
Abstract
Huntington’s disease (HD) is a rare neurodegenerative disease caused due to aggregation of Huntingtin (HTT) protein. This study involves the cloning of 40 DnaJ chaperones from Drosophila, and overexpressing them in yeasts and fly models of HD. Accordingly, DnaJ chaperones were catalogued as enhancers or suppressors based on their growth phenotypes and aggregation properties. 2 of the chaperones that came up as targets were CG5001 and P58IPK. Protein aggregation and slow growth phenotype was rescued in yeasts, S2 cells, and Drosophila transgenic lines of HTT103Q with these overexpressed chaperones. Since DnaJ chaperones have protein sequence similarity across species, they can be used as possible tools to combat the effects of neurodegenerative diseases.
Similar content being viewed by others


Introduction
Neurodegenerative diseases are a class of neurological disorders characterized by gradual dysfunctions associated with the loss of neurons. Protein deposition in the human brain is a phenomenon that is fundamental in most neurodegenerative disorders1. A slow continuous loss of neural cells causes neurodegenerative diseases leading to nervous system dysfunction2. These diseases have diverse pathophysiologies ranging from cognitive impairment to difficulties in performing day-to-day tasks. The clinical features are often paired with underlying physiological mechanisms like reduced growth, protein aggregation, misfolding, mitochondrial dysfunction, oxidative stresses, apoptosis, inflammation, and impaired calcium homeostasis3,4,5,6,7.
Our study focuses on slow growth8 and protein aggregation9 as seen in Huntington’s diseased (HD) mutants. It is supported by our observations showing rescue of these phenotypes in presence of DnaJ domain chaperones. We cloned 40 DnaJ domain chaperones from flies and expressed them in yeasts and transgenic flies to check our findings. Huntington’s disease (HD) results from an expansion of CAG repeats on the gene coding for Huntingtin protein on chromosome 4p16.310. Expansion of CAG repeats in the exon 1 of the Huntingtin gene results in addition of Glutamine (Q) stretches exceeding 40, near the N-terminal of the protein leading to pathogenicities associated with HD11,12. Cleavage of mutant HTT releases N-terminal fragment with increased toxicity and aggregation which is one of the many events that lead to the onset of neurodegeneration13. Expanded poly Q stretches cause the HTT protein to misfold and form aggregates. Although there has been a debate over the role these aggregates play with respect to the disease, where aggregates might sequester essential proteins like transcription factors14,15, proteasomes, or other ubiquitin protease system components16,17, poly Q aggregation is emerging as an early and critical step in the pathogenesis of HD18. Hence, one way to alleviate the pathogenicity associated with HD is to decrease the aggregate load. Inhibition of poly Q oligomerization has been shown to preserve normal cellular protein synthesis and degradation functions, and promote clearance of expanded poly Q repeats in vivo and in vitro12,19. It has been observed that in yeasts, the aggregates in HD mutant are correlated to the length of the poly Q repeats, suggesting that aggregation and pathogenicity are dependent on the increase of the poly Q repeat and could be regulated by the expression of chaperone proteins20.
Chaperone proteins play a crucial role in helping disassemble the protein aggregates or target the proteins for degradation. Thus, they act as a backbone of protein quality control checks. These cellular chaperones regulate the folding and maturation of newly synthesized as well as partially folded proteins and resolve the aggregated misfolded proteins21. Heat shock proteins (Hsps) like Hsp 10, Hsp 40, Hsp 60, Hsp 70 and Hsp 110, are specialized chaperones with the ability to control proteostatic stress22. They manage refolding of misfolded and aggregated proteins23,24,25,26.
In this study, we have generated a library of a novel set of putative Hsp40 and related chaperones from Drosophila and used these to screen a HD model in yeast to look for modifiers of HD related phenotypes. Although there have been reports of using the DnaJ library in yeasts to study the aggregation in Huntington’s disease mutants, we undertook this study by using an extensive overexpression screen to complement the previous studies in a quest to exploit the disaggregase properties of additional DnaJ domain proteins. We further tested these genes in the S2 cell line and fly larval hemocytes to check if the phenotypic traits match those effects found in yeasts. Work done on Hsp40 chaperones in yeast Huntington models report that overexpression of the yeast Hsp40 chaperone, like sis1, decreases polyglutamine aggregate size and toxicity27. Overexpression of Ydj1, another yeast Hsp40 chaperone modulates the physical properties of the HD exon 1 aggregates by suppressing the formation of SDS insoluble HD aggregates28. This class of chaperones helps in protein quality control29 and combats stress induced by heat shock or other parameters in yeasts30.
Hsp40s belong to DnaJ chaperone family. J domain of DnaJ chaperones is a 70 amino acid long region conserved across prokaryotes and eukaryotes29. J protein activates the Hsp70 ATPase in the chaperone cycle31 by transferring the substrate protein to the substrate binding domain of Hsp70. This leads to the folding of the protein. J proteins dissociates from Hsp70 and the substrate protein is released. J proteins have three structural classes across species with a compact helical J-domain present in all 3 of these classes29. Type I-J proteins are called DnaJAs, Type II-J proteins are DnaJBs, and the Type III-J proteins are DnaJCs.32,33.
DnaJ proteins have multiple protein domainswhich assist in protein misfoldings in cells in ER stress mediated diseases34. They also contain specific mitochondrial leader sequences or ER signal peptides targeting them to specific cell organelles that help them combat the misfolding stress due to neurodegenerative diseases34.
Another function of DnaJ chaperones is to prevent protein aggregation and solubilize the aggregates when combined with Hsp70 and nucleotide exchange factors34. A classic interplay of Hsp40 and Hsp70 plays a crucial role in protein quality control in neurodegenerative diseases. Hsp40 chaperones interact with Hsp70 protein family and function as cofactors for different substrates. In some cases, Hsp70 interacts with Hsp40 to act as co-chaperones for translating ribosomes35,36. Drosophila has both the heat shock inducible Hsp70s and the constitutively expressing Hsc70s36,37.
Taking cues from here, we chose to screen the chaperones from flies, which were not investigated earlier, with a role in protein folding activity, according to their prediction models. The functional activity of their human orthologs suggests the same37,38. We screened 35 DnaJ domain chaperones from Drosophila which are: Mrj, CG5001, DnaJ60, CG6693, CG2911, JdpC, CG4164, CG9828, CG2887, CG30156, CG12020, CG10565, Wus, Tpr2, Csp, CG7394, CG8476, CG7130, CG7872, Sec63, Hsc20, P58IPK, Droj2, CG43322, CG7133, CG14650, CG7556, CG32641, CG10375, CG11035, CG3061, CG17187, DnaJ1, CG7387 and CG8531) in the Huntington model of yeast. Along with these, 3 Hsc70 chaperones (Hsc70-1, Hsc70-4, and Hsc70Cb) and 2 Hsp70 chaperones (Hsp70Aa, Hsp70Bb) due to redundancy in their sequence with others) were also screened. These chaperones mostly function in protein unfolding activities37,39.
For example, CG5001 and P58IPK are the two Hsp40 chaperones that were explored further after the screening in yeasts. Multiple human Hsp40s are already known to directly modify Htt toxicity and aggregation28,40,41. However, these two chaperones from Drosophila homologs, have not yet been identified with respect to their activity against Htt and thus they can be adapted for use in humans.
CG5001 has a predicted chaperone binding activity and unfolded protein binding activity37.
It is orthologous to several human genes including DnaJB5 (DnaJ heat shock protein family (Hsp40) member B5) and is located in the cytosol42,43. DnaJB5 acts as a novel biomarker and a possible target for cervical cancer treatment. Knockdown of DnaJB5 decreases proliferation and increases apoptosis of cells of uterine tissue with squamous cell carcinoma. P58IPK is predicted to enable chaperone binding activity and misfolded protein binding activity. It is involved in protein folding in the endoplasmic reticulum and is orthologous to the human DnaJC3 (DnaJ heat shock protein family (Hsp40) member C3). DnaJC3 deficiency in mice causes pancreatic β-cell loss and diabetes. Loss of function mutations in DnaJC3 result in multisystemic neurodegeneration and early-onset diabetes44,45.
HD model in Saccharomyces cerevisiae is developed, by expressing 25Q and 103Q constructs with 25 and 103 CAG repeats respectively46. Similarly, mutants of ALS were made by introducing galactose-inducible plasmids (pRS426 Gal-TDP-43-GFP47 and pAG416 GalFUS-YFP48 expressing TDP43 and FUS in S. cerevisiae.
25Q serves as the control whereas the mutant 103Q expresses the Huntington phenotype46. 25Q grows normally with the protein being diffused throughout the cell cytoplasm. We have chosen the HTT mutant with 103 CAG repeats as it shows growth defects at permissive temperature associated with protein aggregation, but is not lethal. Protein aggregation characteristics of the disease intrigued us to check if the chaperones would play specific roles in disease management. To address the possible pathogenic conditions of HD we looked for potential Hsp40 DnaJ chaperones, which would either aggravate or ameliorate the slow growth and protein aggregation phenotype. This led us to perform an overexpression screen of DnaJ domain proteins of flies and expressing them in the yeast HD diseased mutant (HTT103Q).
The DnaJ domain library being derived from flies, we decided to look at their effects, on HD models of S2 fly cell line and transgenic Drosophila, following the hits obtained from the initial yeast screening data. The fruit fly Drosophila melanogaster has been used extensively for studying the pathophysiology of neurodegenerative diseases49,50, where molecular chaperones have been used to suppress neurodegeneration in Drosophila51,52.
To investigate the mechanism associated with the HD phenotype, we focussed on the two modifiers CG5001 and P58IPK which emerged as potential candidates. The chaperones CG5001 and P58IPK reduced protein aggregation seen in the HD mutant and partially rescued the slow growth phenotype in yeasts validating the effect of these chaperones in the Drosophila system, will help in understanding the disease pathology of neurodegenerative diseases.
