
Abstract
Antibiotic resistance in bacterial pathogens is a major threat to global health, exacerbated by the misuse of antibiotics. In hospital practice, results of bacterial cultures and antibiograms can take several days. Meanwhile, prescribing an empirical antimicrobial treatment is challenging, as clinicians must balance the antibiotic spectrum against the expected probability of susceptibility. We present here a proof of concept study of a machine learning-based system that predicts the probability of antimicrobial susceptibility and explains the contribution of the different cofactors in hospitalized patients, at four different stages prior to the antibiogram (sampling, direct examination, positive culture, and species identification), using only historical bacterial ecology data that can be easily collected from any laboratory information system (LIS) without GDPR restrictions once the data have been anonymised. A comparative analysis of different state-of-the-art machine learning and probabilistic methods was performed using 44,026 instances over 7 years from the Hôpital Européen Marseille, France. Our results show that multilayer dense neural networks and Bayesian models are suitable for early prediction of antibiotic susceptibility, with AUROCs reaching 0.88 at the positive culture stage and 0.92 at the species identification stage, and even 0.82 and 0.92, respectively, for the least frequent situations. Perspectives and potential clinical applications of the system are discussed.
Similar content being viewed by others


Introduction
According to the World Health Organization, antibiotic resistance of bacterial pathogens is “one of the biggest threats to global health” and is accelerated by the misuse of antibiotics1,2. In clinical practice, initial antimicrobial treatment is often prescribed empirically until definite results of bacterial cultures and antibiograms are available. This is a challenging gamble based on probabilistic reasoning, which can get complex, even for experienced clinicians3. To limit the selection of resistant bacteria while effectively treating patients, the aim is to minimize the spectrum width of empiric antibiotics prescribed, while maximizing the likelihood of their activity against suspected germs, depending on the location and severity of the infection and the consequences of treatment failure (Supplementary Fig. S1)4. To mitigate this risk and save broad-spectrum molecules for critical and resistant infections, prescribers are encouraged to follow clinical guidelines such as those edited by earned societies of infectious diseases5,6. They are also supposed to take into account the local microbial ecology, which often proves difficult and gets unrealistic when considering patient subgroups based on demographic or comorbidity characteristics7. Considering that one in three patients in European hospitals receives at least one antimicrobial8, this is a daily issue.
Clinical decision support systems (CDSS) may guide prescribers in their choices of empirical antimicrobial therapy9,10,11 and thus bring precision medicine to hospital-based antimicrobial stewardship programs12. So far, most of these CDSS are expert systems and, in clinical practice, historical antimicrobial resistance data have been mainly used to create institutional or unit-specific antibiograms for surveillance purposes10,13. Nowadays machine learning (ML) based CDSS, ML-CDSS, have grown increasing interest, and some studies have attempted to predict drug resistance at the patient level7,14,15,16,17,18,19,20,21,22,23,24,25,26,27. Based on our systematic review of previously published studies (Supplementary Table S1), focusing on their context, outcome, data used for training, types of ML algorithms, and prediction performance, it appears that most of them required numerous variables that are often difficult to extract from electronic medical records (EMR). Moreover, all these algorithms also included patient’s demographic characteristics, which analysis is restricted in Europe due to the General Data Protection Regulation (GDPR)28.
In this study, we present a proof of concept ML-CDSS designed to assist hospital doctors in selecting empiric antimicrobial therapy for a wide range of infected body sites and bacterial types and species, based on bacterial ecology data that is readily available from any laboratory information system (LIS) and not constrained by GDPR regulations. To do so, we undertook a comprehensive data curation process of the historical bacterial resistance data from the Hôpital Européen Marseille, a French general hospital. We then developed and compared different machine learning, frequentist, and Bayesian algorithms to predict antimicrobial resistance, at the successive stages of the bacteriological process as intermediate laboratory results are routinely used to refine the empiric antimicrobial therapy until the antibiogram is finally available: (1) ‘sampling’ of the clinical specimen; (2) ‘direct’ microscopic examination; (3) characterisation of a positive ‘culture’; and (4) ‘species’ identification (Supplementary Fig. S2). We have developed a prototype ML-CDSS web-based application (Supplementary Fig. S3) including some of the most performing algorithms.
Results
Data description and antibiotic resistance distribution
From January 2014 to December 2020, the bacteriology laboratory of the Hôpital Européen Marseille performed 44,026 antibiograms from clinical specimens (Supplementary Table S2). After removing likely contaminants of bacteria culture, duplicates, screening specimens systematically sampled to identify and isolate the asymptomatic carriers of multidrug-resistant (MDR) bacteria, characterizing the typical aspects on direct microscopic examination after Gram staining and characterizing the typical cultural macroscopic and microscopic features of a positive culture for each bacterial species, we analysed a cleaned dataset of 30,975 antibiograms, isolated from 13,166 patients from 6 different types of wards, mainly the emergency room (25%), critical care (24%), surgery (20%) or medicine (19%). Most bacteria were isolated from urine (41%), lower respiratory tract (23%), blood (14%) or abscesses (12%) specimens. They were mostly Gram-negative rods (71%), including Enterobacteriaceae-like (56%) and non-fermentative (11%) Gram-negative rods, and Gram-positive cocci (27%), including Staphylococcus-like (13%) and Enterococcus-like (10%) Gram-positive cocci (Supplementary Table S2). Major species included Escherichia coli (29%), Staphylococcus aureus (12%), Klebsiella pneumoniae (10%), Enterococcus faecalis (9%) or Pseudomonas aeruginosa (8%) (Supplementary Table S2). The cleaned dataset was divided into a (80% training + 20% testing) dataset of 26,621 antibiograms from January 2014 to December 2019, including 4915 antibiograms in 2019 alone, and a validation dataset of 4354 antibiograms from January to December 2020 (Supplementary Table S2).
Susceptibility to 22 single antibiotics (traditional antibiogram) and 25 antibiotic combinations (combination antibiogram) of clinical interest was interpreted for each bacterial isolate. The mean traditional and combination antibiograms were calculated for each group of isolate (e.g. critical care, urine, Gram-negative rods, or Staphylococcus aureus...) and plotted in Fig. 1 and summarised in Supplementary Table S3. Aspects on direct examination, cultural features and species showed significant heterogeneity in susceptibility to single antibiotics (root-mean-square deviation [RMSD], 33%, 37% and 43%, respectively). Susceptibility rates were also quite heterogeneous between specimen origins too (RMSD, 14%): for example, susceptibility to Amoxicillin-Clavulanate (Amox-Clav) was 56% in urine, but 35% in lower respiratory tract specimens. Global heterogeneity was less important, but still discriminant between ward types (7%): susceptibility to Cefotaxime/Ceftriaxone (CTX/CRO) was only 49% for critical care, but 77% in emergency room isolates. Similarly, the categories of history of MDR bacterial carriage (RMSD, 11%) showed important differences in susceptibility to certain antibiotics: susceptibility to CTX/CRO was only 27% in the case of MDR carriage in the last 3 months, 54% when no MDR carriage was documented for a known patient, and 72% for isolates from previously unknown patients in the hospital. Finally, global susceptibility to single antibiotics appeared stable over time (RMSD between periods, 2%), although there were notable differences for CTX/CRO, Ertapenem, Amikacin, or Trimethoprim-Sulfamethoxazole (TMP-SMX) (Fig. 1 and Supplementary Table S3).

