according to the manufacturer’s protocol. The antibody array was visualized using a chemiluminescence detector, representing the protein expression. (b) The internal positive controls were used to normalize the chemiluminescence signal between membranes, and the relative protein profiles were compared to the DMSO control. (c) Downregulated proteins were clustered based on their KEGG enrichment pathways using the STRING database. The clustered node was identified as steroid hormone biosynthesis (yellow), nitrogen metabolism (green), sphingolipid metabolism (navy), pyrimidine metabolism (red), and glycosaminoglycan biosynthesis (blue), respectively.
Discussion
The Wnt/β-catenin pathway plays a significant role in multiple cancer types, such as colon cancer, hepatocellular carcinoma, ovarian cancer, and others. In cases of breast cancer patients, an increase in Wnt/β-catenin activity has been associated with unfavorable prognoses and metastasis10. This pathway is pivotal in cancer progression, as it facilitates the accumulation of β-catenin, its movement into the nucleus, and the subsequent transcription of genes that influence cell proliferation, differentiation, and viability. Thus, β-catenin has emerged as a promising therapeutic target in cancer and has been extensively studied in various types of inhibitors, including small molecules and natural products31.
In this study, we aimed to study the underlying mechanisms of ergosterol, a sterol derived from mushrooms, against human breast cancer cells. Initially, we conducted a comprehensive examination of the impact of ergosterol on cell behavior, including cell survival, cell cycle progression, and spheroid formation (Fig. 1). Our findings indicated that ergosterol exerted a significant influence on breast cancer cell viability, inducing arrest in the G0/G1 phase of the cell cycle and constraining spheroid formation. Furthermore, we delved into the intricate machinery governing the induction of G0/G1 cell cycle arrest, focusing on pivotal regulatory proteins like cyclin D1. In parallel, we explored the Wnt/β-catenin signaling pathway, recognized for its role in regulating cyclin D1. This pathway involves the accumulation of β-catenin within the cytoplasm, followed by its translocation into the nucleus, which complexes with T-cell factor/lymphoid enhancer factor (TCF/LEF) transcription factors32. This intricate interplay ultimately leads to the transcription of key target genes, including c-Myc and cyclin D1. To further delve into the inhibitory mechanism of ergosterol on β-catenin, we performed a Western blot analysis following 24 h of ergosterol treatment. The results exhibited a dose-dependent reduction in the expression of c-Myc and cyclin D1 in response to ergosterol treatment (Fig. 2A). Additionally, ergosterol treatment led to a noticeable decrease in the nuclear translocation of β-catenin, as evidenced by immunofluorescence analysis (Fig. 2B). To corroborate this observation, we conducted cytoplasmic and nuclear fractionation, which confirmed the influence of ergosterol on β-catenin's translocation dynamics (Fig. 2C). The alignment between the immunofluorescence findings and the nuclear subfraction outcomes further indicated that ergosterol's impact on reducing c-Myc and cyclin D1 expression was attributed to its ability to hinder the nuclear translocation of β-catenin.
In the absence of the Wnt ligand, the destruction complex is activated and phosphorylated β-catenin. This is achieved through the coordinated action of Axin, adenomatous polyposis coli (APC), glycogen synthase kinase 3 (GSK-3) and casein kinase 1α (CK1), which form complex and bound to phosphorylated β-catenin at N-terminus33. The modulation of β-catenin's phosphorylation sites affects this complex. Phosphorylation at Ser 45 by CK1 and Ser 33, Ser 37, and Thr 41 by GSK-3β initiates this process34. Subsequently, the phosphorylated β-catenin undergoes ubiquitination, triggering its degradation through proteasomes. Conversely, when the Wnt signaling pathway is activated, GSK-3β is rendered inactive. This leads to the dissociation of the degradation complex, allowing for the release of β-catenin. Existing evidence suggests that phosphorylation of AKT and GSK-3β at Ser 9 leads to the inactivation of GSK-3β. This process can subsequently lead to the phosphorylation of β-catenin at Ser 552, facilitating its translocation into the nucleus and ultimately triggering the activation of genes regulated by β-catenin24. Our findings demonstrate a notable reduction in AKT phosphorylation and GSK-3β levels upon exposure to ergosterol in both MCF-7 and MDA-MB-231 cells. Moreover, the increase in phosphorylation of β-catenin at Ser 33, Ser 37, Thr 41, and Ser 45, corresponding to recognition by the deconstruction complex and subsequent proteasomal degradation, suggests that ergosterol's impact on β-catenin is achieved by hindering the AKT/GSK-3β axis. To further validate the influence of β-catenin signaling on breast cancer progression, we conducted experiments involving the overexpression of β-catenin. Notably, aberrant β-catenin expression led to significant upregulation of c-Myc, and the presence of ergosterol failed to counteract the overexpression of regulatory proteins in breast cancer cells (Fig. 4). This underscores the pivotal role of the β-catenin pathway in breast cancer, with ergosterol's efficacy being contingent on β-catenin-dependent mechanisms. With a focus on the therapeutic targeting of β-catenin signaling in cancer treatment, particular attention has been directed toward assessing candidate molecules within the pathway, specifically concerning the nuclear translocation of β-catenin and proteasome degradation. In our investigation, we delved into the impact of ergosterol on transcriptional regulation by utilizing the TOP/FOP flash reporter assay. Our findings indicate that ergosterol possesses the ability to suppress the transcriptional activity of β-catenin at the binding site of TCF/LEF. This outcome strongly supports the premise that ergosterol effectively impedes the translocation of β-catenin.
