
Abstract
This study investigated the underlying function and mechanism of genipin in neuroblastoma (NB). Using flow cytometry analysis and cytotoxicity tests, in vitro studies were conducted to assess the effects of genipin on the SK-N-SH cell line. The mechanism of action of genipin was explored through immunofluorescence staining, Western blotting, and caspase-3 activity assays. In addition, we also created a xenograft tumour model to investigate the effects of genipin in vivo. This research confirmed that genipin suppressed cell viability, induced apoptosis, and promoted autophagy, processes that are likely linked to the inhibition of the PI3K/AKT/mTOR signalling pathway. Autophagy inhibition increases the sensitivity of SK-N-SH cells to genipin. Furthermore, combination treatment with a PI3K inhibitor enhanced the therapeutic efficacy of genipin. These results highlight the potential of genipin as a candidate drug for the treatment of NB.
Similar content being viewed by others


Introduction
The cells of origin for neuroblastoma (NB), a common extracranial solid malignant tumour in children, are presumed to be relatively mature but incompletely differentiated precursor cells derived from neural crest tissue. NB can develop at any location along the sympathetic nervous system1,2. More than 7% of malignant tumours in children under the age of 15 are caused by this disease. However, NB contributes to 15% of paediatric cancer mortality. Approximately 90% of NB cases occur in children under the threshold of 10, with 18 months being the median diagnostic age. Patients with NB are classified into different risk groups based on seven prognostic factors, including the age at diagnosis, tumour stage, histological category, tumour differentiation, and molecular changes. The extremely low-risk, low-risk, intermediate-risk, and high-risk groups are among these risk categories. Although patients diagnosed with intermediate- and low-risk NB have an overall survival rate of more than 90%, the total survival rate for the high-risk group remains less than 50%, even with multimodal intensive treatments that include chemotherapy, radiation therapy, immunotherapy, and surgery3,4,5. Furthermore, the standard treatment methods for NB are associated with significant side effects, including severe damage to internal organs, anaemia, impaired fertility, hair loss, stunted growth, and even the development of secondary malignant tumours. As a result, the treatment of NB continues to be an important challenge in paediatric oncology, and new potential treatment strategies must be actively investigated to both mitigate the long-term side effects of conventional treatments and improve the prognosis of high-risk patients who are refractory and relapsed.
Autophagy involves the participation of multiple macromolecules and is a cellular survival mechanism. It captures cytoplasmic components and organelles, transports them to lysosomes, and digests the resulting degradation products to generate new proteins and membranes. This procedure lessens the cellular stress caused by radiation, cytotoxic chemicals, hypoxia, and nutritional restriction6. In cancer, autophagy is a "double-edged sword". Depending on the type of cell and tissue, genetic background, and tumour stage, it can either promote or inhibit the growth of tumours7,8,9,10. Tumour cells occasionally depend more on autophagy than do normal cells to survive. They utilize autophagy as a means to resist drug-induced and γ-ray-induced cell apoptosis11. Autophagy-related genes12 and the PI3K/AKT/mTOR signalling pathway play crucial roles in regulating autophagy13,14. Mammalian target protein of rapamycin, or mTOR, plays a critical role in the regulation of cellular metabolism and the coordination and control of various cellular functions, including survival, proliferation, and autophagy. It is crucial for cancer cells to resist harmful environmental stimuli15,16,17.
The PI3K/AKT/mTOR signalling pathway plays a crucial role in regulating various biological processes, including apoptosis, cell proliferation, cell differentiation, angiogenesis, metabolism, transcription, and translation18,19. Phosphatidylinositol 3-kinases (PI3Ks) are a type of cytoplasmic signalling molecule belonging to a unique and conserved family of intracellular lipid kinases. They can phosphorylate the 3′-hydroxyl group of phosphatidylinositol and phosphoinositides. PI3Ks are classified into three classes (I–III) based on sequence homology and substrate choice. Depending on the receptors with which they interact, class I PI3Ks are further classified into two subfamilies: class IA PI3Ks and class IB PI3Ks. Growth factor receptor tyrosine kinases (RTKs) activate class IA PI3Ks, while G protein-coupled receptors (GPCRs) activate class IB PI3Ks. Class I PI3Ks are activated mainly by the conversion of the lipid substrate PIP2 to PIP3. A group of signalling proteins with pleckstrin homology (PH) domains are recruited to the membrane by PIP3; thereafter, they become activated. The primary target of PIP3 is the protein serine/threonine kinase AKT (also known as protein kinase B, PKB). Activated AKT subsequently activates the downstream target protein mTOR, which promotes anabolism, inhibits catabolism, and participates in various cellular activities20,21,22. In many human cancers, the activity of the PI3K/AKT/mTOR signalling pathway is commonly upregulated, and this pathway plays an irreplaceable role in tumour initiation and progression, making it a promising therapeutic target23,24. Given the potential significance of the PI3K/AKT/mTOR signalling pathway, efforts are currently ongoing to discover various drugs that target this crucial pathway25. Natural products are valuable gifts bestowed upon humanity by nature and are considered a treasure trove for the discovery of novel pharmaceuticals. With their high molecular diversity and novel biological functionalities, they hold great importance in the field of drug development and design. Many natural products have been shown to exert antitumour effects by targeting the PI3K/AKT/mTOR signalling pathway14,26, playing a significant role in enhancing tumour cell chemosensitivity and reducing the occurrence of drug resistance. Furthermore, studying the molecular mechanisms of biologically active compounds from a variety of natural products is crucial given the safety of natural products.
Gardenia jasminoides Ellis, commonly known as Cape jasmine, is a traditional dual-purpose plant used in medicine and food. It is a member of the Rubiaceae family and is a perennial shrub commonly distributed across Asian nations. The dried and mature fruits of this plant are well known and extensively utilized, serving not only as a natural colouring agent in various sectors such as the food and textile industries but also as a crucial traditional Chinese medicinal herb. These fruits are recognized for their significant efficacy in reducing fever, inhibiting inflammation and vasodilation, and improving blood circulation. The major iridoid chemicals identified in G. jasminoides Ellis are geniposide and genipin. The aglycone genipin is created when the bacterial enzyme β-d-glycosidase hydrolyses geniposide in the digestive tract prior to its entry into the bloodstream. It is the main active ingredient with unique pharmacological activities27. Reports have described the therapeutic benefits of genipin in a number of inflammatory illnesses, such as acute liver injury, severe hepatitis28, and acute lung injury29, while also providing protection to the cardiovascular system30,31,32 and nervous system33. Furthermore, genipin is a commonly utilized biocompatible cross-linking agent because of its superior biodegradability, outstanding biocompatibility, and stable cross-linking capabilities34. Previous research has shown the extensive anticancer activity of genipin against different human cancer types35, such as breast cancer, gastric cancer, and liver cancer36. However, currently, no definitive reports are available on the role of genipin in NB.
The objective of this research was to examine the therapeutic effect of genipin and its potential molecular mechanisms in NB. We anticipate that this research will yield novel strategies and insights for the management of NB.
Materials and methods
Materials
DMEM (containing 4.5 g/L d-glucose), foetal bovine serum, penicillin/streptomycin, 0.25% trypsin–EDTA, and phosphate-buffered saline (PBS) were purchased from VivaCell (Shanghai, China). Bax (ET1603-34, 1:1000), Bcl-2 (M1206-4, 1:1000), LC3B (ET1701-65, 1:1000), p62 (HA721171, 1:1000), Beclin 1 (HA721216, 1:1000), PI3K (ET1608-70, 1:1000), p-PI3K (HA721672, 1:1000), AKT (ET1609-51, 1:1000), p-AKT (ET1607-73, 1:1000), mTOR (ET1608-5, 1:1000), p-mTOR (HA600094, 1:1000), and GAPDH (ET1601-4, 1:80,000) antibodies were purchased from HUABIO (Zhejiang, China). Caspase-3 (ab32499, 1:10,000) and cleaved caspase-3 (ab2302, 1:500) antibodies were purchased from Abcam (Cambridge, UK). The cleaved caspase-9 (AF5240, 1:1000) antibody was purchased from Affinity Biosciences (Jiangsu, China). The dilutions mentioned in the parentheses are related to the Western blot experiments. The Ki67 (NBP2-22112) antibody was purchased from Novus Biologicals (Colorado, USA). 3-Methyladenine (3-MA), bafilomycin A1 (Baf A1), LY294002, 740Y-P, and rapamycin were purchased from MedChemExpress (New Jersey, USA). Genipin (purity ≥ 98%), a caspase-3 activity assay kit, protease inhibitors, phosphatase inhibitors, cell lysis buffer, Triton X-100, goat serum, 4% paraformaldehyde, dimethyl sulfoxide (DMSO), and DAPI nuclear stain were purchased from Solarbio (Beijing, China). CCK-8 reagents were obtained from Odyssey (Chengdu, China). The trypsin digestion solution (EDTA-free) and Annexin V-FITC and propidium iodide (PI) apoptosis detection kits were purchased from KeyGEN BioTech (Jiangsu, China). Sodium dodecyl sulfate polyacrylamide gels were purchased from Vazyme (Nanjing, China). TUNEL kits were purchased from Servicebio (Wuhan, China). A diaminobenzidine (DAB) colour development kit was purchased from ZSGB-BIO (Beijing, China).