Mean antibiograms for 22 single antibiotics (A and B, traditional antibiogram) and 25 antibiotic combinations (C and D, combination antibiograms), for isolates grouped by aspects on direct examination, cultural features and species (A and C), and by specimen origins, main ward types, past multidrug bacteria carriage history and periods (B and D). Only the most frequent direct types, culture types and species in the dataset are presented. Color gradient shows the mean antibiotic susceptibility from 0 (always resistant) to 1 (always susceptible). For each group (i.e. direct types, specimen types, period...) the heterogeneity of susceptibility between categories was estimated by the root-mean-square deviation (RMSD). The higher the RMSD, the higher the importance of the corresponding feature should be in the prediction algorithms. Amox-Clav, Amoxicillin-Clavulanate; Pip-Tazo, Piperacillin-Tazobactam; TMP-SMX, Trimethoprim-Sulfamethoxazole; CTX/CRO, Cefotaxime/Ceftriaxone.
Comparison of antibiotic susceptibility prediction models for all isolates
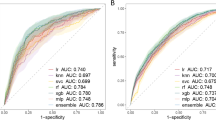
Using the 2014–2019 antibiogram dataset (80% training + 20% testing) with available features (specimen origin, type of ward, previous multidrug resistance [MDR] bacterial carriage and sampling date), we trained different frequentist (FRQ) and Bayesian (BAY) inference models, as well as machine learning (ML) algorithms (Logistic Regression [LR], AdaBoost [ADA], Gradient Boosting [GBS], Extreme Gradient Boosting [XGB], Bagging [BAG], Random Forest [RF] and Neural Networks [NN]) to predict antibiotic susceptibility at each of the intermediate stages of the bacteriological process prior to the antibiogram results (see Supplementary Table S4). These four intermediate stages were: (1) ‘sampling’ stage (models including the specimen origin to provide the specific ecology for a body site); (2) ‘direct’ stage (models including the aspect at direct Gram stain microscopic examination of the specimen); (3) ‘culture’ stage (models including the cultural feature at macroscopic and Gram stain examination of a positive culture); and (4) ‘species’ stage (models including the species bacterial identification of a positive culture). We then used these models to predict the susceptibility probability to each of the 22 single antibiotics and 25 antibiotic combinations for isolates from the 2020 dataset (validation). Mean receiver operator characteristic (ROC) curves for each stage are depicted in Fig. 2 and mean areas under the ROC curves (AUROC) were estimated and detailed in Fig. 2 and Supplementary Table S5. The AUROC for each stage and each antibiotic were also estimated and are shown in Fig. 3.

Mean under the receiver operator characteristic (ROC) curves of frequentist, Bayesian, and machine learning susceptibility prediction models at each stage of the bacteriological process and for all isolates of the 2020 validation dataset. Models: BAY, Bayesian; NN, Neural Network; FRQ, frequentist; XGB, Extreme Gradient Boosting; GBS, Gradient Boosting; ADA, AdaBoost; BAG, Bagging; RF, Random Forest; LR, Logistic regression. AUC, area under the ROC curve.

Distribution of the areas under the receiver operator characteristic curves (AUROC) of frequentist, Bayesian, and machine learning susceptibility prediction models for each antibiotic, at each stage of the bacteriological process and for all isolates of the 2020 validation dataset. Models: BAY, Bayesian; NN, Neural Network; FRQ, frequentist; XGB, Extreme Gradient Boosting; GBS, Gradient Boosting; ADA, AdaBoost; BAG, Bagging; RF, Random Forest; LR, Logistic regression.
The overall mean AUROC of the frequentist, Bayesian, and the seven ML models increased from 0.594 in stage 1 ‘sampling’ to 0.708 in stage 2 ‘direct’, 0.773 in stage 3 ‘culture’, and 0.847 in stage 4 ‘species’, with important differences between models (Fig. 2 and Supplementary Table S5). At all stages, the BAY, NN, FRQ, and XGB models showed much better prediction performance than other models (GBS, ADA, BAG, RF, and LR) (Fig. 2 and Supplementary Table S5). Notably, the NN models showed the highest mean AUROC (0.823) and the highest AUROC at each stage except stage 4, where the AUROC of the BAY model reached 0.918. Conversely, the mean AUROC of the FRQ models was slightly impaired at late stages (Fig. 2 and Supplementary Table S5), as prediction omissions increased for situations in the 2020 validation dataset that were not met in the 2014–2019 training+testing dataset: from 0.7% at stage 1, to 1.4% at stage 2, 2.6% at stage 3, and 10.2% at stage 4. Omission rates remained zero for Bayesian and ML models.
Predictive performance was very heterogeneous between antibiotics (Fig. 3), with antibiotic-specific standard deviations (SD) of AUROC ranging from 0.03 to 0.17, depending on stage and model (Fig. 2). In particular, intermediate stages 2 (‘Direct’) and 3 (‘Culture’) showed the highest variability, with model-specific SD ≥ 0.09. NN models showed the lowest SD (from 0.06 to 0.09 depending on the stage), and the prediction performance of BAY models was also quite consistent between antibiotics (SD from 0.06 to 0.10) (Fig. 2). For example, the AUROC of BAY models ranged from 0.566 for TMP-SMX to 0.850 for Meropenem + Vancomycin in stage 1 and from 0.719 for Levofloxacin to 1.000 for Metronidazole in stage 4.
Comparison of antibiotic susceptibility prediction models for the least frequent situations
To assess the predictive performance of the models for rare situations for which such ML-CDSS should be more clinically useful, the distribution of AUROC for only the least frequent situations (5th percentile of the number of occurrences in the 2014–2019 training+testing dataset) was evaluated and plotted (Fig. 4), and the mean AUROC was estimated for each model and each stage (Fig. 2 and Supplementary Table S5).

Distribution of individual areas under the receiver operator characteristic curves (AUROC) of frequentist, Bayesian, and machine learning susceptibility prediction models for each antibiotic, at each stage of the bacteriological process and for isolates of the 2020 validation dataset corresponding to the least frequent situations only (5th percentile of the number of occurrences in the 2014–2019 dataset). Models: BAY, Bayesian; NN, Neural Network; FRQ, frequentist; XGB, Extreme Gradient Boosting; GBS, Gradient Boosting; ADA, AdaBoost; BAG, Bagging; RF, Random Forest; LR, Logistic regression.