In this report we have also extended the work to the study of another neurodegenerative disease, Amyotrophic Lateral Sclerosis (ALS), which involves a continuous degradation of motor neurons53
Results
Expressing Drosophila Hsp40 and Hsp70 proteins in 25Q and 103Q in yeast
To check for related toxicity in HD, we used 25Q as control and 103Q repeats as HD mutants. We transformed them in yeasts with appropriate plasmids following already published protocols52,54,55. These constructs have a Ura marker and a GFP tag at the C-terminal. The poly Q repeats were under the influence of a galactose-inducible promoter. Huntington’s mutation resulted in slow growth and protein aggregation in yeasts. 25Q repeats did not show lethal phenotype of the HD mutation56. In the 103Q mutant, the growth was slowed down as seen in Supplementary Fig. S2A(a). Growth curves of 25Q and 103Q depicted 103Q growing slower as compared to 25Q based on optical density values obtained from 3 independent biological replicates of each, over a period of 48 h (Supplementary Fig. S2A(b)) (n = 3, p < 0.05, paired t test). Area under the curve, analysis of growth curves of 25Q and 103Q also showed reduced values for 103Q as compared to 25Q (p < 0.05, 1 sample t test) (Supplementary Fig. S2A(c)). Simultaneously, we wanted to check if the Huntington mutant showed any kind of aggregation due to the repeat units. The 25Q strain showed diffused cytoplasmic GFP expression, but the 103Q mutant showed distinct cytoplasmic GFP punctae under confocal microscope, 24 h post induction, of the strain with galactose (Supplementary Fig. S2A(d)). The cells expressing GFP punctae were classified into 3 morphologies, namely multiple punctae phenotype, diffused cytoplasm phenotype, and diffused cytoplasm with punctae. Percentage of cells showing punctae had increased in 103Q whereas percentage of cells showing diffused cytoplasm had decreased in 103Q as compared to 25Q (p < 0.05, 2 way ANOVA) (Supplementary Fig. S2A(e)).
The 103Q mutant strain displayed slow growth and protein aggregation. Taking these into consideration, we screened the HD mutant against a DnaJ domain library of chaperones and co-chaperones to check the growth and protein aggregation phenotype.
Classification of Drosophila chaperones as enhancers or suppressors of HD phenotypes following growth assays in yeast
40 DnaJ domain chaperones and co-chaperones from Drosophila were screened for their effects on HD mutant. Initially, we checked by serial dilution assay where mutant 103Q was transformed individually with these 40 chaperones. The mutant strain was transformed with DnaJ domain chaperones and co-chaperones with an HA epitope tag at the C-terminal. The transformed colonies were selected on media lacking Uracil and Tryptophan (SD Trp- Ura-). These colonies were then serially diluted and spotted onto SD Trp- Ura- and SGal Trp- Ura- plates. Plates with Dextrose (SD) served as control, whereas galactose plates were observed for growth, since the HTT plasmids were under the effect of a galactose-inducible promoter.
Chaperones that made the Huntington mutant grow slower in comparison to the empty vector Luciferase in SGal Trp- Ura- media enhanced the effect of the mutation, hence these were collectively referred to as enhancers. The chaperones that made the HTT mutant grow better than empty vector Luciferase on the SGal Trp- Ura- plates were termed as suppressors of the mutation, since they were suppressing the slow growth phenotype of the mutant. Chaperones P58IPK, CG5001, CG7556, Sec63, CG7133 suppressed the mutation and the chaperones CG12020, CG8531, CG9828, CG43322, CG11035 enhanced the mutation. The results on SGal Trp- Ura- plates are shown in Fig. 1a whereas the rest of the double mutants’ spot dilution assays are shown in Fig. 2. Plates with dextrose medium that served as controls are shown in Supplementary Fig. S3. It is to be noted 25Q HD mutant, in presence of the chaperones, show no change in SD-trp-ura and Sgal-trp-ura media (Suppl. Figs. S4, S5). At the same time wild type strains transformed with selective chaperones also show no change in growth in either dextrose or galactose media Supplementary Fig. S2B. 25Q HD mutant in presence of the chaperones show no change in SD-trp-ura and Sgal-trp-ura media Supplementary Figs. S4, S5. At the same time wild type strains transformed with selective chaperones also show no change in growth in either dextrose or galactose media (Supplementary Fig. S2B).

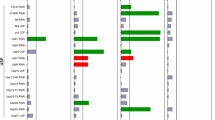
Effect of DnaJ chaperones on the growth of Huntington’s mutant. (a) DnaJ domain chaperones transformed in Saccharomyces cerevisiae model of Huntington’s mutant 103Q serially diluted and spotted on SGal Trp- Ura- plates. 103Q with empty vector Luciferase serves as the control. Others are shown in Fig. 2. (b) 48 h growth assays of mutant 103Q transformed with DnaJ chaperones. Data shown is obtained via 3 biological replicates (n = 3, p < 0.05 paired t test). The blue line indicates the growth of 103Q with empty vector Luciferase whereas the red line indicates the growth curve of 103Q with respective chaperones. Others are shown in Fig. 3a. (c) Classification of DnaJ chaperones as enhancers and suppressors based on area under the curve values of growth curves, determined using GraphPad prism software. Values of area under the curve were converted to a logarithmic scale and were normalized with the slope value of Luciferase. Chaperones to the right of Luciferase are suppressors whereas the ones to the left are enhancers of the mutation. (d) Classification of DnaJ chaperones as enhancers and suppressors on the basis of slope of growth curves determined using GraphPad prism software. Slope values and area under the curve were converted to logarithmic scale and were normalized with the slope value of Luciferase. Chaperones to the right of Luciferase with positive values are suppressors whereas the ones to the left with negative values are enhancers of the mutation.

Effect of chaperones on the growth of mutant (serial dilution assay on galactose medium). Serial dilution assay on SGal Trp- Ura- of remaining Hsp40 chaperones and co-chaperones described in the study (other chaperones are shown in Fig. 1a).
To quantitate the effect of chaperones on the growth of the mutant we monitored a 48 h growth curve. Huntington mutant 103Q with empty vector Luciferase, and those co-expressing the chaperones were induced with 2% galactose, were grown for 48 h. O.D. was measured every 30 min and graphs were plotted using GraphPad software using the data of 3 biological replicates. In Fig. 1b, the blue line indicates the growth curve of HTT103Q with empty vector and the red line denoted growth curve of the mutant in presence of the respective chaperones (n = 3, p < 0.05, paired t test). The area under the curve of all the chaperones including Luciferase was calculated. They were then converted to logarithmic scale. The logarithmic values of chaperones were normalized with the logarithmic values of Luciferase. Chaperones which made the mutant grow faster than empty vector Luciferase showed greater area under the curve in comparison to Luciferase. These were aligned towards the right side of Luciferase with positive values (Fig. 1c). They were categorized as suppressors of the mutation. Chaperones that grew slower than empty vector Luciferase showed a reduced area under the curve. These were aligned on the left side of Luciferase after normalization. These have been shown in Fig. 1c and are collectively termed as enhancers of the mutation. Similarly, the slope values were calculated for all the chaperones to represent the growth rate. In Fig. 1d, suppressors were the ones that had higher slope values than Luciferase and were aligned to the right. Enhancers having reduced slope values than Luciferase were aligned to the left. Growth curves for rest of the 30 chaperones is given in Fig. 3a (n = 3, p < 0.05, paired t test). Classification of remaining 30 chaperones into suppressors and enhancers are based on the area under the curve and slope analysis from the growth curves Fig. 3b,c respectively.

Effect of chaperones on the growth of mutant (growth curve assay). (a) 48 h growth curve assay of the remaining 30 DnaJ chaperones with galactose induction, discussed in the study. Data shown is obtained via 3 biological replicates (Others are shown in Fig. 1b) (n = 3, p < 0.05, paired t test). (b) Classification of these chaperones as enhancers and suppressors based on the area under the curve values of growth curves, determined using GraphPad prism software. Values of area under the curve were converted to a logarithmic scale and were normalized with the slope value of Luciferase. Chaperones to the right of Luciferase are suppressors whereas the ones to the left are enhancers of the mutation. c. Classification of the 30 chaperones as enhancers and suppressors on the basis of slope of growth curves determined using GraphPad prism software. Slope values and area under the curve were converted to a logarithmic scale and were normalized with the slope value of Luciferase. Chaperones to the right of Luciferase with positive values are suppressors whereas the ones to the left with negative values are enhancers of the mutation.
Drosophila chaperones ameliorate mutant HTT aggregation in S. cerevisiae
Mutant 103Q-GFP transformed with chaperones were induced by 2% galactose for switching on the htt genes. The cells were then observed for GFP expression under confocal microscope57. Control 25Q showed GFP diffused all over the cytoplasm whereas mutant 103Q showed punctae of different morphologies, which is an indicator of aggregated proteins. Around 100-150 cells of mutant HTT103Q transformed with each of the chaperones and Luciferase were independently scored. Representative images are shown in Fig. 4a. Based on the confocal images, cells were classified into 3 main morphologies, namely, cells with diffused cytoplasm, cells with punctae, and cells having diffused cytoplasm in addition to punctae. Chaperones, when overexpressed in the mutant strain showed a greater percentage of cells displaying diffused cytoplasm and less cells displaying punctae, when compared to the mutant with empty vector. These were classified as suppressors of the mutation. Whereas, those chaperones that showed less percentage of cells with diffused cytoplasm and more cells with punctae in the chaperone overexpressed strains, were classified as enhancers of the mutation. Here, chaperones CG5001, P58IPK, CG7556, Sec63, and CG7133 are termed as suppressors and CG12020, CG8531, CG9828, CG43322, and CG11035 are the enhancers (Fig. 4b). The remaining chaperones are shown in Fig. 5.