Cell culture and treatment
SK-N-SH cells and HUVECs were obtained from SUNNCELL (Wuhan, China) and were authenticated using short tandem repeat (STR) markers. No mycoplasma contamination was detected in these cells. The cells were cultivated in a humidified atmosphere at 37 °C with 5% CO2 in DMEM containing 10% foetal bovine serum, 100 U/mL penicillin, and 100 μg/mL streptomycin. Cell isolation and passaging were achieved when the cells grew to 70–80% confluence by digestion with 0.25% trypsin–EDTA. In this study, 3–5 generations of SK-N-SH cells and HUVECs were used. Subsequently, the cells were treated with various concentrations of genipin for different durations, or pretreated with culture medium containing inhibitors, activators or vehicle for 2 h, and then genipin or vehicle was added for 24 h of treatment. Genipin, inhibitors, or activators were dissolved in DMSO and then diluted with culture medium to the desired concentration, with a final concentration of DMSO of ≤ 0.1%. As controls, cells that received only DMSO treatment were used.
Cell viability assay
Using the CCK-8 assay, the impact of genipin on cell viability was identified. SK-N-SH cells or HUVECs were cultured in 96-well plates at 100 μL/well (cell density 2 × 104 cells/mL), and the plates were precultured in a cellular thermostat (37 °C, 5% CO2). After cell attachment, the cells were administered the appropriate treatments according to the different groups, and cell viability was determined at each observation time point. After discarding the previous culture media, each well received 100 µL of fresh DMEM and 10 µL of CCK-8 reagent. The wells were then incubated for one hour at 37 °C in the dark. The absorbance was determined with a microplate reader (Bio-Rad, USA) set at 450 nm.
Annexin V/PI staining and flow cytometry
SK-N-SH cells were incubated at a density of 3 × 105 cells/well in 6-well plates. After the appropriate treatment, the old medium was collected, and the cells were detached from the petri dish using trypsin (EDTA-free). The collected cell suspension, along with the old culture medium, was centrifuged at a force of 500g. Afterwards, the cell precipitate was collected and subjected to two PBS washes. Subsequently, binding buffer was added, and the solution was gently mixed. Annexin V-fluorescein isothiocyanate (FITC) and propidium iodide (PI) were added at a 1:1 ratio and incubated for 10 min at room temperature in the dark. Finally, the number of positive cells was detected by flow cytometry (Agilent, USA) at the corresponding wavelength. The number of cells analysed was 26,000. Gating was set based on forwards scatter (FSC) and side scatter (SSC) profiles, and fluorescence compensation was adjusted using single-stained controls and unstained cells.
Western blotting analysis
SK-N-SH cells were cultured in 6-well plates at a density of 4 × 105 cells/well, and the groups of cells were collected after the corresponding treatments. The cells were lysed on ice in cell lysis buffer containing phosphatase and protease inhibitors to extract all of the protein. The protein content of each group was standardized using a BCA assay. Next, identical amounts of denatured proteins isolated from various samples were electrophoretically separated on a sodium dodecyl sulfate polyacrylamide gel before being transferred to a polyvinylidene fluoride membrane (Millipore, USA). The protein transfer membrane was incubated with the primary antibody at 4 °C overnight after being sealed in blocking solution (Tris-buffered saline with 0.1% Tween 20 and 5% fat-free milk) for 1 h. Subsequently, a solution of secondary antibodies labelled with horseradish peroxidase (Fine Test, Wuhan, China) was incubated with the membrane at room temperature for 2 h. Visualization was performed using a ChemiDoc XRS + chemiluminescence detection system (Bio-Rad, USA), and grayscale values were analysed using ImageJ software (Wayne Rasband, NIH, USA).
Caspase-3 activity assay
SK-N-SH cells were incubated at a density of 4 × 105 cells/well in a 6-well plate, and after incubations with different treatments according to the group, the cell suspension was collected and centrifuged to remove the supernatant. Then, lysis buffer was added, and the cells were lysed on ice for 15 min. After centrifuging the cell lysates at 4 °C and 15,000×g, the supernatant was incubated for 2 h at 37 °C with the assay buffer and substrate. A microplate reader was used to measure the absorbance at 405 nm. The reaction substrate was DEVD-pNA (Asp-Glu-Val-Asp-p-nitroanilide). Caspase-3 activity was standardized to the protein content of each sample.
Immunofluorescence staining
SK-N-SH cells were incubated at a density of 1 × 104 cells/well in confocal dishes and subjected to different treatments according to the group. At the end of treatment, the cells were permeabilized with a 0.2% Triton X-100 solution at room temperature for 20 min after fixation with a 4% paraformaldehyde solution for 15 min at room temperature and three PBS washes. After another PBS wash, the cells were treated with 5% goat serum for 30 min at room temperature. The cells were then incubated at 4 °C overnight with an LC3 antibody (1:100); the following day, the primary antibody was withdrawn, and the cells were washed with PBS. Subsequently, an appropriate amount of fluorescein-labelled secondary antibody (Proteintech, Wuhan, China) was added to the cells and incubated in the dark at room temperature for 60 min. Then, the cells were washed three times with PBS and stained with a small amount of DAPI nuclear staining solution for 10 min at room temperature in the dark. Finally, the cells were observed, and images were captured under a laser confocal microscope (SpinSR10, Olympus, Japan). Fifty-four images were captured. The fluorescence signals were analysed using ImageJ software.
Animal experimentation
The animal experiments were approved by the Animal Ethics Committee of North Sichuan Medical College (approval no. 2023-051) and complied with the International Association of Veterinary Editors on the Author’s Guide on Animal Ethics and Welfare. All the methods and treatments were performed in accordance with the ARRIVE guidelines (https://arriveguidelines.org). Male BALB/c nude mice (4–6 weeks old, approximately 20 g) were provided by the Animal Center of North Sichuan Medical College and housed in a specific pathogen-free (SPF) environment with a 12-h light/dark cycle and ad libitum access to food and water. SK-N-SH cells (2 × 107) were resuspended in 200 μL of PBS and subcutaneously injected into the right axillary fossa of the mouse forelimbs to establish a xenograft model of NB37. One week after implantation, the mice were randomly divided into control, LY294002, genipin, and combination treatment groups (n = 5 mice per group) and were intraperitoneally injected with genipin (20 mg/kg) and/or LY294002 (10 mg/kg) every 3 days. The dosage and route of administration of genipin and LY294002 were selected according to previous studies38,39. The control group was injected intraperitoneally with a drug-free vehicle. Mice were weighed every 3 days, and tumour volumes were measured using Vernier callipers. The final measurement of tumour volume was recorded on the 30th day prior to euthanizing the mice. The formula for calculating tumour volume was length × width2/2. At 30 days, all of the mice were euthanized by dislocating their cervical vertebrae after receiving an overdose of 2% pentobarbital sodium. The tumours were removed, weighed, and subjected to pathological analysis.