The combined overall AUROC of all models decreased from 0.731 to 0.656 when restricted to the rarest situations (Fig. 2 and Supplementary Table S5). LR proved to be the worst model at all stages (mean AUROC at 0.554) (Fig. 2, Supplementary Table S5 and Fig. 4). Conversely, the BAY models continued to perform well, with mean AUROCs as high as 0.824 and 0.917 for stages 3 and 4, respectively. Mean AUROCs for NN models were less conserved, especially for stages 1 and 2, where they decreased from 0.693 to 0.512 and from 0.811 to 0.658, respectively. For FRQ models, the mean AUROC at stage 4 fell from 0.893 to 0.662, as many of these rare situations were unmet in the dataset used to train them (Fig. 2, Supplementary Table S5 and Fig. 4).
Overall, based on these comparisons, the best models appeared to be the BAY and NN models.
Contribution of cofactors and model interpretability
To interpret the predictions of the NN models, we used the SHapley Additive exPlanations (SHAP) and first assessed the importance of each feature in the NN models at each stage (Fig. 5). History of MDR bacterial carriage in the last 3 months appeared to be a strong predictor at all stages, with high absolute SHAP values, particularly at stages 1 (‘Sampling’) and 2 (‘Direct’): while a known prior positive carriage of MDR bacteria (in red) was a predictor of decreased antibiotic susceptibility, a known prior negative carriage of MDR bacteria (in blue) was a predictor of increased susceptibility. The direct examination aspect also showed a significant influence on the prediction at stage 2. Unsurprisingly, the cultural feature contributed most to the NN model performance at stage 3 (‘Culture’). At stage 4 (‘Species’), species then cultural features were the strongest contributors to predictions. Conversely, the specimen origin, the ward type and the date had a less significant effect on prediction at all stages.

Relative importance of cofactors in the NN antibiotic susceptibility prediction models, at each stage of the identification process at population level, using the SHapley Additive exPlanations (SHAP). The colour indicates the value of a feature included in the model: for example, red indicates known prior positive carriage of MDR bacteria, blue indicates known prior negative carriage of MDR bacteria and grey indicates unknown prior carriage. A feature with a negative SHAP value is associated with a reduced probability of antibiotic susceptibility.
As an illustration of how features can influence an individual prediction of antibiotic susceptibility by the NN models, we plotted SHAP values at the individual level using a didactic scenario (Fig. 6). In the first example, the predicted susceptibility to CTX/CRO at stage 3 (culture) is 0.87 (Fig. 6A); the culture type (Enterobacteriaceae-like Gram-negative bacteria) and the unknown history of MDR carriage (in red) explain this good probability. In the last scenario, the predicted susceptibility to CTX/CRO at stage 4 (species) is 0.40 (Fig. 6E); the species (Citrobacter koseri), the recent history of a MDR carriage and the critical care ward (in blue) explain this bad probability; they are in part compensated by the Enterobacteriaceae-like Gram-negative bacteria type of this species (in red).

Illustration of the interpretability of the NN and BAY models at individual level, using a didactic scenario with prediction of the susceptibility to Cefotaxime/Ceftriaxone (CTX/CRO) at various stages. (A) A patient unknown from the hospital is managed at the emergency room for a urinary tract infection; a urine specimen is sent to the laboratory and grows an Enterobacteriaceae-like Gram-negative bacteria (stage 3 ‘Culture’). (B) It turns out that she has a known prior negative carriage of MDR bacteria, and the urine specimen grows Escherichia coli (stage 4 ‘Species’). According to the final antibiogram, this E. coli produces an extended-spectrum beta-lactamase (MDR bacteria). (C) One month later, the same patient is admitted in critical care for a septic shock and blood cultures are sampled (stage 1 ‘Sampling’). (D) These blood cultures show a Gram-negative rod (stage 2 ‘Direct’). (E) A Citrobacter koseri is identified (stage 4). For each stage, the panel lists the number of similar situations in the training+testing dataset, the probability of susceptibility to CTX/CRO predicted by the NN and BAY models. For NN models, the waterfall plot shows how each feature contributes to pushing the model output from a base value \(E[f(X)]\) (the average model output over the testing dataset) to the individual output \(f(x)\). Features that increase the probability of the susceptibility to CTX/CRO are shown in red, and those decreasing the probability are shown in blue. For BAY models, each panel presents the likelihood ratio (\(LRT\)) associated with each feature. \(LRT >1\)increase the susceptibility probability higher, while \(LRT <1\) decrease the susceptibility probability.
Using the same scenario, NN and BAY models exhibited close susceptibility predictions, except for least frequent situations (Fig. 6): for the last prediction, the training+testing dataset included only one similar situation, and the predicted susceptibility to CTX/CRO was 40% with NN and 78% with BAY models (Fig. 6E). Covariate-specific SHAP values (NN) and corresponding likelihood ratios (BAY) appeared moderately consistent: on these examples, the overall Spearman’s rank correlation coefficient was 0.57 (p-value, 0.0033). Correlation appeared the best for the MDR carriage history (specific coefficient, 0.92), or the ward (0.89) (Fig. 6). But for culture types and species at stage 4, both NN and BAY showed more contrasted SHAP values and likelihood ratios, as NN models included both cofactors whereas BAY models only included the species (Fig. 6B and E).
Discussion
This study presents an original proof of concept of a ML-CDSS designed to help hospital physicians choose the right empiric antimicrobial therapy, by predicting antimicrobial susceptibility using machine learning and simple historical laboratory data, from a clinically relevant perspective. Indeed, to the best of our knowledge, this work is the first study aimed at predicting the susceptibility to a wide range of antibiotics and antibiotic combinations in a hospital setting across the successive stages of the bacteriological process, from (1) sampling of the clinical specimen, to (2) direct microscopic examination, (3) characterisation of a positive culture, then (4) species identification (see literature review in Supplementary Table S17,14,15,16,17,18,19,20,21,22,23,24,25,26,27). Such intermediate laboratory results are routinely used to refine the empiric antimicrobial therapy until the antibiogram is finally available to adapt the definitive treatment.
To train and validate our models, we used an extensive and heterogeneous dataset, including 44,026 antibiograms from all types of patients and specimens over a 7-year period in a general hospital. The implemented algorithms showed good predictive AUROCs, especially for stages 2 (direct), 3 (culture) and 4 (species) being of 0.811, 0.875 and 0.918, respectively. However, at the early stage 1 (sampling) the mean AUROC was only 0.693. Futhermore AUROCs varied widely between antibiotics, as previously reported (Supplementary Table S1). Recognizing the model’s low performance at the early stage 1 (sampling), we are considering various improvements as future work, such as subsequent stages data imputation using machine learning and probabilistic approaches, as well as enriching the dataset with additional patient information, including comorbidities.
Concerned about of the practical utility of the predictions provided by the system, we undertook a comprehensive data curation process following published guidelines and usual good practices in clinical microbiology and antimicrobial stewardship. This included the interpretation of intrinsic and cross-resistance of bacteria to clinically relevant antibiotic groups and combinations. Before training the models, we also removed likely contaminants, duplicates, and screening specimens that were systematically sampled to identify and isolate the asymptomatic carriers of multidrug-resistant (MDR) bacteria and prevent cross-hospital transmission, as these isolates never warrant antimicrobial prescription. Such important data curation steps were rarely specified in 15 previously published studies (see literature review in Supplementary Table S1).