Effect of DnaJ chaperones on protein aggregation of Huntington’s mutant. (a) Protein aggregation in Saccharomyces cerevisiae model of Huntington’s mutant 103Q transformed with DnaJ chaperones. Aggregates were seen in the form of bright green GFP punctae upon inducing the cells with galactose (2%). Others are shown in Fig. 5a. (b) Different morphologies exhibited by cells with GFP tagged mutant Huntington plasmid 103Q and cotransformed with DnaJ chaperones. Around 100 cells were counted for each chaperone. Others are shown in Fig. 5b.

Effect of chaperones on protein aggregation of the mutant. (a) Aggregates were seen as bright green GFP punctae on inducing the cells with galactose (2%). Rest shown in Fig. 4a. (b) Different morphologies exhibited by cells with GFP tagged mutant Huntington plasmid 103Q and co-transformed with DnaJ chaperones. Around 100 cells were counted for each chaperone. The rest are shown in Fig. 4b.
Selection of best fit candidate enhancers and suppressors for Huntington’s phenotype in yeast
Screening of the best fit enhancers and suppressors was done by combining the results from analysis of growth and data derived from imaging techniques. Accordingly, we arrived at these five potential enhancers and suppressors from all the assays taken together:
CG5001 and P58IPK, as seen in Table 1, were selected for further analyses as they were most effective in rescuing aggregation in addition to recovery of the slow growth property of the HD mutant.
CG5001 and P58IPK reduce protein aggregation in yeast
Semi-denaturing detergent agarose gel electrophoresis (SDD-AGE) is used to characterize large polymers which usually cannot pass through the pores of acryl amide gels. Figure 6a showed the 25Q construct with a monomeric band present on the SDD AGE. On the contrary, HD mutant 103Q showed a polymeric smear. In presence of the chaperones CG5001 and P58IPK, smear of 103Q decreased in intensity in comparison to the mutant Huntington (Fig. 6a). This possibly suggested that the insoluble protein aggregation of the mutant was reduced in presence of these chaperones. Figure 6b is the corresponding western blot which showed the expression of the Huntingtin protein tagged with GFP in strains coexpressing mutant 103Q with CG5001 and P58IPK. Chaperones CG5001 and P58IPK were observed to rescue the slow growth and protein aggregation phenotype of HD mutant. On the basis of these observations, one can possibly draw a relation between the selected chaperones and Huntingtin aggregates. To further explore if the chaperones have a direct effect on the Huntingtin protein, a coimmunoprecipitation assay was carried out.

Effect of chaperones CG5001 and P58IPK on protein aggregation using electrophoretic techniques. (a) Representative semi-denaturing detergent agarose gel electrophoresis (SDD-AGE) showing protein aggregation of the mutant 103Q along with its quantitation ratios of intensities of chaperones obtained from densitometric analysis of SDDAGE blots with respect to 103Q with empty vector control of S. cerevisiae (p < 0.0001). (b) Western blot of mutant 103Q with chaperones CG5001 and P58IPK and empty vector. Luciferase probed with anti-GFP antibody. GAPDH loading control is shown. (c) Representative blot of co-immunoprecipitation of chaperones CG5001 and P58IPK with HTT103Q respectively. (d) Separation of soluble (S) (green) and insoluble (I) (yellow) fractions. Soluble and insoluble fractions of mutant 103Q with empty vector Luciferase and chaperones CG5001 and P58IPK was followed by analysis by silver staining. Ratio of intensity of soluble fractions of 103Q with CG5001 (p = 0.0039) and P58IPK (p = 0.0171) with respect to 103Q with Luciferase, and insoluble fractions of 103Q with CG5001 (p = 0.0004) and P58IPK (p = 0.0023) with respect to 103Q with Luciferase has been shown.
Huntington constructs used in this study have a GFP tag. Chaperone proteins are tagged with HA epitope tags. To check the interaction of Huntingtin protein and chaperones, coimmunoprecipitation was done between 103Q-GFP and the respective HA-tagged chaperones CG5001 and P58IPK. HA bands were seen in the input and IP lanes of mutant 103Q with CG5001 and P58IPK whereas the negative controls show no bands in the IP. This possibly gives an indication to the respective chaperones CG5001 and P58IPK interacting directly with the Huntingtin (103Q) protein in yeasts (Fig. 6c).
CG5001 and P58IPK facilitate protein conversion from insoluble to soluble fraction in yeast
Huntingtin aggregates are usually SDS insoluble in nature58. Silver staining of the corresponding soluble and insoluble protein fractions from the SDS PAGE gel was carried out with HTT103Q with Luciferase and in presence of chaperones CG5001 and P58IPK. The intensities of 103Q soluble fractions increased in presence of CG5001 and P58IPK in comparison to Luciferase. The intensities of insoluble fractions decreased in presence of CG5001 and P58IPK as compared to Luciferase (Fig. 6d). This suggests that the insoluble protein aggregates are being converted to soluble proteins in presence of the chaperones CG5001 and P58IPK.
CG5001 and P58IPK reduce protein aggregation in both S2 cells of Drosophila and in transgenic flies
The chaperones CG5001 and P58IPK are fly chaperones that were initially screened in yeast. The data from yeast prompted us to look at their effects in the fly system. For this purpose, we made Drosophila S2 cell line model system and Drosophila larval hemocyte model of HD. The former HD model was constructed using the Drosophila embryonal S2 cell line. HTT 103Q plasmid and chaperones CG5001 and P58IPK were transfected in S2 cells along with the empty vector (So; Sine oculis) using Gal4, DNA and effectene reagent59. 103Q had a GFP tag and chaperone proteins had an HA tag at the C-terminal. S2 cells expressing 103Q with empty vector, and 103Q with CG5001 and P58IPK were fixed with 4% PFA. Cells were blocked with 5% NGS and probed with anti HA antibody. Immunostaining was carried out using secondary antibody tagged with Alexa fluor 555 (RFP)35,60. Upon staining,
Huntington’s aggregates had a GFP tag, and those with chaperones CG5001 and P58IPK appeared red due to the presence of the HA tag (Shown as magenta in Fig. 7a–c). We observed colocalization of the chaperones CG5001 (Pearson’s correlation coefficient r = 0.8) and P58IPK (Pearson’s correlation coefficient r = 1) with the GFP tagged Huntingtin protein. S2 cells expressing HTT 103Q without chaperones showed the presence of green fluorescent punctae due to the presence of insoluble protein aggregation. These green punctae diminished when HTT103Q was cotransfected with the chaperones CG5001 and P58IPK as seen in Fig. 7d. On the other hand, the percentage of cells displaying diffused cytoplasm increased when 103Q was cotransfected with chaperones CG5001 and P58IPK compared to 103Q with the empty vector (Fig. 7e). Percentage of S2 cells displaying morphology of diffused cytoplasm plus punctae decreased when coexpressed with chaperones CG5001 and P58IPK as compared to control (Fig. 7f).

Effect of CG5001 and P58IPK on Huntington mutation 103Q in Drosophila S2 cells. (a) Transfection of 103Q (green) with empty vector So (Sine oculis) having HA tag (magenta) in S2 cells. (b) Mutant 103Q-S2 cells transfected with CG5001 and immunostained with RFP (shown here in magenta) showed colocalization of GFP and RFP (Pearson’s correlation coefficient r = 0.8). (c) Mutant 103Q-S2 cells transfected with P58IPK and immunostained with RFP (shown here in magenta) showed colocalization of GFP and RFP (Pearson’s correlation coefficient r = 1). (d) Representative graph of S2 cells displaying punctae when transfected with 103Q and chaperones CG5001 and P58IPK. Around 100 cells were counted for each experiment. The data shown is of 3 independent assays. (n = 3, p = 0.04 unpaired t test). (e) Representative graph of S2 cells displaying diffused cytoplasm when transfected with 103Q and chaperones CG5001 and P58IPK. Around 100 cells were counted for each experiment. The data shown is of 3 independent assays. (n = 3, p = 0.02 unpaired t test). (f) Representative graph of S2 cells displaying diffused cytoplasm + punctae phenotype when transfected with 103Q and chaperones CG5001 and P58IPK. Around 100 cells were counted for each experiment. The data shown is of 3 independent assays. (n = 3, p = 0.02 unpaired t test). (g) SDD-AGE analysis of mutant 103Q-S2 cells in presence of chaperones CG5001 and P58IPK and 103Q with empty vector (So) probed with anti-GFP antibody along with ratios of intensities of chaperones obtained from densitometric analysis of SDD-AGE blots (p < 0.0001). (h) Western blot of mutant 103Q-S2 cells transfected with CG5001 and P58IPK respectively probed with anti-GFP antibody. GAPDH loading control is shown. (i) Larval hemocytes from fly crosses of 103Q with empty vector (AP2 Sigma) and 103Q with chaperones CG5001 and P58IPK were observed under fluorescence microscope for different phenotypes. Graphs representing percentage of larval hemocytes expressing punctae (n = 7, p = 0.0246, p = 0.0278 unpaired t test) (j), diffused cytoplasm (n = 7, p = 0.0011, p = 0.0001 unpaired t test) (k), and diffused cytoplasm + punctae (n = 7, p = 0.0047, p = 0.0003 unpaired t test). (l) Around 100 cells were counted for each experiment.
S2 cells were lysed, centrifuged, and resuspended in buffer containing SDS61,62. Gel was prepared using 1X TAE and 0.1% SDS. Proteins were transferred to a nitrocellulose membrane using capillary transfer. The blot was probed with anti-GFP antibody. S2 cells show a polymeric smear of 103Q. The smear intensity decreased in presence of CG5001 and P58IPK in comparison to 103Q alone as shown in Fig. 7g (103Q with CG5001 and P58IPK showed a reduction in smear intensity, compared to control. Quantitation shown in Fig. 7g). This suggested reduced protein aggregation in the presence of chaperones. Immunoblots revealed that the Huntingtin protein is expressed at almost the same level in CG5001 and P58IPK constructs in comparison to the control (Fig. 7h).