Pathological tissue staining
Mouse tumour tissues were obtained, embedded in paraffin, and sectioned at a thickness of 4 μm after being fixed in formalin. After deparaffinization, rehydration and antigen retrieval, the sections were incubated with a 3% H2O2 solution for 10 min at room temperature in the dark. After the sections were washed three times with PBS, they were incubated with 5% goat serum at room temperature for 1 h. Then, the tumour tissue sections were incubated with antibodies against specific markers [including Ki67 (1:200), p-PI3K (1:100), p-AKT (1:100), and p-mTOR (1:100)] at 4 °C overnight. The sections were washed and subsequently incubated with a horseradish peroxidase-labelled secondary antibody solution at room temperature for 30 min and visualized using a DAB colour development kit. After three minutes of staining the nuclei with haematoxylin, images of the histological specimens were captured using a microscope (CX31, Olympus, Japan). A total of 36 photos were captured. A TUNEL kit was used to count the number of apoptotic cells. Tumour tissue slices were treated with proteinase K for 20 min at 37 °C. After washing with PBS, the sections were incubated in TdT incubation buffer (recombinant TdT enzyme:CF488-dUTP labelling mix:equilibration buffer = 1:5:50) for one hour at 37 °C in the dark. The sections were washed three times with PBS before being stained with DAPI nuclear stain at room temperature for 10 min in the dark. Images were acquired with a fluorescence microscope (IX51, Olympus, Japan). Thirty-six images were collected. The acquired images were analysed using ImageJ software.
In vivo toxicological studies
Four groups of animals were established. Intraperitoneal injections of genipin (20 mg/kg) or LY294002 (10 mg/kg) were administered to the animals in the experimental groups, whereas a drug-free vehicle was administered to the control group. At the completion of their therapy, the mice were euthanized. Mouse liver and kidney tissues were fixed, embedded, sectioned, and stained with haematoxylin and eosin to observe toxicity in vivo.
Statistical analysis
Every experiment was conducted at least three times to guarantee the reliability and objectivity of the results. IBM SPSS Statistics 25.0 (IBM Corp, Armonk, NY, USA) was used for statistical analysis, and GraphPad Prism 9.0 (GraphPad Software, San Diego, CA, USA) was used for data visualization. All the data were subjected to normality tests. Two-tailed Student's t tests were used to compare two groups. One-way analysis of variance (ANOVA) or two-way ANOVA was used to compare data across multiple groups. Post hoc multiple comparisons were conducted using the Tukey method. The data are presented as the means ± standard deviations, and p < 0.05 was considered to indicate statistical significance.
Results
Genipin inhibited the growth of NB cells
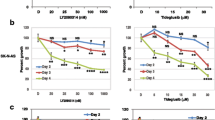
NB cells and HUVECs were exposed to a concentration gradient of genipin ranging from 0 to 400 μM to explore the cytotoxic impact of genipin (Fig. 1a) on NB cells and HUVECs. Using the CCK-8 assay, the impact of genipin on cell viability was evaluated. We found that when the exposure time was less than 48 h or the drug concentration was lower than 400 μM, genipin had no notable impact on HUVECs (Supplementary Fig. S1). As shown in Fig. 1b, genipin decreased the viability of SK-N-SH cells in a concentration- and time-dependent manner. At a concentration of genipin ≥ 100 μM, the viability of SK-N-SH cells was markedly reduced. After 24, 36, and 48 h of exposure, the corresponding IC50 values were 267.2 μM, 197.8 μM, and 148.0 μM, respectively. Therefore, we chose 100, 150, and 200 μM as the three concentrations for the subsequent in vitro experiments.

The effect of genipin on the viability of SK-N-SH cells. (a) The chemical structure of genipin and its molar molecular weight. (b) Effect of genipin on the viability of SK-N-SH cells. SK-N-SH cells were cultured in medium supplemented with vehicle (control group) or 12.5–400 μM genipin (experimental groups) for 24 h, 36 h, or 48 h. Cell viability was assessed using the CCK-8 assay. The percentage of cells in each experimental group relative to the control group was calculated, and the results are presented in a line graph. The data are presented as the means ± SDs. “*” indicates that each experimental group was compared to the corresponding control group at the same time point; *p < 0.05, **p < 0.01, and ***p < 0.001. “#” indicates that the cell viabilities at 36 h and 48 h at the same concentration were compared to the cell viability at 24 h; #p < 0.05, ##p < 0.01, and ###p < 0.001.
Genipin induced apoptosis in NB cells
We performed Annexin V-FITC and PI staining experiments to investigate the effects of genipin on NB cells. After treatment with various concentrations of genipin (100, 150, and 200 μM) for 24 h, we observed that compared with the control, genipin induced SK-N-SH-induced cell death in a concentration-dependent manner (Fig. 2a). We examined the expression of pro- and antiapoptotic proteins to further elucidate the mechanism by which genipin induces SK-N-SH cell death. Genipin markedly upregulated the expression of Bax, cleaved caspase-9, and cleaved caspase-3 proteins while downregulating the expression of Bcl-2 protein (Fig. 2b). In addition, genipin dramatically increased caspase-3 activity in SK-N-SH cells (Fig. 2c). These findings verified that genipin induced NB cell apoptosis.

The effect of genipin on apoptosis in SK-N-SH cells. SK-N-SH cells were incubated in medium containing vehicle (control group) or 100 μM, 150 μM, or 200 μM genipin (experimental groups) for 24 h. (a) Cell death was detected using Annexin V/PI double staining and flow cytometry. The sum of the percentage of cells in quadrants Q2-2 and Q2-4 was calculated and is represented in a histogram. (b) The expression levels of apoptosis-related proteins were detected in SK-N-SH cells using Western blotting. The blots were cropped and the original uncropped ones are shown in Supplementary Fig. S3. (c) Caspase-3 activity was measured, and the percentage in each experimental group relative to the control group was calculated. The results were then represented in a histogram. The density values of the protein bands in the images were measured using ImageJ software, and the results are represented in a histogram. The data are presented as the means ± SDs. *p < 0.05, **p < 0.01, and ***p < 0.001.
Genipin promoted autophagy in NB cells
We used Western blotting to evaluate the expression of autophagy-related proteins in genipin-treated SK-N-SH cells and determine whether autophagy plays a role in the cytotoxicity of the drug. An accepted indicator of autophagosomes in mammalian cells is LC3. Punctate LC3 protein is produced during autophagy, and LC3 transforms from its soluble (LC3-I) form to its lipidated, autophagosome-associated (LC3-II) form. The findings revealed that genipin upregulated the expression of LC3-II in a concentration-dependent manner (Fig. 3a). Nevertheless, we were unable to ascertain whether the increase in LC3-II levels resulted from the activation of autophagy flux or inhibition of autophagolysosomal fusion. Therefore, we also examined the protein expression levels of p62, which is an autophagy-specific substrate. Genipin administration decreased p62 levels in the immunoblot analysis, confirming the activation of autophagy by genipin. Additionally, genipin markedly upregulated the expression of Beclin 1 in SK-N-SH cells (Fig. 3a). Since autophagy is a dynamic process, we utilized the autophagy inhibitor 3-MA to further validate the induction of autophagy by genipin in SK-N-SH cells. Our findings showed that treatment with 3-MA reversed the genipin-induced accumulation of LC3-II (Fig. 3b). Immunofluorescence staining and imaging revealed an increase in the formation of LC3 puncta in SK-N-SH cells treated with genipin. In contrast, we observed a marked decrease in the formation of LC3 puncta induced by genipin when autophagy was inhibited by the autophagy inhibitor 3-MA (Fig. 3d). Simultaneously, we used bafilomycin A1 (Baf A1), a reversible and selective inhibitor of vacuolar H + -ATPase (V-ATPase) that prevents autophagosomes from fusing with lysosomes. According to the immunoblot analysis, compared to genipin treatment alone, bafilomycin A1 significantly enhanced the genipin-induced accumulation of LC3-II (Fig. 3c). This observation was further confirmed by the images of immunofluorescence staining (Fig. 3d). Taken together, our experimental results indicated that treatment with genipin triggered autophagy flux in SK-N-SH cells.