Our study evaluated a wide variety of statistical and state-of-the-art machine learning models as well as “home-made” frequentist (FRQ) and Bayesian (BAY) inference models (Supplementary Table S4), and found heterogeneous prediction performance among them. Neural networks (NN) and BAY models showed the highest mean AUROC for both early (sampling), intermediate (direct and culture) and late (species) stages, even for the least frequent situations for which such prediction algorithms should be more clinically useful. AUROCs of the NN and BAY models also showed the lowest standard deviations between antibiotics. In particular, BAY models kept good and steady AUROCs in rare situations, because these models calculate antibiotic susceptibility probabilities by combining pretest probabilities and likelihood ratios estimated from more general data. But these models were burdensome to program, whereas NN models can be implemented and tuned using open source packages and they are scalable to new input variables. Conversely, the performance of FRQ models was impaired at late stages, as prediction omissions increased in specific situations with previously unmet combinations of features. Logistic regression (LR) was also found to be inaccurate, even at late stages. Ensemble models, such as AdaBoost (ADA), Gradient Boosting (GBS), Bagging (BAG), and Random Forest (RF), but not Extreme Gradient Boosting (XGB), showed poorer performance than BAY and NN models. The superior performance of XGB over GBS can be attributed to the higher regularisation measures, which effectively mitigate overfitting. The poor results obtained by the LR models might be partly due to the choice of label encoding to convert categorical variables into numerical ones. As the other models are not particularly sensitive to this type of encoding, we chose label encoding to make the models more disk space efficient, to avoid sparsity (the variable species had 384 possible values), and to reduce training times. As future work, when more variables are included in the analysis, we will re-examine which of the label encoding or one-hot encoding is more relevant.
Antibiotic susceptibility was very heterogeneous between the different aspects on direct examination, cultural features and species. This supports our decision to develop specific models for each stage of the bacteriological process, although optimising a unique machine learning model covering all stages is an area for further improvement, in order to reduce the complexity when executed by a mobile app. The specimen origin, previous carriage of multidrug resistant (MDR) bacteria, and ward type showed intermediate levels of susceptibility heterogeneity, justifying the inclusion of these features in the models. The global antibiotic susceptibility showed low heterogeneity between period categories, but it remained important to include this temporal feature in the models as resistance of certain bacterial species to certain molecules like Ceftriaxone/Cefotaxime, Ciprofloxacin or TMP-SMX is known to evolve rapidly over time, which was also observed in our dataset. However, properly incorporating the date into the machine learning models remained a challenge due to the concept drift issue29. As future improvements, we plan to consider the month and year as separate variables, as well as use temporal sliding windows for training and testing. We will also train ensembles on different periods and monitor performance with concept drift detectors. Additionally, to keep the model updated with the evolution of antibiogram resistance, we plan to include continuous learning approaches.
Feature heterogeneity was included within the BAY models through the combination of a pre-test probability and feature-specific likelihood ratios, which can provide a direct interpretability of the predictions. For NN and other ML models, we used SHapley Additive exPlanations (SHAP) values to assess and visualise the impact of each feature on the predicted antibiotic susceptibility at both population and individual levels. Notably, all features contributed to the prediction, with direct, culture, species, and history of MDR carriage categories bearing the highest weight, depending on the laboratory stage. The influence of features on prediction was generally consistent between the NN models (SHAP values) and the corresponding BAY models (likelihood ratios). However, the predictions of both models showed some discrepancies, especially for rare situations, which may be due to their very dissimilar learning and predicting strategies. Our results underline the partial interpretability of the ML predictions, a feature that has not consistently been reported in previous studies (Supplementary Table S1), but is essential for instilling confidence to medical doctors using CDSS and ML-CDSS.
We have chosen to present the probability of susceptibility as the output of our models, instead of the most likely outcome (i.e. susceptible or resistant), because we believe that such ML-CDSS should enrich the probabilistic reasoning and risk assessment of antibiotic prescribers, rather than replace their decisions9,10. For example, the minimum probability of susceptibility required by the clinician prescribing an empiric antimicrobial will be higher for a patient with a critical infection than for a mild or moderate infection. The proposed system is not intended to replace practice guidelines either30,31, but to help prescribers to adapt these empiric antimicrobial therapy recommendations to the best available data on bacterial ecology at all stages of the bacteriological process prior to the antibiogram. As part of our future work, we intend to incorporate an assessment of the models’ uncertainty when delivering predictions. This will furnish clinicians with additional information to help them decide whether to consider the provided results or not.
Our prediction models included only a few features such as specimen type, ward type and history of MDR carriage. They did not include demographics, comorbidities and history of antibiotic treatment, although such features have been shown to be predictive of the antibiotic resistance (see literature review in Supplementary Table S1). Compared with the predictive performance of these previously published models, our NN models showed poorer performance in terms of AUROCs at the early stage 1 (sampling) due to the limited number of features included. However, the performance improved at the intermediates stages 2 (direct) and 3 (culture), and was even excellent at the late stage 4 (species) (Supplementary Table S1). For example, the AUROCs reported by Lewin-Epstein et al.26 with a very heterogeneous hospital dataset like ours but including numerous patient characteristics from the EMR, were 0.73–0.79 at stage 1 and 0.80–0.88 at stage 4, whereas the AUROCs of our BAY models were 0.57–0.66 and 0.86–0.97, respectively, for similar antibiotics (data not shown). We therefore plan to include demographic features (such as age, sex, residence location), history of antibiotic consumption or clinical history in NN and other ML models, and evaluate predictive gains, especially at the earliest stage 1 (sampling) for which our algorithms have so far provided insufficient performance for clinical decision support.
However, it is worth noting that such patient-related sensitive data are now strictly controlled in Europe by the GDPR28. Besides, they are typically stored in electronic medical record (EMR) systems with heterogeneous formats, and are not readily accessible in all hospital facilities. Conversely, our models maintained good performance despite using only simple bacterial ecology data, which can be easily extracted from any laboratory information system (LIS) without any GDPR restrictions after on-site anonymisation. Of course, these AMR predictions and model performances are specific to our dataset and reflect the specific bacterial ecology of Hôpital Européen Marseille (HEM). However, we hypothesize that the overall architecture of these algorithms is generalisable and that similar models could be easily adapted to other hospitals, trained with specific bacteriological data. This will be specifically evaluated in an ongoing multi-centre study aimed at testing AMR prediction models in six French hospitals from different regions, including HEM. Through an interoperable health data warehouse being set up in eight French hospitals including HEM, we also plan to further develop our AMR prediction models and assess the impact of data pooling and updating on prediction performance, while taking into account the temporal and spatial heterogeneity of antimicrobial resistance. A federated learning approach, in which only the model parameters are shared between these hospitals rather than data, will also be evaluated and compared with a centralized approach and a local one.