Following the results of the chaperones CG5001 and P58IPK in Drosophila S2 cell lines, we decided to observe their effects on the protein aggregation in Huntington’s disease mutant flies. For this purpose, a transgene Drosophila line with 103Q was coexpressed with CG5001 and P58IPK respectively. The Cg Gal4 line was used to express the transgenes in the hemocytes using Gal4-UAS system. Larval hemocytes were dissected and observed under a fluorescence microscope. We found three different morphologies of aggregates in the mutant 103Q and those expressing CG5001 and P58IPK respectively (Fig. 7i). Upto 7 independent biological replicates were imaged for quantitation for each set of transgene cross. The number of puncta (in both categories, solely puncta and puncta with diffused cytoplasm morphology) was reduced in HD cell lines with chaperones as compared to HD cell lines with empty vector (AP2 sigma) (Fig. 7j, l), HD cell lines with chaperones showed greater percentage of diffused cytoplasm (Fig. 7k). This indicates reduction of protein aggregation phenotype of HD due to the effect of P58IPK and CG5001. The scores assigned were evaluated using GraphPad, and the results were found to be statistically significant (p < 0.05). The reduction of GFP punctae in S2 cells and larval hemocytes implied a decrease in the aggregates.
CG5001 also rescues growth defects in ALS mutant strains of S. cerevisiae
ALS strains of S. cerevisiae with TDP-43 and FUS showed slow growth in spot dilution assay compared to empty vector Luciferase. When CG5001 was overexpressed in these mutant strains, the slow growth phenotype was partially rescued Supplementary Fig. S7. DnaJB5, the human ortholog of CG5001 needs to be explored further, to find the possible mechanism of suppression of toxicity related to neurodegenerative diseases.
Discussion
Members of the Hsp40 protein family play a role in numerous processes involving protein folding and refolding. Liberek et al., first reported that function of DnaJ proteins is to assist DnaK (bacterial Hsp70) in increasing its ATPase activity. It was previously reported, that DnaJ helps Hsp70 along with DnaK in disaggregation of proteins in vitro54,55,63,64. A study by Acebron et al.59,65 also reports DnaJ working together with DnaK and nucleotide exchange factor GrpE prevents protein aggregation. DnaJ has an essential role in almost all Hsp70 chaperone activities. DnaJ chaperones act as the first blockade against protein aggregation as Hsp40 recruits Hsp70 in stressed conditions. These studies were followed by other researchers to check the toxicity caused due to protein aggregation diseases using DnaJ chaperones. Cytosolic members of DnaJA and DnaJB subfamilies have been reported to suppress Parkin C289G aggregation in Hsp70 dependent manner63,66. Overexpression of DnaJB1 and its yeast homolog Sis1 decreases TDP-43 mediated toxicity by affecting cell growth, cell shape, and ubiquitin–proteasome system inhibition59,67. On the other hand, overexpression of DnaJB2 improves muscle performance and motor neuron survival in mutant SOD1, carried out in in vitro models of ALS66,68.
DnaJ/Hsp40 class actively works against disease toxicity in different models like E. coli, Drosophila melanogaster, Saccharomyces cerevisiae, Mus musculus, and human cell line HEK29340,50,68,69. A study from Drosophila sheds light on Hsp40 and its co-chaperone Hsp70 in vitro, suppressing the formation of large detergent insoluble poly Q HTT aggregates. The activity of Hsp40 and Hsp70 results in accumulation of detergent soluble inclusions. Thus, these chaperones have an ability to shield toxic forms of poly Q proteins and direct them into non-toxic aggregates28. DnaJ1/HDJ1, which is the Drosophila ortholog of human HDJ1 suppresses neurotoxicity in a fly model of HD69,70. Full length DnaJB14 and DnaJB12 both have protective properties against mutant FUS aggregation in an Hsp70 dependent manner. Overexpression of Drosophila DnaJ1 which encodes a protein homologous to human chaperone HDJ1 suppresses poly Q toxicity in fly model of spinocerebellar ataxia type-171,72. Drosophila Hsp40 chaperone Mrj and interacts with Hsp70 proteins and prevents aggregation of pathogenic Huntingtin40. Poly Q toxicity in Saccharomyces cerevisiae is mediated by physical crosstalk within a network of cellular Poly Q and prion like proteins and might be caused by sequestration of crucial nucleolar and mitochondrial proteins72,73. Since HD is also translationally controlled52,73, we hypothesize that these chaperones may possibly play a role in reducing the translational54 and nucleolar defects9,74,75 seen in HD46,76. We wanted to check if the diseased phenotype shown by the HD mutant in yeast would be rescued or aggravated in the presence of overexpression of specific DnaJ domain chaperone proteins. There is a high degree of sequence similarity in the yeast Hsp40 proteins with Drosophila and their mammalian orthologs. Drosophila genome lacks Hsp104 homologs40. Drosophila has 39 Hsp40/DnaJ domain protein genes37, seven Hsp70, and six Hsc70 chaperones35,60. In this study, Hsp40/DnaJ proteins were selected based on their ability to bind unfolded proteins, as reported for their human ortholog counterpart or from predicted modeling studies in Drosophila. Each of the Hsp70 proteins was selected based on their sequence variability from each other. Out of the 40 Hsp40 chaperones and co-chaperones screened, two chaperones CG5001 and P58IPK emerged as potential suppressors of HD related phenotypes in yeast. They showed consistent suppression of the slow growth and protein aggregation phenotype of the HD mutant. The 2 chaperones also enhanced the conversion of insoluble protein fractions to soluble protein fractions in the mutant yeast. Co-immunoprecipitation experiments showed a direct interaction between the chaperones and HTT aggregates. For further validation, we decided to check the effects of these 2 chaperones in the fly system.
Both CG5001 and P58IPK chaperones from Drosophila were selected from a class of DnaJ chaperones as they emerged as potential candidates in rescuing the growth defects and aggregation of the proteins in the screening assay of the yeast model of HD. The hypothetical model has been illustrated in Fig. 8 to show the mechanism of action. CG5001 and P58IPK were found to rescue the protein aggregation phenotype of HD mutant in S2 cells as well as in larval hemocytes of transgenic Drosophila lines. Based on these findings, it can be considered that CG5001 and P58IPK are suppressors that help to control the protein aggregates seen in HD. The human orthologs for CG5001 and P58IPK are DNAJB5 and DNAJC3 respectively77,78,79. It is interesting to note here that DnaJB class of chaperones have been reported to play a crucial role in protection against neurodegeneration by maintaining neuronal proteostasis both in vitro and in vivo61,79.

Hypothetical model. Schematic of the model originating from this work. We hypothesize association of DnaJ domain chaperones with the misfolded proteins that are formed due to repetitive CAG (103Q) repeats in the HD mutant. The misfolded proteins bind with the chaperones CG5001 and P58IPK causing them to act as an inhibitor of protein aggregation related to multiple CAG repeats.
To summarize, our findings from the initial screen in the Huntington mutant in yeast, we can hypothesize that certain DnaJ domain chaperone proteins are effective in reducing the load of aggregated proteins in HD, as observed in other ER stress mediated diseases80. In presence of the chaperones CG5001 and P58IPK we noticed a decrease in the insoluble fraction and an increase of the soluble fraction (Fig. 6d). Out of which, selective ones like CG5001 and P58IPK, with findings from both yeasts and flies show that these 2 candidates have a potential in reducing the possible pathogenicity in HD. Predicted function of CG5001 from modeling studies reveals that it acts as a chaperone cofactor assisting in protein refolding37,38 in the cytosol. CG5001 has a role in ER stress response. PERK is a Protein kinase R like eIF2α kinase which is normally activated in ER stress response. It works by attenuating protein synthesis and reducing ER client protein load. P58IPK is known to inhibit PERK and plays a functional role as a downstream marker of PERK activity in the later phase of ER stress response81,82. This is specifically interesting as it has been shown that Huntingtin is translationally regulated and the translation initiation factor eIF2α is upregulated in the fly cell lines52. Considering these earlier works, we need to explore along these lines to find if other translation associated parameters can be regulated by these two chaperone modifiers in neurodegenerative diseases like Huntington, ALS, Parkinson’s etc.
Multiple sequence alignments of these chaperones reveal that many of these chaperones have protein sequences conserved across species of yeasts, Drosophila, and humans Supplementary Fig. S8. This makes these chaperones a potential target for gene therapy83.
Structural and material properties of HTT fragments of protein in intracellular aggregates can form liquid like reversible assemblies. These assemblies are driven by Huntington’s poly Q tract and proline rich regions. These assemblies are converted to solid fibrillar like structures in cells and in vitro84,85. It can be hypothesized that CG5001 and P58IPK can possibly prevent phase separation in similar model studies.
These chaperones should also be explored further for their ability to manage the effects of other neurodegenerative diseases like ALS, Parkinson's, Spinocerebellar ataxia (SCA), and other toxic Poly Q mutations. Huntington N terminal fragments have been reported to yield different phases based on aggregate morphologies and sizes86. Gaining more insights, into the mechanism of formation of different phases of the Huntington’s aggregates and its association with the chaperone network of proteins will provide a better understanding of the pathophysiology of HD and other neurodegenerative diseases. Since these chaperones also help to reduce ER stress, it can be investigated to check the recovery of ER stress and associated translational defects.
Materials and methods
Cloning of 35 Hsp40, 3 Hsc70, and 2 Hsp70 co-chaperones’ library for expression in yeast and flies
Drosophila Hsp40 and Hsp70 proteins were cloned by Topo-D-Entr cloning kit (Invitrogen) into Topo-D-Entr vector. This was done post PCR amplification using gene specific primers. Constructs were confirmed by sequencing and were transferred to destination vector pUASgHA for Hsp40 and pUASt-ccdB-FLAG (pTWF) for Hsp70. Destination vector pUASg-HA had 3X HA tag at C-terminal51,52,87 and pUASt-ccdB-FLAG (pTWF) had 3X FLAG tag at the C-terminal. This was done for S2 cell based assays using LR clonase. For expression of Hsp40 and Hsp70 in yeast cells, Topo-D-Entr clones were cloned in destination vector pAG424GALccdB-HA with 3X HA tag at the C-terminal43. Protein expression of each isoform was confirmed by immunoblotting using either an anti-HA antibody or an anti-FLAG antibody accordingly.