The effect of genipin on autophagy in SK-N-SH cells. (a) SK-N-SH cells were incubated in medium containing vehicle (control group) or 100 μM, 150 μM, or 200 μM genipin (experimental groups) for 24 h. We performed Western blotting to assess the expression levels of autophagy-related proteins in SK-N-SH cells. The blots were cropped and the original uncropped ones are shown in Supplementary Fig. S4a. (b) The cells were preincubated with culture medium containing either 5 mM 3-MA or vehicle for 2 h, followed by the addition of 150 μM genipin or vehicle for 24 h of incubation. Western blotting was performed to measure the protein levels of LC3-I and LC3-II. The blots were cropped and the original uncropped ones are shown in Supplementary Fig. S4b. (c) The cells were incubated with medium containing 150 μM genipin or vehicle in the presence or absence of 10 nM Baf A1 for 24 h. Western blotting was performed to measure the protein levels of LC3-I and LC3-II. The blots were cropped and the original uncropped ones are shown in Supplementary Fig. S4c. (d) Immunofluorescence staining was performed to detect the formation of LC3 puncta in SK-N-SH cells. LC3 puncta were visualized as green fluorescence, while the cell nuclei were stained with blue fluorescence. Scale bar: 50 μm. The last row shows magnified images of a specific region. Scale bar: 20 μm. The relative expression levels in the images were measured using ImageJ software, and the results are represented in a histogram. The data are presented as the means ± SDs. *p < 0.05, **p < 0.01, and ***p < 0.001.
Inhibition of autophagy enhanced the proapoptotic effect of genipin on NB cells
Autophagy is a physiological mechanism that promotes cell survival and is efficiently utilized by tumour cells as a means to evade cell death and develop drug resistance. Therefore, we used the autophagy-specific inhibitor 3-MA to elucidate the role of genipin-induced autophagy in SK-N-SH cells. The increased expression of autophagy markers induced by genipin was considerably reduced by pretreatment with 3-MA. Additionally, 3-MA pretreatment increased the expression of cleaved caspase-3, cleaved caspase-9, and Bax induced by genipin, while decreasing the expression of Bcl-2 (Fig. 4a). Furthermore, the results from the CCK-8 assay showed that pretreatment with the autophagy inhibitor 3-MA significantly enhanced the inhibitory effect of genipin on cell growth (Fig. 4b). Similarly, the inhibition of autophagy in SK-N-SH cells significantly augmented genipin-induced caspase-3 activity (Fig. 4c). Furthermore, compared to treatment with genipin alone, cotreatment with genipin and 3-MA markedly increased the quantity of Annexin V/PI-positive cells (Fig. 4d). In conclusion, these results indicated that SK-N-SH cells utilized autophagy as a defence mechanism against genipin-induced cell apoptosis. Inhibition of autophagy significantly enhanced the proapoptotic effect of genipin on cells. These findings highlighted the potential connection between autophagy and apoptosis triggered by genipin in SK-N-SH cells.

The role of genipin-induced autophagy in SK-N-SH cells. The cells were preincubated with culture medium containing either 5 mM 3-MA or vehicle for 2 h, followed by the addition of 150 μM genipin or vehicle for 24 h of incubation. (a) Western blotting was performed to detect the levels of LC3-I, LC3-II, and apoptosis-related proteins. The blots were cropped and the original uncropped ones are shown in Supplementary Fig. S5. (b) Cell viability was assessed using the CCK-8 assay, and the percentage of viable cells in each experimental group compared to the control group was calculated. The results are represented using a histogram. (c) Caspase-3 activity was measured, and the percentage in each experimental group compared to the control group was calculated. The results are represented using a histogram. (d) Cell apoptosis was detected using the Annexin V/PI double staining method and flow cytometry. The sum of the percentages of cells in quadrants Q2-2 and Q2-4 was calculated, and the results are represented in a histogram. The density values of the protein bands in the images were measured using ImageJ software, and the results are represented in a histogram. The data are presented as the means ± SDs. *p < 0.05, **p < 0.01, and ***p < 0.001.
Genipin induced apoptosis and autophagy by inhibiting the PI3K/AKT/mTOR pathway
The role of the PI3K/AKT/mTOR signalling pathway in controlling a number of biological processes, including cell signalling, proliferation, apoptosis, the cell cycle, and autophagy, is widely recognized. Many human malignancies often activate the PI3K/AKT/mTOR signalling pathway, which is thought to be a prospective target for therapy. Immunoblotting assays were used to measure the levels of p-PI3K, p-AKT, and p-mTOR as a method to determine whether genipin had an impact on the PI3K/AKT/mTOR signalling pathway (Fig. 5a). The administration of genipin to SK-N-SH cells resulted in a concentration-dependent decrease in the levels of phosphorylated PI3K, AKT, and mTOR compared to those in the control group. We first treated the cells with the PI3K inhibitor LY294002 prior to exposure to genipin to verify the involvement of the PI3K/AKT/mTOR signalling pathway in the anticancer effect of genipin on NB cells. Compared with genipin treatment alone, pretreatment with LY294002 substantially enhanced the reduction in p-PI3K, p-AKT, and p-mTOR protein levels induced by genipin (Fig. 5b). In addition, pretreatment with LY294002 significantly increased the accumulation of the autophagy marker LC3-II compared to that in cells incubated with genipin alone (Fig. 5b). The results of the CCK-8 assay showed that the combination of genipin and LY294002 significantly enhanced the inhibitory effect of genipin on the viability of SK-N-SH cells (Fig. 5c). Similarly, pretreatment of cells with the PI3K inhibitor significantly enhanced the caspase-3 activity induced by genipin (Fig. 5d). In addition, the increased number of Annexin V/PI-positive cells (Fig. 5e) and the elevated expression levels of the proapoptotic protein Bax (Fig. 5b) confirmed that combination treatment with a PI3K inhibitor and genipin significantly enhanced the ability of genipin to induce cell apoptosis. Next, we pretreated the cells with 740Y-P, an effective and cell-permeable PI3K activator, to further confirm the relationship between the PI3K/AKT/mTOR signalling pathway and the anticancer activity of genipin. As shown in Supplementary Fig. S2a, 740Y-P attenuated the inhibitory effect of genipin on the phosphorylation of proteins in this pathway. Moreover, as shown in Supplementary Fig. S2a–d, the proautophagic, antiproliferative, and proapoptotic effects of genipin were reversed by 740Y-P. Next, we pretreated SK-N-SH cells with rapamycin to validate whether mTOR was involved in the anticancer effect of genipin. Compared to cells treated with genipin alone, cells cotreated with rapamycin and genipin exhibited significantly decreased levels of p-mTOR proteins, significantly increased levels of autophagy markers, a significantly increased expression of proapoptotic proteins (Fig. 5f), a significantly enhanced inhibition of cell viability (Fig. 5g), a significantly elevated caspase-3 activity (Fig. 5h), and a significantly greater number of apoptotic cells (Fig. 5i). The above results showed that genipin decreased the viability of SK-N-SH cells, promoted cellular autophagy, and induced cell apoptosis, and these mechanisms were associated with the inhibition of the PI3K/AKT/mTOR pathway.


Regulatory effect of genipin on the PI3K/AKT/mTOR pathway in NB cells. (a) SK-N-SH cells were cultured in medium supplemented with vehicle (control group) or 100, 150, or 200 μM genipin (experimental groups) for 24 h. Western blotting was used to detect the protein expression levels of p-PI3K, PI3K, p-AKT, AKT, p-mTOR, and mTOR in SK-N-SH cells. The blots were cropped and the original uncropped ones are shown in Supplementary Fig. S6a. The cells were preincubated with culture medium containing either 10 μM LY294002 or vehicle for 2 h, followed by the addition of 150 μM genipin or vehicle for 24 h of incubation. (b) Western blotting was used to detect the protein levels of p-PI3K, PI3K, p-AKT, AKT, p-mTOR, mTOR, LC3-I, LC3-II, and Bax in SK-N-SH cells. The blots were cropped and the original uncropped ones are shown in Supplementary Fig. S6b. (c) Cell viability was assessed using the CCK-8 assay, and the percentage of viable cells in each experimental group compared to the control group was calculated. The results are represented using a histogram. (d) Caspase-3 activity was measured, and the percentage in each experimental group compared to the control group was calculated. The results are represented using a histogram. (e) Flow cytometry was used to detect cell apoptosis. The sum of the percentages of cells in quadrants Q2-2 and Q2-4 was calculated, and the results are represented in a histogram. The cells were preincubated with culture medium containing either 20 nM rapamycin or vehicle for 2 h, followed by the addition of 150 μM genipin or vehicle for 24 h of incubation. (f) Western blotting was performed to detect the protein levels of p-mTOR, mTOR, LC3-I, LC3-II, and Bax in SK-N-SH cells. The blots were cropped and the original uncropped ones are shown in Supplementary Fig. S6d. (g) A CCK-8 assay was used to measure cell viability. (h) Caspase-3 activity was evaluated. (i) Flow cytometry was used to assess cell apoptosis. The density values of the protein bands in the images were measured using ImageJ software, and the results are represented in a histogram. The data are presented as the means ± SDs. *p < 0.05, **p < 0.01, and ***p < 0.001.