In parallel, several studies are underway to assess the potential utility of such algorithms in clinical microbiology and antimicrobial stewardship. In the six aforementioned hospitals, the susceptibility probability and spectrum width of empiric antimicrobial therapies actually prescribed will be retrospectively described to assess the potential efficacy gains (beneficial adaptations) and spectrum width reductions (safe de-escalations) that prediction algorithms may have induced. In addition, these actual therapies as well as empiric antimicrobial therapies recommended a posteriori by an expert panel before and after the use of prediction algorithms, will be compared overall and with the final expert recommended therapies after adaptation to the antimicrobial susceptibility results (antibiograms). Further intervention studies, followed by implementation science will also be needed to best integrate these modern tools into clinical practice32,33. The proposed AMR prediction algorithms could then be embedded in a standalone application that could be used by clinicians at the patient’s bedside, without the need to complete lengthy online forms. The models could also be embedded directly into the laboratory information management system (LIMS) to provide preliminary probabilistic antibiograms, or into EMR systems or health data warehouses as an ML-CDSS for prescribers, or as a dashboard flagging antimicrobial prescriptions with low susceptibility probability or useless broad-spectrum activity for antimicrobial stewardship teams.
In conclusion, we believe that the regular use of such predictive algorithms trained with simple laboratory data could significantly improve the relevance of empiric antimicrobial prescriptions in hospitals, even at intermediate stages of the bacteriological process. It could reduce the risk of treatment inadequacy and failure, save broad-spectrum molecules, prevent bacterial resistance selection and significantly improve the current standard of care34. Such algorithms may be particularly useful for on-call infectiologists who are asked to provide antimicrobial stewardship advice in unfamiliar hospitals, or in countries where antibiograms are not routinely available and where antimicrobial resistance creates the most serious problems35. A prototype web application based on the Gradio Python package36 and hosted on a private Huggingface space (https://huggingface.co/) is being tested at HEM (see Supplementary Fig. S3). We hope that this pilot work, as well as ongoing and planned studies to assess the potential utility of such algorithms, will contribute to the fight against the pandemic antibiotic resistance and the emergence of a post-antibiotic era.
Methods
Study design and settings
The aim of this contribution is to present a monocentric retrospective proof-concept study to develop and evaluate antimicrobial susceptibility prediction models that could be incorporated into a ML-CDSS to assist clinicians in selecting empirical antimicrobial therapy for a wide range of infected body sites and bacterial types and species, and at the successive intermediate stages of the bacteriological process until the antibiogram is finally available in the laboratory : (1) ‘sampling‘ of the clinical specimen; (2) ‘direct‘ microscopic examination; (3) characterisation of a positive ‘culture’; and (4) ‘species’ identification (Supplementary Fig. S2). Bacterial ecology data (i.e., positive bacterial cultures and antimicrobial susceptibility—antibiograms) from the Hôpital Européen Marseille, a French general hospital located in the popular central districts of a city of nearly 900,000 people in southeastern France, were analysed between January 2014 and December 2020.
After a comprehensive data curation process to generate clinically relevant features, we developed several frequentist, Bayesian, and state-of-the-art machine learning models to predict antimicrobial resistance. We then evaluated the performance of these models, and illustrated their interpretability (Fig. 7). The study was conducted in accordance with the recommendations of the TRIPOD statement on multivariate prediction models for individual prognosis or diagnosis (see Supplementary Table S6)37.

Overall process of the data curation, features preparation, development of antibiotic susceptibility prediction models, and evaluation of their performance and interpretability..
Data
All antibiograms since the opening of the Hôpital Européen Marseille were extracted from the bacteriology laboratory information system, between January 2014 and December 2020, in compliance with the General Data Protection Regulation (GDPR). The specimen were cultured following the latest guidelines of the French learned societies of microbiology30. Bacterial identification was carried out using matrix-assisted laser desorption ionisation/time of flight mass spectrometry (MALDI/ToF MS) with VITEK® MS (bioMérieux France, Craponne, France). Antibiotic susceptibility testing was performed using the automated VITEK® 2 system (bioMérieux France) or diffusion techniques on agar plates when appropriate, and minimal inhibitory concentrations (MICs) were prospectively interpreted using SIRxpert Master® software (i2a, Montpellier, France) based on the prevalent French (CA-SFM) and European (EUCAST) guidelines38,39.
Feature definition and data preparation
Based on the metadata and results of each antibiogram, we characterised: (1) the type of ward (e.g. emergency room, intensive care, medicine, surgery, day care unit...); (2) the body site origin of the specimen (e.g. blood or intravenous catheter, urine, lower respiratory tract, joint/bone, digestive, genital, cerebrospinal..); (3) the history of a previous multidrug resistant (MDR) bacteria carriage for the same patient within the last three months (i.e. usual threshold for risk factors of MDR bacterial infections)40; (4) the clinical relevance of the bacterial isolates taking into account the body site (i.e. relevant or likely contaminants such as a single blood culture positive for common bacteria of the skin flora such as Staphylococcus epidermidis)41; (5) duplicates (i.e. recurrent similar isolates and antibiogram within the last two days). To provide antibiotic susceptibility predictions based on information available at the ‘direct’ and ‘culture’ stages prior to the species identification (see Supplementary Fig. S2), we also characterised for each bacterial species: (6) the typical aspect on direct Gram stain examination of a specimen (i.e. Gram-positive cocci, Gram-negative rods...); and (7) the typical cultural feature on macroscopic and Gram stain examination of a positive culture (i.e. Staphylococcus-like Gram-positive cocci, Enterobacteriaceae-like Gram-negative rods, non-fermentative Gram-negative rods...) (see Supplementary Table S7). Finally, (8) we analysed the susceptibility (i.e. susceptible or resistant) of each bacterial isolate to 22 single antibiotics (traditional antibiogram) and 25 antibiotic combinations (combination antibiogram) of clinical interest (Table 1). Isolates with “intermediate” susceptibility were considered as resistant. In our lab, the “Susceptible, Increased Exposure” category was only introduced in 2021, after the study period. If susceptibility to a specific antibiotic was not available, we used recommended expert rules on intrinsic and cross-resistance30,42. These data preparation steps aimed to be clinically relevant and grounded in the practical issues encountered in the field of clinical microbiology and antimicrobial stewardship. The chosen criteria that did not rely on published studies or guidelines but on usual good clinical practices were validated by infectious diseases clinicians of the digital infectiology working group (Groupe d’Infectiologie digitale, GID) of the French infectious diseases earned society (SPILF)43.