Construction of yeast 25Q, 103Q, TDP-43 and FUS strains followed by overexpression of DnaJ chaperones and co-chaperones in these strain backgrounds
Bacterial transformation
DH5α cells were made competent using the calcium chloride method. Galactose inducible Huntington plasmid (25Q and 103Q) was added to competent cells. Transformation was done with the help of heat shock method. The transformed cells were plated on LB plates containing ampicillin. ALS plasmids pRS426 Gal TDP-43 GFP and 416 Gal-FUS-YFP were also transformed using similar methods. These are Addgene plasmids numbered 27,476 and 29,593 respectively and are deposited by the Gitler lab.
Plasmid isolation
Transformed bacterial culture pellet was resuspended in solution I containing Glucose (50 mM), EDTA (10 mM), and Tris–Hcl (25 mM). Lysis was done using solution II containing SDS (1%) and NaOH (0.2N). pH was neutralized with solution III containing Potassium acetate (5 M) and Glacial acetic acid. Supernatant was subjected to Phenol: Chloroform: Isoamyl alcohol treatment. Alcohol (100%) was used to precipitate out the DNA.
Yeast transformation
Saccharomyces cerevisiae cells (w303a, MATa, ade2, his3, leu2, trp1, ura3) were grown in a primary culture of YPD (yeast extract, peptone, and dextrose) media until mid-log phase. Cells were resuspended in 100 mM Lithium Acetate (LiOAc) and divided into aliquots which were treated with Polyethylene Glycol (50%), Lithium Acetate (1 M), ssDNA (5ul), and plasmid (5 μl) at 42 °C88. The plasmids used were galactose inducible Huntington’s 25Q, 103Q, TDP43, and FUS with the Ura- marker. Cells were plated on SD Ura- media (yeast nitrogen base 0.67%, Dextrose 2%, agar 2%, and Amino acid mixture lacking Uracil ~ 0.07%)86,89. The media used are from Clontech Laboratories, Mountainview, CA.
Double transformations were carried out in these mutant backgrounds with different DnaJ chaperone plasmids with a Trp- marker. The final plating was done in SD Trp- Ura- media (yeast nitrogen base 0.67%, Dextrose 2%, agar 2%, and Amino acid mixture lacking Uracil and Tryptophan ~ 0.07%)87,88. For every double transformant, 2 representative clones were selected for further assays.
Serial dilution assay
25Q and 103Q strains were grown in SD Ura- medium. They were then washed and grown in Raf Ura- medium (yeast nitrogen base 0.67%, Raffinose (Raf) 2%, and amino acid mixture lacking Uracil ~ 0.07%). Optical Density (O.D.) was adjusted to 0.2 and the culture was serially diluted and spotted on SD Ura- and SGal Ura- plates (yeast nitrogen base 0.67%, galactose 2%, agar 2%, and amino acid mixture lacking Uracil ~ 0.07%). Pictures were taken 24 h post serial dilution assay on galactose medium.
In case of double transformations, at least 2 clones from each of the double transformants of HTT 103Q with the chaperones were spotted onto SD Trp- Ura- and SGal Trp- Ura- medium in presence of HTT 103Q with the empty vector Luciferase.
Growth assay
Double transformants of HTT 103Q yeasts with and without chaperones were grown in SD Trp- Ura-. This was carried out in 3 independent biological replicates with 2 different transformants of Huntington mutant carrying a copy of the different chaperones. Cells were subjected to Raf Trp- Ura- (yeast nitrogen base 0.67%, Raffinose 2%, and Amino acid mixture lacking Uracil and Tryptophan ~ 0.07%) wash and were grown till they reach mid-log phase.
Galactose (2%) was added to the media and cultures were grown in a 96 well plate in triplicates90. O.D. was recorded every 30 min. for 48 h at 30 °C with continuous shaking in the Epoch2 BioTek Spectrophotometer89,90. The data of triplicates was acquired and growth curve was plotted using GraphPad software.
Microscopy
25Q and 103Q Huntington mutant yeast strains were grown overnight in S.D. Ura- medium. O.D. was adjusted to midlog phase in Raf Ura- and induced with galactose (2%). After 24 h, cells were observed under Nikon Ti Eclipse microscope with GFP laser using 60 × and 100 × objectives. All the images were acquired at 5× zoom using 100× plan Apo objective with 1.49 numerical aperture, using the manufacturer’s software. Images were processed using FIJI software88,91. For the HTT103Q strain coexpressing the chaperones, cells were grown in SD Trp- Ura- and Raf Trp- Ura- followed by induction with galactose and were used for further processing. Number of cells showing morphologies relating to variable toxicities were calculated from 3 different biological replicates with cell counts ranging from 100 to 120 in each category. Diffused cytoplasm was considered to be the normal phenotype and multiple punctae was considered to be the toxic phenotype. Cells displaying both were considered to be intermediate on the level of toxicity.
Quantitation of image analysis
Mutant strains transformed with different chaperones, after image acquisition, showed cells displaying morphologies of diffused cytoplasm, punctae, and diffused cytoplasm with punctae.
These were counted from representative images of 3 sets of biological replicates for each strain. Chaperones, when overexpressed in the mutant strain showed a higher percentage of cells displaying diffused cytoplasm morphology and less cells displaying punctae as compared to the mutant with the empty vector. These were classified as suppressors of the mutation. Whereas, those chaperones that showed less percentage of cells displaying diffused cytoplasm and more of the punctae when overexpressed in the mutant were classified as enhancers of the mutation.
SDD-AGE (semi denaturing detergent agarose gel electrophoresis)
Galactose induced yeast cultures were grown till mid log phase. Yeast spheroplasts were made using spheroplasting solution (D-Sorbitol 3 M, MgCl2 1 M, Tris 1 M, β-mercaptoethanol, Zymolyase) Lysis was done using lysis buffer (tris 1 M, β-mercaptoethanol, Benzonase, triton-X, PMSF). Protein was quantified using Bradford’s reagent. Gel was prepared using agarose (1.5%) in 1 × TAE and SDS (0.1%)86,89. Samples were loaded and run at 55 V for around 3 h. Nitrocellulose membrane capillary transfer was set up overnight62,92. Primary antibody used was anti-GFP monoclonal antibody, mouse (Elabscience, catalog number EAB20090, 1:10,000). Secondary antibody used was anti-mouse IgG HRP linked antibody (Cell Signalling Technology, catalog number 7076, 1:10,000) Blot was developed using Pierce™ ECL substrate (Thermo Fisher Scientific, catalog number 32106) and developed using Amersham Imager 600 (GE Healthcare Life Sciences). For SDD AGE of S2 cells, they were lysed using SDS and S2 lysis buffer (Tris Hcl 50 mM, Nacl 150 mM, Nonidet P-40 1%), sonicated, and then loaded in the gel.
Western blot
Galactose induced yeast cultures were pelleted down after 24 h of induction. Cell pellet was lysed via bead beating (lysing matrix C, MP Biomed, catalog number 116912050-CF) using lysis buffer (Tris 50 mM, Nacl 150 mM, Nonidet P-40 0.1%, DTT 1 mM, glycerol 10%, and protease inhibitor cocktail Roche). The protein was equalized using Bradford reagent and separated using SDS PAGE gel (12%) at 120 V for around 1.5 h. The gel was transferred to PVDF (Poly vinylidene fluoride) membrane by electrical transfer at 80 V for around 3 h. Primary antibody used was anti-GFP monoclonal antibody, mouse (Elabscience, catalog number E-AB-20090, 1:10,000). Secondary antibody used was anti-mouse IgG HRP linked antibody (Cell Signalling Technology, catalog number 7076, 1:10,000). Blot was developed using Pierce™ ECL substrate (Thermo Fisher Scientific, catalog number 32106) and Amersham Imager 600 (GE Healthcare Life Sciences)54. For Western blot of S2 cells, they were lysed using SDS and S2 lysis buffer (Tris Hcl 50 mM, Nacl 150 mM, Nonidet P-40 1%), sonicated, and then loaded in the gel.
S2 cell culture and transfection
S2 cells were grown in Schneider’s media supplemented with fetal bovine serum (FBS) (10%). Transfections were done using effectene and gal4 using manufacturer’s protocol90,93. S2 cells were transfected with 103Q with empty vector So (Sine oculis), 103Q with CG5001, and 103Q with P58IPK respectively. Immunostaining: Transfected S2 cells were added to concanavalin A coated dish and allowed to adhere for 10 min. Excess media was removed and cells were fixed with Paraformaldehyde (PFA) (4%) solution. PBST (phosphate buffer saline with tween 20) washes were given to the cells. Cells were blocked with normal goat serum (NGS) (5%) for an hour. Primary antibody anti-HA (mouse, BioLegend, catalog number 901503, 1:5,000) was added to 103Q transfected with CG5001 and with P58IPK and cells were kept overnight. They were washed with PBST thrice, and as secondary antibody, Alexa fluor 555 (RFP) (anti-mouse, Life Technologies, catalog number A21424, 1:5000) was added. Cells were washed again with PBST and were imaged under a Nikon A1 confocal microscope using 60 × and 100 × objectives. In case of chaperones, the 103Q was GFP tagged whereas the antibody was HA specific, which was later stained with RFP. The cells showing GFP and RFP colocalization were observed and quantitated35,68. Pearson’s correlation coefficient (r) was calculated with the help of a colocalization plugin (JaCoP) of FIJI software. GraphPad Prism (9) was used to analyze and plot the data.