Beyond its nuclear translocation, the proteasome degradation process plays a pivotal role in maintaining cellular equilibrium. Under normal circumstances, ubiquitinated β-catenin is identified by the proteasome, a cellular machinery tasked with protein degradation. This orchestrated action results in β-catenin breakdown into smaller peptides, preventing its accumulation within the cell. However, in cancer, certain ubiquitin-specific proteases (USPs) like USP47 and USP9x induce the stabilization of β-catenin, thereby influencing its regulatory functions35,36,37. Our investigation also closely examined the influence of ergosterol on the global protein synthesis of breast cancer cells, along with a key protein synthesis marker, p4EBP1. The outcomes highlight ergosterol's ability to suppress protein synthesis in both cell types. Interestingly, a reduction in the expression of USP47 and USP9x was observed, implying that ergosterol plays a role in diminishing the cytoplasmic stabilization of β-catenin.
Cancer metabolism also plays a role in increased cellular energy demands, biosynthetic needs, and redox balance38. From the metabolic antibody array result, ergosterol influenced the overall metabolism of MDA-MB-231 cells by downregulating critical metabolic pathways, including steroid hormone biosynthesis, nitrogen metabolism, sphingolipid metabolism, pyrimidine metabolism, and glycosaminoglycan biosynthesis (Fig. 6). Previous studies reported that these metabolic pathways are potential therapeutic opportunities for developing targeted treatments28,29,39,40.
Overall, our research offers compelling insights into the intrinsic mechanisms underlying the anti-tumor potential of ergosterol. Notably, ergosterol's anti-tumor efficacy has been previously documented across various models. For instance, in a study focused on rat bladder carcinogenesis, ergosterol exhibited the ability to downregulate the cyclin D1 gene, mitigating its effects20. In the context of breast cancer, ergosterol derivatives derived from Ganoderma lucidum demonstrated the capacity to impede cell cycle progression at the G0/G1 phase, along with targeting Akt, BCL-XL, cyclin D1, and c-Myc, colony formation inhibition21. Additionally, ergosterol sourced from Amauroderma rude inhibits breast cancer cell migration, invasion, colony formation and induces apoptosis by upregulating Foxo3, thereby activating its downstream effectors, Bim and Fas22. Our study reaffirms these earlier findings by consistently demonstrating anti-proliferative, cell cycle-arresting, and spheroid formation inhibition of cancer cells. According to previous studies about ergosterol's effect on cancer, anti-proliferation and metastasis were investigated. Our present study exposed the promising therapeutic effects of ergosterol on cancer stemness by targeting β-catenin pathway. Cancer stemness refers to cancer cells' self-renewal and differentiation ability, which uncontrollably maintains homeostasis. Some types of cancer harbor the mutation of downstream genes such as APC or absence of E-cadherin, a cell protein that bind with β-catenin and maintain cell adhesion, like cholangiocarcinoma41, colon42 and breast cancer43. In breast cancer cells, especially those with triple-negative breast cancer, loss of E-cadherin resulted in β-catenin accumulation in the cytoplasm, nuclear translocation, and EMT process and enhanced the migration and metastasis of cancer43. Our result from ergosterol treatment on breast cancer cells demonstrated the inhibitory effect of ergosterol on β-catenin translocation, downregulation of Wnt/β-catenin targets, and subsequently repressed cell proliferation, and spheroid formation, which reflexed their self-renewal ability, as summarized in Fig. 7. Notably, this study is the first to highlight the effects of ergosterol in the β-catenin signaling pathway.

The summary figure of ergosterol effect via AKT/GSK-3/β-catenin on human breast cancer cells.
In conclusion, our data provide comprehensive insights into the potential of ergosterol as a therapeutic candidate for breast cancer. Through cellular and molecular experiments, the study establishes ergosterol’s ability to inhibit cancer cell growth, suppress β-catenin signaling, and influence metabolic pathways. These findings pave the way for further exploration of ergosterol-based treatments and promise to advance breast cancer therapy.