After removing potential contaminants, duplicates, and rectal and nasal swab screening specimens for multidrug-resistant (MDR) bacterial carriage, we calculated the average antibiotic susceptibility rates and constructed mean antibiograms for individual antibiotics (traditional antibiograms) or combinations of antibiotics (combined antibiograms)44, for each type of each category of isolate: bacterial species; aspect on direct examination; cultural feature; specimen origin; ward type; and past history of MDR carriage. Mean antibiograms were plotted to illustrate the differences in susceptibility rates between each category of isolate. Root mean square deviations (RMSD) were also estimated for each category, using the mean susceptibility rates of all isolates as a reference.
For ML models, we chose the best way to convert categorical variables after comparing one-hot encoding and label encoding, considering that for certain models, such as logistic regression, one-hot encoding would be more appropriate, while for tree-based models, both encoding techniques would be acceptable. Both approaches gave similar results. We therefore decided to use label encoding as it is more space efficient, avoids dealing with sparse datasets and reduces the training times. For example, the variable ‘species’ had 384 possible values, so encoding it with a one-hot encoding way would not have been space efficient. As AMR evolves over time, and in order to give more weight to recent observations, the date was normalised between 0 and 1, with 1 being the most recent date.
The dataset was then divided into three parts: training, testing, and validation. The training and the testing datasets comprised 80% and 20% of the data registered between 2014 and 2019, respectively. The validation dataset consisted of all the data collected in 2020.
Global architecture of the antimicrobial susceptibility prediction models
The models were designed to predict antibiotic susceptibility predictions across the successive intermediate stages of the bacteriological process before the antibiogram is finally available in the laboratory, typically on Day 2–4 (see Supplementary Fig. S2). These four stages included : (1) the ‘sampling’ stage (models included the origin of the clinical specimen to provide the specific ecology for a body site, which is known before clinicians prescribe an empiric antimicrobial therapy on Day 0); (2) ‘direct’ stage (models included the aspect at direct Gram stain microscopic examination of the specimen, which feature can be provided by the laboratory on Day 0–1); (3) ‘culture’ stage (models included the cultural feature at macroscopic and Gram stain examination of a positive culture, which is usually communicated by the laboratory on Day 1–2 and allows clinicians to refine their empiric prescriptions); and (4) ‘species’ stage (models included the species bacterial identification of a positive culture, which is usually available on Day 2–3).
Cofactors or other features included in each stage are summarised in Supplementary Table S4. All models considered the specimen origin, the type of ward, and the past history of a previous MDR bacterial carriage. Stage 4 ‘species’ machine-learning (ML) models also included the culture type, as this feature improved predictions for rare situations, probably because the antibiotic susceptibility of rare species is generally close to that of species from the same culture type (e.g., non-fermentative Gram-negative rods). However, we did not include the direct type as a feature in stages 3 ‘culture’ and 4 ‘species’ ML models, because a direct type (e.g., Gram-positive cocci) can include heterogeneous species with contrasting susceptibility patterns.
A multifaceted approach was developed, which included frequentist inference, Bayesian inference, ensemble-based machine learning techniques, and dense neural networks. All models were trained using Python v3.6, with the Scikit-Learn Python library for ensemble-based machine learning models45, and the Keras library and KerasClassifier, a scikit-learn API wrapper, for the neural networks46. To ensure the generalisation of the machine learning approaches, the models’ hyperparameters were chosen using an exhaustive grid search methodology with 10-fold cross-validation over the training dataset. The models were then tested using the testing dataset and validated using the validation dataset. The results presented in this manuscript are those obtained with the validation dataset to ensure the generalisabilty of our models. Details of each model are provided in the following sections.
Frequentist inference models
For each antibiotic and each stage preceding antibiogram results, we first trained very simple frequentist inference models (FRQ) using the last year of the training+testing dataset (2019) and validated them using the validation dataset (2020). These frequentist models directly estimated the posterior susceptibility probability, as the proportion of antibiotic susceptibility within isolates with similar features within the last year of the training+testing dataset (2019) (Eq. 1, an example for the ‘species’ stage).
where \(P(S | \text {Species, Specimen, Ward, MDR})_{2019}\) is the probability of susceptibility P(S) to a given antibiotic group or combination within the last year of the training+testing dataset (2019), given the species of bacteria grown from positive cultures, the specimen origin, the type of hospital ward, and the previous history of MDR carriage (Supplementary Table S4). In case no similar situation was available in the training+testing dataset, we chose to attribute a probability of susceptibility of 0.5, so that these isolates were not excluded from AUROC estimations. With this kind of “intention to treat” analysis, AUROCs were thus impaired by prediction omissions, whereas a kind of “per protocol” analysis excluding prediction omissions would have artificially overestimated AUROCs.
Bayesian inference models
Using the whole training+testing dataset (2014–2019), we trained one Bayesian inference model (BAY) per antibiotic and stage and validated them using the validation dataset (2020). We first estimated likelihood ratios, \(LRT\), from prior and posterior susceptibility probabilities, after converting probabilities P(S) into odds O(S) (Eqs. 2 and 3). For all combinations of isolates features, posterior probabilities were then approximated, using prior probabilities and corresponding likelihood ratios (Eq. 4), and conversions from odds O(S) to probabilities P(S) (Eq. 5).
where \(O(S)_{2014-19}\) is the prior probability of susceptibility S to a given antibiotic group or combination, within the whole training+testing dataset (2014–2019), \(LRT_{2019}\) is the likelihood ratio for the last year of the training+testing dataset (2019), and \(LRT_{\text {Species}}\), \(LRT_{\text {Specimen}}\), \(LRT_{\text {Ward}}\), and \(LRT_{\text {MDR}}\) are the likelihood ratios for the species, the specimen, the hospital ward, and the previous MDR carriage, respectively (Supplementary Table S4).
Machine learning models
State-of-the-art supervised machine learning models were developed, trained and tested using the training+testing dataset (2014–2019). The models were then validated with the validation dataset (2020). Logistic Regression (LR) was used as a baseline. Various ensemble models were used, including AdaBoost (ADA)47,48, Gradient Boosting (GBS)49, Extreme Gradient Boosting (XGB)50, Bagging (BAG)51, Random Forest (RF)47 and neural networks (NN)46,52, were utilized. Support-vector machine (SVM) models were excluded in the exploratory analysis due to their poor performance, which was consistent with the limited number of categorical features in our dataset.
Logistic Regression (LR) is a type of generalised linear model used to estimate the probability of an instance belonging to a particular class, by computing a weighted sum of input features, along with a bias term.
Ensemble models are a group of learning methods that combine weak learners into a strong learner, providing a decision based on majority voting. In this study, decision tree learners were chosen, given their suitability for handling categorical variables. Three boosting methods were considered: AdaBoost (ADA), that focuses on underfit cases47,48; Gradient Boosting (GBS), that fits the new predictor to residual errors49; and Extreme Gradient Boosting (XGB), that uses a more regularized model to control overfitting50. Bagging (BAG) applies the same training model to every predictor, training each on different random subsets of the training set51. Random Forest (RF) is considered an improvement that changes the way sub-trees are learned to reduce correlations47.