Fly crosses (screening chaperone lines against Huntington’s disease in Drosophila melanogaster)
The Gal4/UAS system was used to drive gene expression with the following lines: Cg-Gal4 (BDSC stock no. 7011) and UAS-HttQ103-GFP (a kind gift from Prof. Norbert Perrimon’s lab). The Cg Gal4 was used to express the transgenes in the hemocytes. Virgin females of CgGal4 were crossed with males of the UAS-HttQ103-GFP line. The GFP positive virgin females from the progeny were next crossed with the males of two test chaperone lines, UAS CG-5001 and UAS P58IPK, and a control, UAS- AP2 sigma (HA tag). Hemocyte dissection was carried out in Schneider 2 (S2) cell media in the GFP-positive larvae of each of these crosses and observed under the fluorescence microscope. For fixing, larvae were dissected in Schneider 2 (S2) cell media, plated on a dish, and kept aside for 30 min for the cells to adhere.
The media was then removed and PFA (4%) was added and kept for 15 min. The PFA was then pipetted out and the dishes were washed and stored at 4 °C in PBST (1X). A heterogeneous population was found in the cells. The fields were further quantitated into three categories: cells showing distinct puncta, puncta in a pool of diffused cytoplasm, and cells showing diffused cytoplasm without any puncta. For each trial, around 60–100 cells were observed for each cross.
Co-immunoprecipitation
Protein A lysate (Repligen, catalog number IPA400HC) were blocked with TE (tris EDTA) and BSA (bovine serum albumin) overnight at 4 °C. Anti-GFP purified antibody (mouse, Elabscience, catalog number E-AB-20090) was added to the beads (1:1000) and it was kept for binding overnight at 4 °C. Yeast cell lysate was added to the mixture of beads and antibody for binding overnight at 4 °C. Beads were washed thrice with yeast lysis buffer. Elution was done by the addition of Glycine (pH 2) (0.2 M) and further neutralization was done with Tris (pH 8.8) (1.5 M). The eluates were mixed with loading dye and processed for western blots52,54. A mixture of blocked beads and lysate without the antibody was used as a negative control. The membrane was blocked and probed with anti-HA antibody (rabbit, Proteintech) overnight at 40C and developed using Amersham Imager 600 (GE Healthcare Life Sciences).
Separation of soluble and insoluble yeast HTT103Q fractions
HTT103Q yeast cells were grown till mid log phase for 24 h post galactose induction. Yeast cell pellets were washed with aggregate lysis buffer (ALB). It consists of potassium phosphate (50 mM), EDTA (1 mM), glycerol (5%), PMSF (1 mM), and complete mini protease inhibitor cocktail (Roche). Lysates were then incubated with ALB and zymolase (1 mg/ml) (20 T, MP Biomedicals LLC, catalog number 320921). Lysates were then sonicated, centrifuged and the supernatant was separated as soluble fraction. Pellet was resuspended in ALB containing NP40 and subjected to sonication. Pellet was washed with ALB and resuspended by sonication. It was then centrifuged and the supernatant was stored as the insoluble fraction80.
Silver staining
Soluble and insoluble fractions were run on SDS gel (12%) at 120 V. Gel was washed in water and fixed using fixing solution (methanol 50%, acetic acid 12%, formaldehyde 0.05%) for 1 h. The gel was washed thrice with ethanol (50%) and then washed in sodium thiosulphate (0.8 mM) for 2 min for sensitization with Sodium thiosulfate. Sodium thiosulfate was removed by a water wash and the gel was kept for staining in silver nitrate solution (AgNO3 2 mg/ml, formaldehyde 0.08%) for 15 min in the dark. It was then washed with water and developed using developing solution (Na2CO3 6% w/v, 2% sodium thiosulfate 0.8 mM, and formaldehyde 0.01%). The gel was put in stop solution (methanol 50%, acetic acid 12%) for storage94 (Table 2).
Data availability
The data that support the findings of this study are available in the methods section for the yeast strains and plasmid construction. Some of the plasmids used are commercially available on Addgene47,48,95 and others that have been newly incorporated are available on request to the corresponding authors. ALS plasmids used are pRS426 Gal TDP-43 GFP (Addgene plasmid # 27476) and 416 Gal-FUS-YFP (Addgene plasmid # 29593). Primers were previously used for Topo D Entr cloning of fly chaperones56 and were transferred in yeast vector via LR reaction. These primers are mentioned in the supplementary table S1. Supplementary Fig. S4 is a phylogenetic tree constructed using coding sequences (CDS) of chaperone genes and their respective human and yeast orthologs. Datasets analysed in this figure are available in Flybase and NCBI Gene databases. The gene IDs used for the same are given in Table 2.
References
Kovacs, G. Molecular pathological classification of neurodegenerative diseases: Turning towards precision medicine. Int. J. Mol. Sci. 17, 189. https://doi.org/10.3390/ijms17020189 (2016).
Brown, R. C., Lockwood, A. H. & Sonawane, B. R. Neurodegenerative diseases: An overview of environmental risk factors. Environ. Health Perspect. 113, 1250–1256. https://doi.org/10.1289/ehp.7567 (2005).
Sathasivam, S., Grierson, A. J. & Shaw, P. J. Characterization of the caspase cascade in a cell culture model of SOD1-related familial amyotrophic lateral sclerosis: Expression, activation and therapeutic effects of inhibition. Neuropathol. Appl. Neurobiol. 31, 467–485. https://doi.org/10.1111/j.1365-2990.2005.00658.x (2005).
Borthwick, G. M., Johnson, M. A., Ince, P. G., Shaw, P. J. & Turnbull, D. M. Mitochondrial enzyme activity in amyotrophic lateral sclerosis: implications for the role of mitochondria in neuronal cell death. Ann. Neurol. 46, 787–790. https://doi.org/10.1002/1531-8249(199911)46:5%3c787::aidana17%3e3.0.co;2-8 (1999).
Williams, R. E. et al. Cultured glial cells are resistant to the effects of motor neurone disease-associated SOD1 mutations. Neurosci. Lett. 302, 146–150. https://doi.org/10.1016/S0304-3940(01)01686-X (2001).
Lyras, L. A., Evans, P. J., Shaw, P. J., Ince, P. G. & Halliwell, B. Oxidative damage and motor neurone disease difficulties in the measurement of protein carbonyls in human brain tissue. Free Radic. Res. 24, 397–406. https://doi.org/10.3109/10715769609088038 (1996).
Loncke, J., Vervliet, T., Parys, J. B., Kaasik, A. & Bultynck, G. Uniting the divergent Wolfram syndrome–linked proteins WFS1 and CISD2 as modulators of Ca 2+ signaling. Sci. Signal. 14, 1. https://doi.org/10.1126/scisignal.abc6165 (2021).
Tauber, E. et al. Functional gene expression profiling in yeast implicates translational dysfunction in mutant huntingtin toxicity. J. Biol. Chem. 286, 410–419. https://doi.org/10.1074/jbc.M110.101527 (2011).
Meriin, A. B. et al. Huntingtin toxicity in yeast model depends on polyglutamine aggregation mediated by a prion-like protein Rnq1. J. Cell Biol. 157, 997–1004. https://doi.org/10.1083/jcb.200112104 (2002).
Macdonald, M. A novel gene containing a trinucleotide repeat that is expanded and unstable on Huntington’s disease chromosomes. Cell. 72, 971–983. https://doi.org/10.1016/00928674(93)90585-E (1993).
Cardoso, F. Huntington disease and other choreas. Neurol. Clin. 27, 719–736. https://doi.org/10.1016/j.ncl.2009.04.001 (2009).
Martindale, D. et al. Length of huntingtin and its polyglutamine tract influences localization and frequency of intracellular aggregates. Nat. Genet. 18, 150–154. https://doi.org/10.1038/ng0298-150 (1998).
Wellington, C. L. et al. Caspase cleavage of mutant huntingtin precedes neurodegeneration in Huntington’s disease. J. Neurosci. 22, 7862–7872. https://doi.org/10.1523/JNEUROSCI.22-18-07862.2002 (2002).
McCampbell, A. CREB-binding protein sequestration by expanded polyglutamine. Hum. Mol. Genet. 9, 2197–2202. https://doi.org/10.1093/hmg/9.14.2197 (2000).
Nucifora, F. C. et al. Interference by huntingtin and atrophin-1 with CBP-mediated transcription leading to cellular toxicity. Science 2001(291), 2423–2428. https://doi.org/10.1126/science.1056784 (1979).
Cummings, C. J. et al. Chaperone suppression of aggregation and altered subcellular proteasome localization imply protein misfolding in SCA1. Nat. Genet. 19, 148–154. https://doi.org/10.1038/502 (1998).
Donaldson, K. M. et al. Ubiquitin-mediated sequestration of normal cellular proteins into polyglutamine aggregates. Proc. Natl. Acad. Sci. 100, 8892–8897. https://doi.org/10.1073/pnas.1530212100 (2003).
Bates, G. Huntingtin aggregation and toxicity in Huntington’s disease. The Lancet. 361, 1642–1644. https://doi.org/10.1016/S0140-6736(03)13304-1 (2003).
Sánchez, I., Mahlke, C. & Yuan, J. Pivotal role of oligomerization in expanded polyglutamine neurodegenerative disorders. Nature. 421, 373–379. https://doi.org/10.1038/nature01301 (2003).
Krobitsch, S. & Lindquist, S. Aggregation of huntingtin in yeast varies with the length of the polyglutamine expansion and the expression of chaperone proteins. Proc. Natl. Acad. Sci. 97, 1589–1594. https://doi.org/10.1073/pnas.97.4.1589 (2000).
Vogel, M., Bukau, B. & Mayer, M. P. Allosteric regulation of Hsp70 chaperones by a proline switch. Mol. Cell. 21, 359–367. https://doi.org/10.1016/j.molcel.2005.12.017 (2006).
Vashist, S. & Ng, D. T. W. Misfolded proteins are sorted by a sequential checkpoint mechanism of ER quality control. J. Cell Biol. 165, 41–52. https://doi.org/10.1083/jcb.200309132 (2004).