Genipin inhibited tumour growth in vivo
In conjunction with in vitro experiments, we used SK-N-SH cells to establish a xenograft tumour model and determine the anticancer activity of genipin in vivo (Fig. 6a). As shown in Fig. 6b, compared to the control group, the genipin-treated group exhibited restricted tumour growth, with a decreased tumour weight and volume (Fig. 6c,d). Before administering genipin, we intraperitoneally injected animals with LY294002. Compared to the group treated with genipin alone, the inhibitory effect of the combination treatment on tumour growth was greater (Fig. 6b), with significant decreases in tumour weight and volume (Fig. 6c,d). The immunoblotting results showed that, compared to the control group, genipin promoted the accumulation of LC3-II (Fig. 6e). Moreover, compared to the group treated with genipin alone, in the combination treatment group, the levels of autophagy markers were significantly increased. The expression of the proapoptotic protein Bax was upregulated in the group that received genipin treatment, compared to the control group. Moreover, compared to the group treated with genipin alone, the combination of genipin and LY294002 dramatically increased the expression level of the Bax protein (Fig. 6e). Additionally, immunoblotting and immunohistochemistry results showed that the levels of phosphorylated PI3K, AKT, and mTOR in the genipin treatment group were lower than those in the control group, while the combination treatment group exhibited significantly lower levels of phosphorylated PI3K, AKT, and mTOR than those in the group treated with genipin alone (Fig. 6e,f). The immunohistochemistry results showed that, compared to the control group, the level of Ki67 protein decreased in the genipin treatment group. Compared to the genipin treatment group, the combination treatment group showed significant downregulation of Ki67 protein expression, indicating that the combination of LY294002 with genipin significantly enhanced the inhibitory effect of genipin on cancer cell proliferation (Fig. 6f). The results of the TUNEL staining assay showed that, compared to the control group, the genipin treatment group had an increased number of apoptotic cells. Compared to the genipin treatment group, the combination treatment group had a significantly higher number of apoptotic cells, indicating that the combination of LY294002 with genipin significantly enhanced genipin's induction of apoptosis in cancer cells (Fig. 6g). Figure 7 displays the results of body weights and the histopathological analysis of the mice in the control group and treatment group. After treatment with genipin at a dose of 20 mg/kg and/or LY294002 at a dose of 10 mg/kg for a certain period, no abnormalities or toxic reactions were observed in the liver or kidney tissue sections from the mice in each group. No significant differences in body weight were observed among the groups of mice. The evidence indicated that genipin inhibited tumour growth in vivo, promoted apoptosis and autophagy, downregulated the phosphorylation levels of PI3K, AKT, and mTOR, and did not produce any adverse effects on the animals at the therapeutic dose. LY294002 enhanced the therapeutic effect of genipin.


Genipin restricts tumour growth in vivo. (a) A xenograft tumour model was established by subcutaneously injecting SK-N-SH cells (2 × 107) into the armpits of nude mice. After 7 days, the nude mice were randomly divided into a control group (n = 5) and treatment groups (n = 5 per group). The treatment group received LY294002 (10 mg/kg) and/or genipin (20 mg/kg), while the control group received a drug-free vehicle. The administration route was intraperitoneal injection, with a frequency of once every 3 days. This figure was generated using FigDraw. (b) Images of tumours at the endpoint of treatment. (c) Tumour weights. The results are represented in a histogram. (d) Tumour volume. The results are represented in a line graph and a histogram. The line graph “*” indicates that the LY294002 treatment group or the genipin treatment group was compared with the control group at the same time point; *p < 0.05, **p < 0.01, and ***p < 0.001. “#” indicates that the combination treatment group was compared with the genipin treatment group at the same time point; #p < 0.05, ##p < 0.01, and ###p < 0.001. (e) Western blotting was performed to detect the protein levels of p-PI3K, p-AKT, p-mTOR, LC3-I, LC3-II, and Bax. The blots were cropped and the original uncropped ones are shown in Supplementary Fig. S7. (f) Immunohistochemistry was performed to assess the expression of the p-PI3K, p-AKT, p-mTOR, and Ki67 proteins in tumour tissues. Scale bar: 50 μm. (g) The TUNEL assay was employed to assess the level of cellular apoptosis in tumour tissues. Apoptotic cells were visualized as green, while the cell nuclei were stained blue. Scale bar: 50 μm. The relative expression levels in the images were measured using ImageJ software, and the results are represented in a histogram. The data are presented as the means ± SDs. *p < 0.05, **p < 0.01, and ***p < 0.001.

Effects of different treatments on the mouse tissue structure. (a) Liver and kidney sections of mice from the control group (n = 5) and treatment groups (n = 5 per group) were stained with haematoxylin and eosin. The sections showed no signs of hepatotoxicity (indicated by arrows) and no evidence of nephrotoxicity (indicated by circles). Scale bar: 50 μm. (b) Changes in the body weight of each group of mice. The results are represented in histograms and line graphs. The data are presented as the means ± SDs. *p < 0.05, **p < 0.01, ***p < 0.001, and ns, not significant.
Discussion
According to prior research, NB is the most prevalent cancer diagnosed before the age of 12 months. Treatment and management of this disease are based on risk stratification. In young infants with favourable biological characteristics, their tumours can spontaneously regress without the need for treatment, even in the presence of metastatic disease. However, overall survival rates for children diagnosed after the age of 18 months who have either unfavourable biological characteristics (defined as adverse pathology and/or MYCN amplification) or metastatic or unresectable disease remain low even with multimodal combination treatments such as surgery, radiation therapy, chemotherapy, and autologous stem cell transplantation4. The unique clinical and biological heterogeneity of NB, the propensity for relapse in high-risk patients, the limitations of surgery, and the long-term side effects of radiation and chemotherapy have inspired in-depth research into its biological behaviour and the exploration of novel therapeutic approaches. Our findings revealed that genipin promoted cell apoptosis, suppressed the growth of NB cells, and triggered autophagy. The suppression of the PI3K/AKT/mTOR signalling pathway was linked to this process.
Natural products are an important source of new therapeutic drugs, as their unique molecular characteristics can provide superior efficacy and safety. Compounds derived from plants, such as resveratrol40, curcumin41, ginsenosides42, cynaropicrin43, and apigenin44, have been extensively studied and are known to have antitumour activities in NB via various pathways. Genipin is a type of iridoid substance extracted from the plant G. jasminoides Ellis35. Feng and colleagues45 reported that genipin suppresses the growth of human leukaemia K562 cells and causes apoptosis through the activation of JNK and the induction of the Fas ligand. According to Kim et al.38, the combination of genipin and oxaliplatin has synergistic antitumour effects on colorectal cancer via the reactive oxygen species (ROS)/endoplasmic reticulum (ER) stress/BIM pathway both in vitro and in vivo. In our research, the natural compound genipin suppressed the growth of NB cells, promoted apoptosis, and did not cause significant toxicity in HUVECs. Similarly, in animal experiments, genipin limited tumour growth, reduced the tumour volume, and decreased the tumour weight. Following the administration of the medication, neither the control nor the experimental mouse groups exhibited any notable pathological alterations or signs of toxicity in their liver or kidney tissues, and no statistically significant differences in body weight were observed among the groups. These outcomes showed that genipin exhibited anti-cancer activity in NB and possessed good biocompatibility.