Artificial Neural Networks (NN) are based on connected unit functions or nodes called artificial neurons46,52. One dense multilayer NN classifier with one output per antibiotic was implemented for each stage. Their architecture comprised (Supplementary Fig. S4): (1) an input layer that received relevant features and information of the data; (2) three hidden dense layers that captured complex patterns and relationships between features; (3) batch normalization layers, which were introduced before each hidden dense layer to standardize inputs and stabilize training; (4) dropout layers after each hidden dense layer to mitigate overfitting; and (5) a final output layer providing specific susceptibility predictions, with one node per antibiotic or combination of antibiotics. The hidden layers contained between 50 and 150 nodes. The intermediate layers used ReLU (Rectified Linear Unit) activation, while the final layer used sigmoid activation to generate an output between 0 and 1 corresponding to the probability of susceptibility to each antibiotic or antibiotic combination.
Evaluation of model performance
The prediction performance of frequentist (FRQ), Bayesian (BAY), and machine learning models was compared by conducting a receiver operator characteristic (ROC) analysis, using the validation dataset. Classifiers were antibiotic susceptibility probabilities, and binary outcomes were ‘susceptible’ or ‘resistant’ antibiogram results. Areas under the ROC curve (AUROC) were estimated for each antibiotic and antibiotic combination at each stage. The mean AUROC was calculated for each model at each stage, and mean ROC curves, along with AUROC distributions, were plotted. Models were compared based on mean AUROC globally and specifically for the least frequent situations (e.g., rare species in a rare specimen in a rare ward) represented by the 5th percentile of occurrences in the 2014–2019 dataset. For FRQ models, the prediction omission rate (i.e. ratio of the number of prediction omissions to the total number of predictions) was also calculated. The best model family was then chosen.
Model interpretability
Understanding model predictions is crucial in health-related applications. The FRQ and BAY models, which reflect the mean antibiograms (Fig. 1 and Supplementary Table S3), combine features directly (FRQ) or use feature-specific likelihood ratios (BAY). In LR, feature influence is included in odds ratios. RF models also allow for feature interpretation by providing the degree of contribution of each variable to the final decision. However, interpretation can be more difficult for other ML methods that are not directly explainable and are often considered as “black boxes”, such as neural networks (NN). To interpret NN predictions, we thus used the SHapley Additive exPlanations (SHAP), as previously used in other machine learning implementations for health prediction purposes33,53. The distribution of feature-specific SHAP values was plotted for each stage of the identification process at a population level using the SHAP Python package32. Moreover, to illustrate how features influence the prediction of antibiotic susceptibility at a patient level, we presented a didactic scenario in which the main features that influence the NN predictions (feature-specific SHAP values) were compared with the corresponding BAY predictions (likelihood ratios).
Data availability
The bacterial ecology data used in this study are specific to the Hôpital Européen Marseille and are not publicly available. However, these anonymised data can be obtained from the author S. Rebaudet upon reasonable request and signing a material transfer agreement. The code used for data analysis is available upon request to the author R. Urena.
References
World Health Organization (WHO). Antimicrobial resistance, https://www.who.int/news-room/fact-sheets/detail/antibiotic-resistance (2023).
Stracy, M. et al. Minimizing treatment-induced emergence of antibiotic resistance in bacterial infections. Science 375, 889–894. https://doi.org/10.1126/science.abg9868 (2022).
Strich, J. R., Heil, E. L. & Masur, H. Considerations for empiric antimicrobial therapy in sepsis and septic shock in an era of antimicrobial resistance. J. Infect. Dis. 222, S119–S131. https://doi.org/10.1093/infdis/jiaa221 (2020).
Raman, G., Avendano, E., Berger, S. & Menon, V. Appropriate initial antibiotic therapy in hospitalized patients with gram-negative infections: Systematic review and meta-analysis. BMC Infect. Dis. 15, 395. https://doi.org/10.1186/s12879-015-1123-5 (2015).
Infectious Diseases Society of America (IDSA). Idsa practice guidelines, https://www.idsociety.org/practiceguidelines#/name_na_str/ASC/0/+ (2023).
Société de Pathologie Infectieuse de Langue Française (SPILF). Recommandations, https://www.infectiologie.com/fr/recommandations.html (2023).
Yelin, I. et al. Personal clinical history predicts antibiotic resistance of urinary tract infections. Nat. Med. 25, 1143. https://doi.org/10.1038/s41591-019-0503-6 (2019).
Plachouras, D. et al. Antimicrobial use in European acute care hospitals: Results from the second point prevalence survey (PPS) of healthcare-associated infections and antimicrobial use, 2016 to 2017. Eurosurveillance 23, 1800393. https://doi.org/10.2807/1560-7917.ES.23.46.1800393 (2018).
Bremmer, D. N., Trienski, T. L., Walsh, T. L. & Moffa, M. A. Role of technology in antimicrobial stewardship. Med. Clin. North Am. 102, 955–963. https://doi.org/10.1016/j.mcna.2018.05.007 (2018).
Peiffer-Smadja, N. et al. Machine learning for clinical decision support in infectious diseases: A narrative review of current applications. Clin. Microbiol. Infect. https://doi.org/10.1016/j.cmi.2019.09.009 (2020).
Van Dort, B. A., Penm, J., Ritchie, A. & Baysari, M. T. The impact of digital interventions on antimicrobial stewardship in hospitals: A qualitative synthesis of systematic reviews. J. Antimicrob. Chemother. 77, 1828–1837. https://doi.org/10.1093/jac/dkac112 (2022).
Watkins, R. R. Antibiotic stewardship in the era of precision medicine. JAC Antimicrob. Resist. 4, dlac066. https://doi.org/10.1093/jacamr/dlac066 (2022).
Forrest, G. N. et al. Use of electronic health records and clinical decision support systems for antimicrobial stewardship. Clin. Infect. Dis. 59, S122-133. https://doi.org/10.1093/cid/ciu565 (2014).
Dan, S. et al. Prediction of fluoroquinolone resistance in gram-negative bacteria causing bloodstream infections. Antimicrob. Agents Chemother. 60, 2265–2272. https://doi.org/10.1128/AAC.02728-15 (2016).
Vazquez-Guillamet, M., Vazquez, R., Micek, S. & Kollef, M. Predicting resistance to piperacillin-tazobactam, cefepime and meropenem in septic patients with bloodstream infection due to gram-negative bacteria. Clin. Infect. Dis. 65, 1607–1614. https://doi.org/10.1093/cid/cix612 (2017).
Sullivan, T., Ichikawa, O., Dudley, J., Li, L. & Aberg, J. The rapid prediction of carbapenem resistance in patients with Klebsiella pneumoniae bacteremia using electronic medical record data. Open Forum Infect. Dis. 5, ofy091. https://doi.org/10.1093/ofid/ofy091 (2018).
Oonsivilai, M. et al. Using machine learning to guide targeted and locally-tailored empiric antibiotic prescribing in a children's hospital in cambodia. Wellcome Open Res. 3, 131. https://doi.org/10.12688/wellcomeopenres.14847.1 (2018).