Joshi, B. S. et al. DNAJB6b-enriched small extracellular vesicles decrease polyglutamine aggregation in in vitro and in vivo models of Huntington disease. Science. 24, 103282. https://doi.org/10.1016/j.isci.2021.103282 (2021).
Månsson, C. et al. Conserved S/T residues of the human chaperone DNAJB6 are required for effective inhibition of Aβ42 amyloid fibril formation. Biochemistry. 57, 4891–4902. https://doi.org/10.1021/acs.biochem.8b00353 (2018).
McMahon, S., Bergink, S., Kampinga, H. H. & Ecroyd, H. DNAJB chaperones suppress destabilised protein aggregation via a region distinct from that used to inhibit amyloidogenesis. J. Cell Sci. 134, 1. https://doi.org/10.1242/jcs.255596 (2021).
Kumar, J., Reidy, M. & Masison, D. C. Yeast J-protein Sis1 prevents prion toxicity by moderating depletion of prion protein. Genetics. 219, 1. https://doi.org/10.1093/genetics/iyab129 (2021).
Gokhale, K. C., Newnam, G. P., Sherman, M. Y. & Chernoff, Y. O. Modulation of prion-dependent polyglutamine aggregation and toxicity by chaperone proteins in the yeast model. J. Biol. Chem. 280, 22809–22818. https://doi.org/10.1074/jbc.M500390200 (2005).
Muchowski, P. J. et al. Hsp70 and Hsp40 chaperones can inhibit self-assembly of polyglutamine proteins into amyloid-like fibrils. Proc. Natl. Acad. Sci. 97, 7841–7846. https://doi.org/10.1073/pnas.140202897 (2000).
Cheetham, M. E. & Caplan, A. J. Structure, function and evolution of DnaJ: conservation and adaptation of chaperone function. Cell Stress Chaperones. 3, 28. https://doi.org/10.1379/1466-1268(1998)003%3c0028:SFAEOD%3e2.3.CO;2 (1998).
Cabrera, M. et al. Chaperone-facilitated aggregation of thermo-sensitive proteins shields them from degradation during heat stress. Cell Rep. 30, 2430-2443.e4. https://doi.org/10.1016/j.celrep.2020.01.077 (2020).
Hageman, J. & Kampinga, H. H. Computational analysis of the human HSPH/HSPA/DNAJ family and cloning of a human HSPH/HSPA/DNAJ expression library. Cell Stress Chaperones. 14, 1–21. https://doi.org/10.1007/s12192-008-0060-2 (2009).
Walsh, P., Bursać, D., Law, Y. C., Cyr, D. & Lithgow, T. The J-protein family: Modulating protein assembly, disassembly and translocation. EMBO Rep. 5, 567–571. https://doi.org/10.1038/sj.embor.7400172 (2004).
Dilliott, A. A. et al. DnaJC7 in amyotrophic lateral sclerosis. Int. J. Mol. Sci. 23, 4076. https://doi.org/10.3390/ijms23084076 (2022).
Gässler, C. S., Wiederkehr, T., Brehmer, D., Bukau, B. & Mayer, M. P. Bag-1M accelerates nucleotide release for human Hsc70 and Hsp70 and can act concentration-dependent as positive and negative cofactor. J. Biol. Chem. 276, 32538–32544. https://doi.org/10.1074/jbc.M105328200 (2001).
Horton, L. E., James, P., Craig, E. A. & Hensold, J. O. The Yeast hsp70 Homologue Ssa is required for translation and interacts with Sis1 and Pab1 on translating ribosomes. J. Biol. Chem. 276, 14426–14433. https://doi.org/10.1074/jbc.M100266200 (2001).
Fontaine, S. N. et al. DnaJ/Hsc70 chaperone complexes control the extracellular release of neurodegenerative-associated proteins. EMBO J. 35, 1537–1549. https://doi.org/10.15252/embj.201593489 (2016).
Gaudet, P., Livstone, M. S., Lewis, S. E. & Thomas, P. D. Phylogenetic-based propagation of functional annotations within the Gene Ontology consortium. Brief. Bioinform. 12, 449–462. https://doi.org/10.1093/bib/bbr042 (2011).
Sinha, D. et al. HSPIR: A manually annotated heat shock protein information resource. Bioinformatics. 28, 2853–2855. https://doi.org/10.1093/bioinformatics/bts520 (2012).
Li, Y. et al. Cloning and analysis of DnaJ family members in the silkworm, Bombyx mori. Gene. 576, 88–98. https://doi.org/10.1016/j.gene.2015.09.079 (2016).
Chuang, J.-Z. et al. Characterization of a brain-enriched chaperone, MRJ, that inhibits huntingtin aggregation and toxicity independently. J. Biol. Chem. 277, 19831–19838. https://doi.org/10.1074/jbc.M109613200 (2002).
Jana, N. R. Polyglutamine length-dependent interaction of Hsp40 and Hsp70 family chaperones with truncated N-terminal huntingtin: Their role in suppression of aggregation and cellular toxicity. Hum. Mol. Genet. 9, 2009–2018. https://doi.org/10.1093/hmg/9.13.2009 (2000).
Hu, Y. DNAJB5 (hsc40) gene as a novel biomarker for cervical cancer. Eur. J. Gynaecol. Oncol. https://doi.org/10.22514/ejgo.2022.023 (2022).
Hageman, J., van Waarde, M. A. W. H., Zylicz, A., Walerych, D. & Kampinga, H. H. The diverse members of the mammalian HSP70 machine show distinct chaperone-like activities. Biochem. J. 435, 127–142. https://doi.org/10.1042/BJ20101247 (2011).
Lytrivi, M. et al. DNAJC3 deficiency induces β-cell mitochondrial apoptosis and causes syndromic young-onset diabetes. Eur. J. Endocrinol. 184, 455–468. https://doi.org/10.1530/EJE-20-0636 (2021).
Gao, D. et al. ERp29 induces breast cancer cell growth arrest and survival through modulation of activation of p38 and upregulation of ER stress protein p58IPK. Lab. Investig. 92, 200–213. https://doi.org/10.1038/labinvest.2011.163 (2012).
Solans, A., Zambrano, A., Rodríguez, M. & Barrientos, A. Cytotoxicity of a mutant huntingtin fragment in yeast involves early alterations in mitochondrial OXPHOS complexes II and III. Hum. Mol. Genet. 15, 3063–3081. https://doi.org/10.1093/hmg/ddl248 (2006).
Johnson, B. S., McCaffery, J. M., Lindquist, S. & Gitler, A. D. A yeast TDP-43 proteinopathy model: Exploring the molecular determinants of TDP-43 aggregation and cellular toxicity. Proc. Natl. Acad. Sci. 105, 6439–6444. https://doi.org/10.1073/pnas.0802082105 (2008).
Sun, Z. et al. Molecular determinants and genetic modifiers of aggregation and toxicity for the ALS disease protein FUS/TLS. PLoS Biol. 9, e1000614. https://doi.org/10.1371/journal.pbio.1000614 (2011).
Rogers, S. L. & Rogers, G. C. Culture of Drosophila S2 cells and their use for RNAi-mediated lossof-function studies and immunofluorescence microscopy. Nat. Protoc. 3, 606–611. https://doi.org/10.1038/nprot.2008.18 (2008).
Fernández-Moreno, M. A., Farr, C. L., Kaguni, L. S., & Garesse, R. Drosophila melanogaster as a model system to study mitochondrial biology. pp. 33–49. https://doi.org/10.1007/978-1-59745-365-3_3 (2007).
Warrick, J. M. et al. Suppression of polyglutamine-mediated neurodegeneration in Drosophila by the molecular chaperone HSP70. Nat. Genet. 23, 425–428. https://doi.org/10.1038/70532 (1999).
Joag, H. et al. A role of cellular translation regulation associated with toxic Huntingtin protein. Cell. Mol. Life Sci. 77, 3657–3670. https://doi.org/10.1007/s00018-019-03392-y (2020).
Robberecht, W. & Philips, T. The changing scene of amyotrophic lateral sclerosis. Nat. Rev. Neurosci. 14, 248–264. https://doi.org/10.1038/nrn3430 (2013).
Mogk, A. Identification of thermolabile Escherichia coli proteins: Prevention and reversion of aggregation by DnaK and ClpB. EMBO J. 18, 6934–6949. https://doi.org/10.1093/emboj/18.24.6934 (1999).
Tomoyasu, T., Mogk, A., Langen, H., Goloubinoff, P. & Bukau, B. Genetic dissection of the roles of chaperones and proteases in protein folding and degradation in the Escherichia coli cytosol. Mol. Microbiol. 40, 397–413. https://doi.org/10.1046/j.1365-2958.2001.02383.x (2001).
Gusella, J. F. & MacDonald, M. E. Molecular genetics: Unmasking polyglutamine triggers in neurodegenerative disease. Nat. Rev. Neurosci. 1, 109–115. https://doi.org/10.1038/35039051 (2000).
Li, L. et al. Real-time imaging of Huntingtin aggregates diverting target search and gene transcription. Elife. 5, 1. https://doi.org/10.7554/eLife.17056 (2016).
Kim, Y. E. et al. Soluble oligomers of PolyQExpanded huntingtin target a multiplicity of key cellular factors. Mol. Cell. 63, 951–964. https://doi.org/10.1016/j.molcel.2016.07.022 (2016).
Park, S.-K. et al. Overexpression of the essential Sis1 chaperone reduces TDP-43 effects on toxicity and proteolysis. PLoS Genet. 13, e1006805. https://doi.org/10.1371/journal.pgen.1006805 (2017).
Desai, M. et al. Mrj is a chaperone of the Hsp40 family that regulates Orb2 oligomerization and long-term memory in Drosophila. PLoS Biol. 22, e3002585. https://doi.org/10.1371/journal.pbio.3002585 (2024).