Autophagy is a highly conserved catabolic process in which faulty proteins and organelles are enclosed within autophagosomes, which then merge with lysosomes for subsequent decomposition and recycling. It is involved in various cellular biological activities6. In addition to its function in regular physiological activities, autophagy also plays a vital role in pathological conditions such as cancer. Indeed, autophagy has a complex role in both the initiation and progression of tumours. Research has shown that defects in cellular autophagy are involved in tumour occurrence. Qu et al.46 and Yue et al.47 found that a heterozygous disruption in the beclin 1 gene increases the occurrence of spontaneous malignancy by constructing a monoallelic deletion model in beclin 1 mice. The above results indicate that beclin 1 acts as a haploinsufficient gene for tumour suppression. According to Takamura et al.48, mice with liver-specific Atg7−/− and systemic mosaic deletion of Atg5 develop benign hepatic adenomas. In contrast, autophagy is essential for the survival and progression of tumours49. The autophagic response supports the growth and progression of established tumours by increasing the tolerance of tumour cells to cell-intrinsic and microenvironmental stimuli that normally promote tumour cell death, such as nutrient deprivation, hypoxia, and therapeutic stress9. Defects in autophagy may result in inflammation, reactive oxygen species (ROS) production, genomic instability, and DNA damage, which can promote tumorigenesis, and excess levels of these factors are unfavourable for tumour growth. Hence, tumours might reactivate or upregulate autophagy to counteract this damage and sustain their growth. As reported by Xie et al.50, Atg7 facilitates the growth of melanoma cells driven by Braf V600E by reducing oxidative stress and bypassing cellular senescence. Furthermore, many types of tumours exhibit an increase in basal levels of autophagy, and this increase can occur in a cell-autonomous manner, thereby sustaining tumour growth. Yang et al. confirmed that pancreatic cancer cells autonomously increase basal autophagy and rely on this autophagy process to sustain continuous growth. Guo and colleagues reported that Ras gene-driven tumour formation leads to an elevated level of basal autophagy51,52,53,54. The above evidence suggests that autophagy is a dynamic process in tumour development and acts as a barrier that limits tumour initiation. Autophagy maintains cellular homeostasis by limiting genomic damage and clearing oncogenic substrates, thereby inhibiting tumour formation. However, once the tumour is established, adaptive changes can occur, leading to a protumorigenic role of autophagy in tumour progression and subsequent tumour maintenance. Previous studies have shown that certain anticancer medications can induce autophagy. The coadministration of imipramine, a tricyclic antidepressant (TCA), and ticlopidine has been shown to trigger autophagy in glioma cells, leading to autophagy-related cell death55. Moreover, Sun et al.56 reported that epirubicin can trigger a defensive autophagic response in breast cancer cells to resist the apoptosis induced by this drug. In the present study, we explored for the first time the impact of genipin on autophagy in NB. In our in vitro experiments, we observed that the administration of genipin upregulated the protein expression of LC3-II and Beclin 1, and this effect was attenuated by pretreatment with 3-MA. Moreover, our in vivo experiments confirmed that the protein expression levels of LC3-II were increased by genipin treatment. We assessed the level of the selective autophagy substrate p62 to determine whether genipin increased autophagosome formation or blocked autophagosome degradation in SK-N-SH cells. The findings revealed that the p62 protein level was decreased by genipin. Additionally, we used the autophagy‒lysosome fusion inhibitor Baf A1 to assess autophagy flux57. We observed that compared with genipin treatment alone, treatment with genipin in combination with Baf A1 significantly enhanced the accumulation of LC3-II. Based on these findings, we can infer that genipin enhanced autophagy flux in NB cells. Previous studies have indicated that autophagy inhibitors have synergistic effects with various anticancer drugs49,58,59. Our research showed that cell apoptosis and autophagy were induced by genipin in a dose-dependent manner. Autophagy inhibition resulted in enhanced growth inhibition and the induction of apoptosis in genipin-treated SK-N-SH cells. According to the aforementioned findings, autophagy flux increased in SK-N-SH cells exposed to genipin, potentially acting as a shield to keep cancer cells alive and resist apoptosis. The combination of genipin and autophagy inhibitors could be a beneficial strategy for NB treatment.
Numerous molecules are involved in the processes of cell proliferation, apoptosis, metabolism, and other life activities, and the interactions between these molecules form an intricate network. One of the pathways commonly activated in human cancer is the PI3K/AKT/mTOR signalling pathway, and medications that target this pathway can effectively suppress tumour growth and trigger cancer cell apoptosis19,60,61. The PI3K/AKT/mTOR pathway, which includes increased AKT phosphorylation, upregulated PI3K expression, and aberrant receptor tyrosine kinase (RTK) activity, is abnormally activated in the majority of NBs and plays a significant role in the survival and progression of NB. This pathway can serve as a therapeutic target for NB62,63. In this study, following the administration of genipin to NB cells, the levels of phosphorylated PI3K, AKT, and mTOR decreased, indicating the inactivation of the PI3K/AKT/mTOR signalling pathway. We speculate that the antitumour effect of genipin may be associated with inactivation of this pathway. Next, we utilized LY294002, a first-generation broad-spectrum PI3K inhibitor developed in the early 1990s64, as well as rapamycin, an mTOR-targeting agent65, to investigate the relationship between this pathway and the antitumour effect of genipin. mTOR is a serine/threonine protein kinase that binds to different partner proteins and assembles into two separate complexes: mTOR complex 1 (mTORC1) and mTOR complex 2 (mTORC2). Among them, mTORC1, which is sensitive to rapamycin, plays a role in regulating numerous cellular processes, such as apoptosis and autophagy66. We observed that the downregulation of phosphorylated PI3K, AKT, and mTOR enhanced the sensitivity of SK-N-SH cells to genipin and upregulated the expression of the autophagy-associated protein LC3-II. We established a xenograft tumour model to evaluate the anticancer effect of genipin in vivo. In addition to treating mice with genipin alone, mice were treated with LY294002 in combination with genipin. The experimental data showed that the genipin treatment group had limited tumour growth, down-regulated the level of pathway-related protein phosphorylation, and increased the level of autophagy-related and pro-apoptotic proteins. Compared to treatment with genipin alone, combination treatment with LY294002 and genipin significantly suppressed tumour growth and reduced the tumour volume and weight. The phosphorylation levels of related proteins in the pathway significantly decreased, while the expression levels of autophagy-associated proteins and proapoptotic proteins further increased. These findings were corroborated by immunohistochemistry and TUNEL staining. The results of the in vivo study were consistent with those of the in vitro study. In addition, we used the PI3K activator 740Y-P to further validate that genipin exerts therapeutic effects on NB by regulating the PI3K/AKT/mTOR signalling axis67. We observed that pretreatment with 740Y-P reversed the anticancer effect of genipin. The above results indicated that genipin suppressed the PI3K/AKT/mTOR signalling pathway in NB cells, which hindered cell growth, induced apoptosis, and promoted autophagy. Furthermore, combining genipin with the PI3K inhibitor LY294002 significantly enhanced its anticancer effects both in vitro and in vivo.
In summary, these findings establish a promising foundation for the potential application of genipin in the treatment of NB. The combination of genipin with other drugs may be a potential strategy to enhance the anticancer efficacy of genipin. However, further research is needed to determine the optimal drug combinations, dosages, and relevant molecular mechanisms. Notably, in animal studies, no significant toxic reactions or adverse events were observed in mice treated with genipin. This result indicated that genipin exhibited good biocompatibility at therapeutic doses. Nonetheless, further toxicological studies are still needed to confirm its safety. This study has several limitations. We have not conducted in-depth research on the direct mechanism of genipin. Considering that molecular interactions form a complex network, future research needs to delve into the molecular level for a more detailed discussion and to comprehensively evaluate the effectiveness of genipin. In addition, given the heterogeneity among different NB cell lines, future studies should be conducted on a variety of cell lines to explore the heterogeneity of the effects of genipin treatment on NB, to ensure the generalizability of the treatment strategies, and to provide a more substantial theoretical basis for clinical practice.
Conclusions
Overall, this study indicated that genipin could serve as a safe and effective anticancer agent by restricting cell growth, inducing apoptosis, and promoting autophagy both in vitro and in vivo. This mechanism may be associated with the inhibition of the PI3K/AKT/mTOR signalling pathway (Fig. 8). Inhibiting autophagy enhanced the sensitivity of SK-N-SH cells to genipin. Combination therapy consisting of genipin and PI3K inhibitors holds promise as an innovative and effective approach for treating NB. These findings provide preliminary evidence for the further development and application of genipin as a therapeutic drug for NB.