Data availability
The datasets used and/or analyzed in this study are available from the corresponding authors on reasonable request.
References
Sung, H. et al. Global cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 71, 209–249 (2021).
Lumachi, F., Santeufemia, D. A. & Basso, S. M. Current medical treatment of estrogen receptor-positive breast cancer. World J. Biol. Chem. 6, 231 (2015).
Hortobagyi, G. N. Treatment of breast cancer. New Engl. J. Med. 339, 974–984 (1998).
Howard, F. M. & Olopade, O. I. Epidemiology of triple-negative breast cancer: A review. Cancer J. 27, 8–16 (2021).
Shapiro, C. L. & Recht, A. Side effects of adjuvant treatment of breast cancer. New Engl. J. Med. 344, 1997–2008 (2001).
Hong, D. et al. Epithelial-to-mesenchymal transition and cancer stem cells contribute to breast cancer heterogeneity. J. Cell. Physiol. 233, 9136–9144 (2018).
Geyer, F. C. et al. β-Catenin pathway activation in breast cancer is associated with triple-negative phenotype but not with CTNNB1 mutation. Modern Pathol. 24, 209–231 (2011).
Ram Makena, M. et al. Wnt/β-catenin signaling: The culprit in pancreatic carcinogenesis and therapeutic resistance. Int. J. Mol. Sci. 20, 4242 (2019).
Zhang, Y. & Wang, X. Targeting the Wnt/β-catenin signaling pathway in cancer. J. Hematol. Oncol. 13, 1–16 (2020).
Wang, Z. et al. Clinical implications of β-catenin protein expression in breast cancer. Int. J. Clin. Exp. Pathol. 8, 14989 (2015).
Huang, M., Lu, J.-J. & Ding, J. Natural products in cancer therapy: Past, present and future. Nat. Prod. Bioprospect. 11, 5–13 (2021).
Dupont, S. et al. Antioxidant properties of ergosterol and its role in yeast resistance to oxidation. Antioxidants 10, 1024 (2021).
Xiong, M. et al. Antidiabetic activity of ergosterol from Pleurotus ostreatus in KK-Ay mice with spontaneous type 2 diabetes mellitus. Mol. Nutr. Food Res. 62, 1700444 (2018).
Mbambo, B., Odhav, B. & Mohanlall, V. Antifungal activity of stigmasterol, sitosterol and ergosterol from Bulbine natalensis Baker (Asphodelaceae). J. Med. Plants Res. 6, 5135–5141 (2012).
Sillapachaiyaporn, C., Nilkhet, S., Ung, A. T. & Chuchawankul, S. Anti-HIV-1 protease activity of the crude extracts and isolated compounds from Auricularia polytricha. BMC Complement. Altern. Med. 19, 1–10 (2019).
Sillapachaiyaporn, C., Mongkolpobsin, K., Chuchawankul, S., Tencomnao, T. & Baek, S. J. Neuroprotective effects of ergosterol against TNF-α-induced HT-22 hippocampal cell injury. Biomed. Pharmacother. 154, 113596 (2022).
Takaku, T., Kimura, Y. & Okuda, H. Isolation of an antitumor compound from Agaricus blazei Murill and its mechanism of action. J. Nutr. 131, 1409–1413 (2001).
Chen, S. et al. Anti-tumor and anti-angiogenic ergosterols from Ganoderma lucidum. Front. Chem. 5, 85 (2017).
Wu, H.-Y. et al. Ergosterol peroxide from marine fungus Phoma sp. induces ROS-dependent apoptosis and autophagy in human lung adenocarcinoma cells. Sci. Rep. 8, 17956 (2018).
Ikarashi, N. et al. A mechanism by which ergosterol inhibits the promotion of bladder carcinogenesis in rats. Biomedicines 8, 180 (2020).
Martínez-Montemayor, M. M. et al. Identification of biologically active Ganoderma lucidum compounds and synthesis of improved derivatives that confer anti-cancer activities in vitro. Front. Pharmacol. 10, 115 (2019).
Li, X. et al. Ergosterol purified from medicinal mushroom Amauroderma rude inhibits cancer growth in vitro and in vivo by up-regulating multiple tumor suppressors. Oncotarget 6, 17832 (2015).
Barth, A. I., Stewart, D. B. & Nelson, W. J. T cell factor-activated transcription is not sufficient to induce anchorage-independent growth of epithelial cells expressing mutant β-catenin. Proc. Natl. Acad. Sci. 96, 4947–4952 (1999).
Fang, D. et al. Phosphorylation of β-catenin by AKT promotes β-catenin transcriptional activity. J. Biol. Chem. 282, 11221–11229 (2007).