Smith, H. L., Li, W. & Cheetham, M. E. Molecular chaperones and neuronal proteostasis. Semin. Cell Dev. Biol. 40, 142–152. https://doi.org/10.1016/j.semcdb.2015.03.003 (2015).
Alberti, S., Halfmann, R., & Lindquist, S. Biochemical, Cell biological, and genetic assays to analyze amyloid and prion aggregation in yeast, pp. 709–734. https://doi.org/10.1016/S00766879(10)70030-6.
Kakkar, V., Kuiper, E. F. E., Pandey, A., Braakman, I. & Kampinga, H. H. Versatile members of the DNAJ family show Hsp70 dependent anti-aggregation activity on RING1 mutant parkin C289G. Sci. Rep. 6, 34830. https://doi.org/10.1038/srep34830 (2016).
Farrawell, N. E. et al. Distinct partitioning of ALS associated TDP-43, FUS and SOD1 mutants into cellular inclusions. Sci. Rep. 5, 13416. https://doi.org/10.1038/srep13416 (2015).
Acebrón, S. P., Fernández-Sáiz, V., Taneva, S. G., Moro, F. & Muga, A. DnaJ recruits DnaK to protein aggregates. J. Biol. Chem. 283, 1381–1390. https://doi.org/10.1074/jbc.M706189200 (2008).
Novoselov, S. S. et al. Molecular chaperone mediated late-stage neuroprotection in the SOD1G93A mouse model of amyotrophic lateral sclerosis. PLoS One. 8, e73944. https://doi.org/10.1371/journal.pone.0073944 (2013).
Desai, M., Deo, A., Naik, J., Bose, T., & Majumdar, A. Mrj an Hsp40 family chaperone regulates the oligomerization of Orb2 and long-term memory. https://doi.org/10.1101/2022.10.16.512122.
Mercado, G. & Hetz, C. Drug repurposing to target proteostasis and prevent neurodegeneration: Accelerating translational efforts. Brain. 140, 1544–1547. https://doi.org/10.1093/brain/awx107 (2017).
Chan, H. Y. E. Mechanisms of chaperone suppression of polyglutamine disease: Selectivity, synergy and modulation of protein solubility in Drosophila. Hum. Mol. Genet. 9, 2811–2820. https://doi.org/10.1093/hmg/9.19.2811 (2000).
Kryndushkin, D. S., Smirnov, V. N., Ter-Avanesyan, M. D. & Kushnirov, V. V. Increased expression of Hsp40 chaperones, transcriptional factors, and ribosomal protein Rpp0 can cure yeast prions. J. Biol. Chem. 277, 23702–23708. https://doi.org/10.1074/jbc.M111547200 (2002).
Fernandez-Funez, P. et al. Identification of genes that modify ataxin-1-induced neurodegeneration. Nature. 408, 101–106. https://doi.org/10.1038/35040584 (2000).
Zhao, Y. et al. Comparative analysis of mutant huntingtin binding partners in yeast species. Sci. Rep. 8, 9554. https://doi.org/10.1038/s41598-018-27900-5 (2018).
Eshraghi, M. et al. Mutant Huntingtin stalls ribosomes and represses protein synthesis in a cellular model of Huntington disease. Nat. Commun. 12, 1461. https://doi.org/10.1038/s41467-021-21637-y (2021).
Lee, J., Hwang, Y. J., Ryu, H., Kowall, N. W. & Ryu, H. Nucleolar dysfunction in Huntington’s disease. Biochim. Biophys. Acta (BBA) 1842, 785–790. https://doi.org/10.1016/j.bbadis.2013.09.017 (2014).
Sönmez, A. et al. Nucleolar stress controls mutant Huntington toxicity and monitors Huntington’s disease progression. Cell Death Dis. 12, 1139. https://doi.org/10.1038/s41419-021-04432-x (2021).
Rodríguez-González, C., Lin, S., Arkan, S. & Hansen, C. Co-chaperones DNAJA1 and DNAJB6 are critical for regulation of polyglutamine aggregation. Sci. Rep. 10, 8130. https://doi.org/10.1038/s41598-020-65046-5 (2020).
Juenemann, K., Wiemhoefer, A. & Reits, E. A. Detection of ubiquitinated huntingtin species in intracellular aggregates. Front. Mol. Neurosci. 8, 1. https://doi.org/10.3389/fnmol.2015.00001 (2015).
Gonzaga-Jauregui, C. et al. Exome sequence analysis suggests that genetic burden contributes to phenotypic variability and complex neuropathy. Cell Rep. 12, 1169–1183. https://doi.org/10.1016/j.celrep.2015.07.023 (2015).
Synofzik, M. et al. Absence of BiP Co-chaperone DNAJC3 Causes Diabetes Mellitus and Multisystemic Neurodegeneration. Am. J. Hum. Genet. 95, 689–697. https://doi.org/10.1016/j.ajhg.2014.10.013 (2014).
Hamdan, N., Kritsiligkou, P. & Grant, C. M. ER stress causes widespread protein aggregation and prion formation. J. Cell Biol. 216, 2295–2304. https://doi.org/10.1083/jcb.201612165 (2017).
Tsou, W.-L. et al. The deubiquitinase ataxin-3 requires Rad23 and DnaJ-1 for its neuroprotective role in Drosophila melanogaster. Neurobiol. Dis. 82, 12–21. https://doi.org/10.1016/j.nbd.2015.05.010 (2015).
Yan, W. et al. Control of PERK eIF2α kinase activity by the endoplasmic reticulum stress-induced molecular chaperone P58 IPK. Proc. Natl. Acad. Sci. 99, 15920–15925. https://doi.org/10.1073/pnas.252341799 (2002).
Popiel, H. A. et al. Hsp40 gene therapy exerts therapeutic effects on polyglutamine disease mice via a non-cell autonomous mechanism. PLoS One. 7, e51069. https://doi.org/10.1371/journal.pone.0051069 (2012).
Carmichael, J. et al. Bacterial and yeast chaperones reduce both aggregate formation and cell death in mammalian cell models of Huntington’s disease. Proc. Natl. Acad. Sci. 97, 9701–9705. https://doi.org/10.1073/pnas.170280697 (2000).
Peskett, T. R. et al. A liquid to solid phase transition underlying pathological huntingtin exon1 aggregation. Mol. Cell. 70, 588601.e6. https://doi.org/10.1016/j.molcel.2018.04.007 (2018).
Posey, A. E. et al. Profilin reduces aggregation and phase separation of huntingtin N-terminal fragments by preferentially binding to soluble monomers and oligomers. J. Biol. Chem. 293, 3734–3746. https://doi.org/10.1074/jbc.RA117.000357 (2018).
Bischof, J. et al. A versatile platform for creating a comprehensive UAS-ORFeome library in Drosophila. Development. 140, 2434–2442. https://doi.org/10.1242/dev.088757 (2013).
Ito, H., Fukuda, Y., Murata, K. & Kimura, A. Transformation of intact yeast cells treated with alkali cations. J. Bacteriol. 153, 163–168. https://doi.org/10.1128/jb.153.1.163-168.1983 (1983).
Douglas, P. M. et al. Chaperone-dependent amyloid assembly protects cells from prion toxicity. Proc. Natl. Acad. Sci. 105, 7206–7211. https://doi.org/10.1073/pnas.0802593105 (2008).
Marešová, L. & Sychrová, H. Applications of a microplate reader in yeast physiology research. Biotechniques. 43, 667–672. https://doi.org/10.2144/000112620 (2007).
Huang, D. & Shusta, E. V. Secretion and surface display of green fluorescent protein using the yeast Saccharomyces cerevisiae. Biotechnol. Prog. 21, 349–357. https://doi.org/10.1021/bp0497482 (2008).
Xu, X., Lambrecht, A. D., & Xiao, W. Yeast survival and growth assays, pp. 183–191. https://doi.org/10.1007/978-1-4939-0799-1_13 (2014).
Mosher, J. T. Effectene TM reagent yields high transfection efficiencies with Drosophila melanogaster S2 cells.
Wray, W., Boulikas, T., Wray, V. P. & Hancock, R. Silver staining of proteins in polyacrylamide gels. Anal. Biochem. 118, 197–203. https://doi.org/10.1016/0003-2697(81)90179-2 (1981).
Meriin, A. B. et al. Huntington toxicity in yeast model depends on polyglutamine aggregation mediated by a prion-like protein Rnq1. J. Cell Biol. 157, 997–1004. https://doi.org/10.1083/jcb.200112104 (2002).
Acknowledgements
We would like to thank Dr. Renu Mohan for helpful suggestions on preparing the MS. We would like to thank Prof. Michael Sherman for the 25Q and 103Q plasmids and Dr. Jennifer Gerton for sharing yeast strains. We would like to thank Meghal Desai for the plasmid library construction. We would like to thank Surya Bansi Singh, and Neelanjana Das for their help regarding imaging experiments.
Funding
This work was supported by a research grant from the Department of Biotechnology (DBTIYBA) (BT/09/IYBA/2015/03), (DBT RLS) (BT/RLF/Re-entry/54/2013) to TB and Wellcome Trust India Alliance Intermediate fellowship to AM (IA/I/13/2/501030). AD, SA, and SM are recipients of the Indian Council of Medical Research (ICMR) senior research fellowship (SRF) (5/3/8/79/ITR-F/2020-ITR, 5/3/8/89/ITR-F/2020-ITR, and 45/12/2022HUM/BMS respectively).
Author information
Authors and Affiliations
Contributions
The experiments were designed by TB and AM. The yeast experiments were performed by AD, TB, SM, and SA. The fly experiments were performed by RG and AM. MS was prepared by TB, AM, AD, SM, SA and RG.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Deo, A., Ghosh, R., Ahire, S. et al. Two novel DnaJ chaperone proteins CG5001 and P58IPK regulate the pathogenicity of Huntington’s disease related aggregates. Sci Rep 14, 20867 (2024). https://doi.org/10.1038/s41598-024-71065-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-024-71065-3
- Springer Nature Limited