Schematic representation of the role of the genipin in NB. We investigated the impact of genipin on NB both in vitro and in vivo. The PI3K/AKT/mTOR signalling pathway plays a crucial role in regulating cellular processes such as autophagy, growth, and apoptosis and is commonly activated in human cancers. In our study, genipin restricted the growth of NB cells in vitro and in vivo, triggered the formation of autophagic flux, and induced apoptosis, and its mechanism was associated with the inhibition of the PI3K/AKT/mTOR signalling axis activation. Our findings provide new perspectives for the treatment of NB. This figure was generated using FigDraw.
Data availability
This published article contains all of the data generated or analysed during the course of this study.
Abbreviations
- NB:
-
Neuroblastoma
- SK-N-SH:
-
SK-N-SH neuroblastoma cells
- HUVECs:
-
Human umbilical vein endothelial cells
- DMEM:
-
Dulbecco’s Modified Eagle’s Medium
- EDTA:
-
Ethylenediaminetetraacetic acid
- CCK-8:
-
Cell Counting Kit-8
- FITC:
-
Fluorescein isothiocyanate
- PI:
-
Propidium iodide
- Bax:
-
Bcl-2-associated X protein
- Bcl-2:
-
B-cell lymphoma-2
- LC3:
-
Microtubule-associated protein 1 light chain 3
- p62:
-
Sequestosome 1
- 3-MA:
-
3-Methyladenine
- AKT:
-
Protein kinase B
- Baf A1:
-
Bafilomycin A1
- PI3K:
-
Phosphatidylinositol 3-kinases
- p-PI3K:
-
Phosphorylated phosphoinositide 3-kinase
- p-AKT:
-
Phosphorylated protein kinase B
- mTOR:
-
Mechanistic target of rapamycin
- p-mTOR:
-
Phosphorylated mechanistic target of rapamycin
- PIP2 :
-
Phosphatidylinositol-4,5-bisphosphate
- PIP3 :
-
Phosphatidylinositol-3,4,5-trisphosphate
- IOD:
-
Integrated optical density
- TUNEL:
-
TdT-mediated dUTP nick-end labeling
References
Ponzoni, M. et al. Recent advances in the developmental origin of neuroblastoma: An overview. J. Exp. Clin. Cancer Res. 41, 92 (2022).
Maris, J. M. Recent advances in neuroblastoma. N. Engl. J. Med. 362, 2202–2211 (2010).
Maris, J. M., Hogarty, M. D., Bagatell, R. & Cohn, S. L. Neuroblastoma. Lancet 369, 2106–2120 (2007).
Matthay, K. K. et al. Neuroblastoma. Nat. Rev. Dis. Primers 2, 16078 (2016).
Cohn, S. L. et al. The International Neuroblastoma Risk Group (INRG) classification system: An INRG Task Force report. J. Clin. Oncol. 27, 289–297 (2009).
Rabinowitz, J. D. & White, E. Autophagy and metabolism. Science 330, 1344–1348 (2010).
Degenhardt, K. et al. Autophagy promotes tumor cell survival and restricts necrosis, inflammation, and tumorigenesis. Cancer Cell 10, 51–64 (2006).
Mathew, R. et al. Autophagy suppresses tumorigenesis through elimination of p62. Cell 137, 1062–1075 (2009).
Galluzzi, L. et al. Autophagy in malignant transformation and cancer progression. EMBO J. 34, 856–880 (2015).
White, E. & DiPaola, R. S. The double-edged sword of autophagy modulation in cancer. Clin. Cancer Res. 15, 5308–5316 (2009).
Apel, A., Herr, I., Schwarz, H., Rodemann, H. P. & Mayer, A. Blocked autophagy sensitizes resistant carcinoma cells to radiation therapy. Cancer Res. 68, 1485–1494 (2008).
Li, X., He, S. & Ma, B. Autophagy and autophagy-related proteins in cancer. Mol. Cancer 19, 12 (2020).
Peng, Y., Wang, Y., Zhou, C., Mei, W. & Zeng, C. PI3K/Akt/mTOR pathway and its role in cancer therapeutics: Are we making headway?. Front. Oncol. 12, 819128 (2022).
Xu, Z. et al. Targeting PI3K/AKT/mTOR-mediated autophagy for tumor therapy. Appl. Microbiol. Biotechnol. 104, 575–587 (2020).
Kim, Y. C. & Guan, K.-L. mTOR: A pharmacologic target for autophagy regulation. J. Clin. Investig. 125, 25–32 (2015).
Chiarini, F., Evangelisti, C., McCubrey, J. A. & Martelli, A. M. Current treatment strategies for inhibiting mTOR in cancer. Trends Pharmacol. Sci. 36, 124–135 (2015).
Mathew, R., Karantza-Wadsworth, V. & White, E. Role of autophagy in cancer. Nat. Rev. Cancer 7, 961–967 (2007).
Hoxhaj, G. & Manning, B. D. The PI3K-AKT network at the interface of oncogenic signalling and cancer metabolism. Nat. Rev. Cancer 20, 74–88 (2020).
Ediriweera, M. K., Tennekoon, K. H. & Samarakoon, S. R. Role of the PI3K/AKT/mTOR signaling pathway in ovarian cancer: Biological and therapeutic significance. Semin. Cancer Biol. 59, 147–160 (2019).
Klippel, A., Kavanaugh, W. M., Pot, D. & Williams, L. T. A specific product of phosphatidylinositol 3-kinase directly activates the protein kinase Akt through its pleckstrin homology domain. Mol. Cell Biol. 17, 338–344 (1997).
Franke, T. F., Kaplan, D. R., Cantley, L. C. & Toker, A. Direct regulation of the Akt proto-oncogene product by phosphatidylinositol-3,4-bisphosphate. Science 275, 665–668 (1997).
Engelman, J. A., Luo, J. & Cantley, L. C. The evolution of phosphatidylinositol 3-kinases as regulators of growth and metabolism. Nat. Rev. Genet. 7, 606–619 (2006).
Aoki, M. & Fujishita, T. Oncogenic roles of the PI3K/AKT/mTOR axis. Curr. Top. Microbiol. Immunol. 407, 153–189 (2017).
Mayer, I. A. & Arteaga, C. L. The PI3K/AKT pathway as a target for cancer treatment. Annu. Rev. Med. 67, 11–28 (2016).
He, Y. et al. Targeting PI3K/Akt signal transduction for cancer therapy. Signal Transduct. Target Ther. 6, 1–17 (2021).
Tewari, D., Patni, P., Bishayee, A., Sah, A. N. & Bishayee, A. Natural products targeting the PI3K-Akt-mTOR signaling pathway in cancer: A novel therapeutic strategy. Semin. Cancer Biol. 80, 1–17 (2022).
Chen, L. et al. Gardenia jasminoides Ellis: Ethnopharmacology, phytochemistry, and pharmacological and industrial applications of an important traditional Chinese medicine. J. Ethnopharmacol. 257, 112829 (2020).
Fan, X. et al. Therapeutic potential of genipin in various acute liver injury, fulminant hepatitis, NAFLD and other non-cancer liver diseases: More friend than foe. Pharmacol. Res. 159, 104945 (2020).
Luo, X. et al. Genipin attenuates mitochondrial-dependent apoptosis, endoplasmic reticulum stress, and inflammation via the PI3K/AKT pathway in acute lung injury. Int. Immunopharmacol. 76, 105842 (2019).
Zhong, H. et al. Genipin alleviates high-fat diet-induced hyperlipidemia and hepatic lipid accumulation in mice via miR-142a-5p/SREBP-1c axis. FEBS J. 285, 501–517 (2018).
Zhao, L. et al. Potential cardioprotective effect of genipin via cyclooxidase 2 suppression and p53 signal pathway attenuation in induced myocardial infarction in rats. Shock 58, 457–463 (2022).
Shumin, C. et al. Genipin alleviates vascular hyperpermeability following hemorrhagic shock by up-regulation of SIRT3/autophagy. Cell Death Discov. 4, 52 (2018).
Li, Y., Li, L. & Hölscher, C. Therapeutic potential of genipin in central neurodegenerative diseases. CNS Drugs 30, 889–897 (2016).
Yu, Y., Xu, S., Li, S. & Pan, H. Genipin-cross-linked hydrogels based on biomaterials for drug delivery: A review. Biomater. Sci. 9, 1583–1597 (2021).