Pai, S. G. et al. Wnt/beta-catenin pathway: Modulating anticancer immune response. J. Hematol. Oncol. 10, 1–12 (2017).
Wang, L., Zhang, S. & Wang, X. The metabolic mechanisms of breast cancer metastasis. Front. Oncol. 10, 602416 (2021).
Sanderson, J. T. The steroid hormone biosynthesis pathway as a target for endocrine-disrupting chemicals. Toxicol. Sci. 94, 3–21 (2006).
Ogretmen, B. Sphingolipid metabolism in cancer signalling and therapy. Nat. Rev. Cancer 18, 33–50 (2018).
Wang, W., Cui, J., Ma, H., Lu, W. & Huang, J. Targeting pyrimidine metabolism in the era of precision cancer medicine. Front. Oncol. 11, 684961 (2021).
Berdiaki, A. et al. Glycosaminoglycans: Carriers and targets for tailored anti-cancer therapy. Biomolecules 11, 395 (2021).
Liu, D. et al. Small molecules from natural products targeting the Wnt/β-catenin pathway as a therapeutic strategy. Biomed. Pharmacother. 117, 108990 (2019).
Huber, O. et al. Nuclear localization of β-catenin by interaction with transcription factor LEF-1. Mech. Dev. 59, 3–10 (1996).
Hagen, T. & Vidal-Puig, A. Characterisation of the phosphorylation of β-catenin at the GSK-3 priming site Ser45. Biochem. Biophys. Res. Commun. 294, 324–328 (2002).
Liu, C. et al. Control of β-catenin phosphorylation/degradation by a dual-kinase mechanism. Cell 108, 837–847 (2002).
Shi, J. et al. Deubiquitinase USP47/UBP64E regulates β-catenin ubiquitination and degradation and plays a positive role in Wnt signaling. Mol. Cell. Biol. 35, 3301–3311 (2015).
Yang, B. et al. Deubiquitinase USP9X deubiquitinates β-catenin and promotes high grade glioma cell growth. Oncotarget 7, 79515 (2016).
Park, H.-B., Kim, J.-W. & Baek, K.-H. Regulation of Wnt signaling through ubiquitination and deubiquitination in cancers. Int. J. Mol. Sci. 21, 3904 (2020).
DeBerardinis, R. J. & Chandel, N. S. Fundamentals of cancer metabolism. Sci. Adv. 2(5), e1600200 (2016).
Capper, C. P., Rae, J. M. & Auchus, R. J. The metabolism, analysis, and targeting of steroid hormones in breast and prostate cancer. Hormones Cancer 7, 149–164 (2016).
Wei, J., Hu, M., Huang, K., Lin, S. & Du, H. Roles of proteoglycans and glycosaminoglycans in cancer development and progression. Int. J. Mol. Sci. 21, 5983 (2020).
Abuetabh, Y. et al. Expression of E-cadherin and β-catenin in two cholangiocarcinoma cell lines (OZ and HuCCT1) with different degree of invasiveness of the primary tumor. Annals Clin. Lab. Sci. 41, 217–223 (2011).
Disoma, C., Zhou, Y., Li, S., Peng, J. & Xia, Z. Wnt/β-catenin signaling in colorectal cancer: Is therapeutic targeting even possible?. Biochimie 195, 39–53 (2022).
Xu, X., Zhang, M., Xu, F. & Jiang, S. Wnt signaling in breast cancer: Biological mechanisms, challenges and opportunities. Mol. Cancer 19, 1–35 (2020).
Acknowledgements
We would like to give our gratitude to the Second Century Fund (C2F) scholarship from Chulalongkorn University for supporting S.N.’s Ph.D. scholarship and conducting research abroad at SNU. This work was funded by Thailand Science Research and Innovation Fund Chulalongkorn University (HEA663700088) and the 90th anniversary Chulalongkorn University Fund (Ratchadaphiseksomphot Endowment Fund, GCUGR1125652057D). This work was also supported by the National Research Foundation of Korea (NRF) grant, funded by the Korean government (NRF-2018R1A2B2002923 and 2021K2A9A1A2037773).
Author information
Authors and Affiliations
Contributions
S.N. conducted experiments and wrote the original draft, W.V. investigated and analyzed data. P.L. validated and interpreted the data. A.P. and T.T. reviewed and revised the manuscript. S.C. supervised, reviewed and revised the manuscript. S.J.B. supervised, provided resources, reviewed and revised the manuscript. All authors reviewed the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Nilkhet, S., Vongthip, W., Lertpatipanpong, P. et al. Ergosterol inhibits the proliferation of breast cancer cells by suppressing AKT/GSK-3beta/beta-catenin pathway. Sci Rep 14, 19664 (2024). https://doi.org/10.1038/s41598-024-70516-1
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-024-70516-1
- Springer Nature Limited