Shanmugam, M. K. et al. Potential role of genipin in cancer therapy. Pharmacol. Res. 133, 195–200 (2018).
Cho, Y. S. Genipin, an inhibitor of UCP2 as a promising new anticancer agent: A review of the literature. Int. J. Mol. Sci. 23, 5637 (2022).
Helson, L., Das, S. K. & Hajdu, S. I. Human neuroblastoma in nude mice. Cancer Res. 35, 2594–2599 (1975).
Kim, B. R. et al. Genipin enhances the therapeutic effects of oxaliplatin by upregulating BIM in colorectal cancer. Mol. Cancer Ther. 18, 751–761 (2019).
Jiang, H., Fan, D., Zhou, G., Li, X. & Deng, H. Phosphatidylinositol 3-kinase inhibitor(LY294002) induces apoptosis of human nasopharyngeal carcinoma in vitro and in vivo. J. Exp. Clin. Cancer Res. 29, 34 (2010).
Graham, R. M. et al. Resveratrol augments ER stress and the cytotoxic effects of glycolytic inhibition in neuroblastoma by downregulating Akt in a mechanism independent of SIRT1. Exp. Mol. Med. 48, e210 (2016).
Caruso Bavisotto, C. et al. Curcumin affects HSP60 folding activity and levels in neuroblastoma cells. Int. J. Mol. Sci. 21, 661 (2020).
Oh, J.-M., Kim, E. & Chun, S. Ginsenoside compound K induces Ros-mediated apoptosis and autophagic inhibition in human neuroblastoma cells in vitro and in vivo. Int. J. Mol. Sci. 20, 4279 (2019).
Yang, R. et al. Suppression of endoplasmic reticulum stress-dependent autophagy enhances cynaropicrin-induced apoptosis via attenuation of the P62/Keap1/Nrf2 pathways in neuroblastoma. Front. Pharmacol. 13, 977622 (2022).
Das, A., Banik, N. L. & Ray, S. K. Mechanism of apoptosis with the involvement of calpain and caspase cascades in human malignant neuroblastoma SH-SY5Y cells exposed to flavonoids. Int. J. Cancer 119, 2575–2585 (2006).
Feng, Q. et al. Apoptosis induced by genipin in human leukemia K562 cells: Involvement of c-Jun N-terminal kinase in G2/M arrest. Acta Pharmacol. Sin. 32, 519–527 (2011).
Qu, X. et al. Promotion of tumorigenesis by heterozygous disruption of the beclin 1 autophagy gene. J. Clin. Investig. 112, 1809–1820 (2003).
Yue, Z., Jin, S., Yang, C., Levine, A. J. & Heintz, N. Beclin 1, an autophagy gene essential for early embryonic development, is a haploinsufficient tumor suppressor. Proc. Natl. Acad. Sci. U. S. A. 100, 15077–15082 (2003).
Takamura, A. et al. Autophagy-deficient mice develop multiple liver tumors. Genes Dev. 25, 795–800 (2011).
Xia, H., Green, D. R. & Zou, W. Autophagy in tumour immunity and therapy. Nat. Rev. Cancer 21, 281–297 (2021).
Xie, X., Koh, J. Y., Price, S., White, E. & Mehnert, J. M. Atg7 overcomes senescence and promotes growth of BrafV600E-driven melanoma. Cancer Discov. 5, 410–423 (2015).
Kimmelman, A. C. The dynamic nature of autophagy in cancer. Genes Dev. 25, 1999–2010 (2011).
Guo, J. Y. et al. Activated Ras requires autophagy to maintain oxidative metabolism and tumorigenesis. Genes Dev. 25, 460–470 (2011).
Yang, S. & Kimmelman, A. C. A critical role for autophagy in pancreatic cancer. Autophagy 7, 912–913 (2011).
Yang, S. et al. Pancreatic cancers require autophagy for tumor growth. Genes Dev. 25, 717–729 (2011).
Shchors, K., Massaras, A. & Hanahan, D. Dual targeting of the autophagic regulatory circuitry in gliomas with repurposed drugs elicits cell-lethal autophagy and therapeutic benefit. Cancer Cell 28, 456–471 (2015).
Sun, W.-L., Chen, J., Wang, Y.-P. & Zheng, H. Autophagy protects breast cancer cells from epirubicin-induced apoptosis and facilitates epirubicin-resistance development. Autophagy 7, 1035–1044 (2011).
Yamamoto, A. et al. Bafilomycin A1 prevents maturation of autophagic vacuoles by inhibiting fusion between autophagosomes and lysosomes in rat hepatoma cell line, H-4-II-E cells. Cell Struct. Funct. 23, 33–42 (1998).
Ferreira, P. M. P., de Sousa, R. W. R., de Ferreira, J. R. O., Militão, G. C. G. & Bezerra, D. P. Chloroquine and hydroxychloroquine in antitumor therapies based on autophagy-related mechanisms. Pharmacol. Res. 168, 105582 (2021).
Levy, J. M. M. et al. Autophagy inhibition improves chemosensitivity in BRAF(V600E) brain tumors. Cancer Discov. 4, 773–780 (2014).
Alzahrani, A. S. PI3K/Akt/mTOR inhibitors in cancer: At the bench and bedside. Semin. Cancer Biol. 59, 125–132 (2019).
Tian, L.-Y., Smit, D. J. & Jücker, M. The role of PI3K/AKT/mTOR signaling in hepatocellular carcinoma metabolism. Int. J. Mol. Sci. 24, 2652 (2023).
Zafar, A. et al. Molecular targeting therapies for neuroblastoma: Progress and challenges. Med. Res. Rev. 41, 961–1021 (2021).
King, D., Yeomanson, D. & Bryant, H. E. PI3King the lock: Targeting the PI3K/Akt/mTOR pathway as a novel therapeutic strategy in neuroblastoma. J. Pediatr. Hematol. Oncol. 37, 245–251 (2015).
Vlahos, C. J., Matter, W. F., Hui, K. Y. & Brown, R. F. A specific inhibitor of phosphatidylinositol 3-kinase, 2-(4-morpholinyl)-8-phenyl-4H-1-benzopyran-4-one (LY294002). J. Biol. Chem. 269, 5241–5248 (1994).
Niu, H., Wang, J., Li, H. & He, P. Rapamycin potentiates cytotoxicity by docetaxel possibly through downregulation of Survivin in lung cancer cells. J. Exp. Clin. Cancer Res. 30, 28 (2011).
Laplante, M. & Sabatini, D. M. mTOR signaling in growth control and disease. Cell 149, 274–293 (2012).
Derossi, D., Williams, E. J., Green, P. J., Dunican, D. J. & Doherty, P. Stimulation of mitogenesis by a cell-permeable PI 3-kinase binding peptide. Biochem. Biophys. Res. Commun. 251, 148–152 (1998).
Acknowledgements
We thank the Science and Technology Innovation Centre, the Institute of Hepatobiliary Research and the Institute of Rheumatology and Immunology of North Sichuan Medical College for providing the experimental sites and equipment.
Funding
This study was supported by the project of Scientific Research and Development Programme of North Sichuan Medical College, China (Grant No. CBY23-QNA15).
Author information
Authors and Affiliations
Contributions
X.L. conceived the study, conducted the formal analysis and investigation, performed the data visualization, and drafted the initial manuscript. X.L. also contributed to the data analysis, validation, and methodology. C.Z. and B.C. contributed to the review and editing of the initial manuscript and provided supervisory support. Y.X., Q.Z., and E.W. provided supervision for the initial manuscript, confirming the authenticity of all the raw data. Y.H. conceived and supervised the study and contributed to the writing, reviewing, and editing of the manuscript. All authors have read and approved the final manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Ethical approval
The animal experimentation procedures in this study followed the guiding principles of the Basel Declaration. This research received approval from the Animal Ethics Committee of North Sichuan Medical College (approval no. 2023-051).
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Liu, X., Zhou, C., Cheng, B. et al. Genipin promotes the apoptosis and autophagy of neuroblastoma cells by suppressing the PI3K/AKT/mTOR pathway. Sci Rep 14, 20231 (2024). https://doi.org/10.1038/s41598-024-71123-w
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-024-71123-w
- Springer Nature Limited