Tandan, M., Timilsina, M., Cormican, M. & Vellinga, A. Role of patient descriptors in predicting antimicrobial resistance in urinary tract infections using a decision tree approach: A retrospective cohort study. Int. J. Med. Inform. 127, 127–133. https://doi.org/10.1016/j.ijmedinf.2019.04.020 (2019).
MacFadden, D. R. et al. Decision-support models for empiric antibiotic selection in gram-negative bloodstream infections. Clin. Microbiol. Infect. 25(108), e1-108.e7. https://doi.org/10.1016/j.cmi.2018.03.029 (2019).
Moran, E., Robinson, E., Green, C., Keeling, M. & Collyer, B. Towards personalized guidelines: Using machine-learning algorithms to guide antimicrobial selection. J. Antimicrob. Chemother. 75, 2677–2680. https://doi.org/10.1093/jac/dkaa222 (2020).
Kanjilal, S. et al. A decision algorithm to promote outpatient antimicrobial stewardship for uncomplicated urinary tract infection. Sci. Transl. Med. https://doi.org/10.1126/scitranslmed.aay5067 (2020).
Feretzakis, G. et al. Using machine learning techniques to aid empirical antibiotic therapy decisions in the intensive care unit of a general hospital in greece. Antibiotics 9, 50. https://doi.org/10.3390/antibiotics9020050 (2020).
Feretzakis, G. et al. Using machine learning algorithms to predict antimicrobial resistance and assist empirical treatment. Stud. Health Technol. Inform. 272, 75–78. https://doi.org/10.3233/SHTI200497 (2020).
Feretzakis, G. et al. Machine learning for antibiotic resistance prediction: A prototype using off-the-shelf techniques and entry-level data to guide empiric antimicrobial therapy. Healthc. Inform. Res. 27, 214–221. https://doi.org/10.4258/hir.2021.27.3.214 (2021).
Feretzakis, G. et al. Using machine learning to predict antimicrobial resistance of Acinetobacter baumannii, Klebsiella pneumoniae and Pseudomonas aeruginosa strains. Stud. Health Technol. Inform. 281, 43–47. https://doi.org/10.3233/SHTI210117 (2021).
Lewin-Epstein, O., Baruch, S., Hadany, L., Stein, G. Y. & Obolski, U. Predicting antibiotic resistance in hospitalized patients by applying machine learning to electronic medical records. Clin. Infect. Dis. 72, e848–e855. https://doi.org/10.1093/cid/ciaa1576 (2021).
Tzelves, L. et al. Using machine learning techniques to predict antimicrobial resistance in stone disease patients. World J. Urol. 40, 1731–1736. https://doi.org/10.1007/s00345-022-04043-x (2022).
Chico, V. The impact of the general data protection regulation on health research. Br. Med. Bull. 128, 109–118. https://doi.org/10.1093/bmb/ldy038 (2018).
Gama, J., Žliobaitė, I., Bifet, A., Pechenizkiy, M. & Bouchachia, A. A survey on concept drift adaptation. ACM Comput. Surv. (CSUR) 46, 1–37. https://doi.org/10.1145/2523813 (2014).
Société Française de Microbiologie. CASFM/EUCAST Recommandations 2021 V.1.0 Avril (2021).
Société Française de Microbiologie (SFM) and Société Française de Mycologie Médicale (SFMM) and Société Française de Parasitologie. Rémic - Référentiel en microbiologie Médicale, 6.2 edn. (2018).
Lundberg, S. M. & Lee, S.-I. A unified approach to interpreting model predictions. In Advances in Neural Information Processing Systems, vol. 30 (Curran Associates, Inc., 2017).
Bibault, J.-E. et al. Development and validation of an interpretable artificial intelligence model to predict 10-year prostate cancer mortality. Cancers https://doi.org/10.3390/cancers13123064 (2021).
van der Werf, T. S. Artificial intelligence to guide empirical antimicrobial therapy-ready for prime time?. Clin. Infect. Dis. 72, e856–e858. https://doi.org/10.1093/cid/ciaa1585 (2021).
Antimicrobial Resistance Collaborators. Global burden of bacterial antimicrobial resistance in 2019: A systematic analysis. Lancet 399, 629–655. https://doi.org/10.1016/S0140-6736(21)02724-0 (2022).
Abid, A. et al. Gradio: Hassle-free sharing and testing of ml models in the wild (2019). arXiv:1906.02569.
Collins, G. S., Reitsma, J. B., Altman, D. G. & Moons, K. G. M. Transparent reporting of a multivariable prediction model for individual prognosis or diagnosis (tripod): The tripod statement. J. Clin. Epidemiol. 68, 134–143. https://doi.org/10.1016/j.jclinepi.2014.11.010 (2015).
The European Committee on Antimicrobial Susceptibility Testing - EUCAST, https://www.eucast.org/.
Comité de l’Antibiogramme de la Société Française de Microbiologie - CA-SFM, https://www.sfm-microbiologie.org/presentation-de-la-sfm/sections-et-groupes-de-travail/comite-de-lantibiogramme/.
Haute Autorité de Santé. Antibiothérapie des infections à entérobactéries et à pseudomonas aeruginosa chez l’adulte: place des carbapénèmes et de leurs alternatives. recommandation de bonne pratique, https://has-sante.fr/jcms/c_2968915/fr/antibiotherapie-des-infections-a-enterobacteries-et-a-pseudomonas-aeruginosa-chez-l-adulte-place-des-carbapenemes-et-de-leurs-alternatives (2019).
Murray, P. R. The clinician and the microbiology laboratory. In Mandell, Douglas, and Bennett’s Principles and Practice of Infectious Diseases, 8th ed, 191–223, https://doi.org/10.1016/B978-1-4557-4801-3.00016-3 (Saunders, 2015).
European Committee on Antimicrobial Susceptibility Testing (EUCAST). Eucast: Expert rules and intrinsic resistance, http://www.eucast.org/expert_rules_and_intrinsic_resistance/ (2023).
Groupe infectiologie digitale (GID) de la Société de Pathologie Infectieuse de Langue Française (SPILF), https://www.infectiologie.com/fr/groupe-infectiologie-digitale.html.
Klinker, K. P. et al. Antimicrobial stewardship and antibiograms: Importance of moving beyond traditional antibiograms. Ther. Adv. Infect. Dis. 8, 20499361211011372. https://doi.org/10.1177/20499361211011373 (2021).
Pedregosa, F. et al. Scikit-learn: Machine learning in python. J. Mach. Learn. Res. 12, 2825–2830 (2011).
Keras. Keras: Deep learning for humans, https://keras.io/ (2023).
Ho, T. K. Random decision forests. In Proceedings of 3rd International Conference on Document Analysis and Recognition, vol. 1, 278–282 (1995).
Schapire, R. E. Explaining adaboost. In Empirical Inference: Festschrift in Honor of Vladimir N. Vapnik, 37–52, https://doi.org/10.1007/978-3-642-41136-6_5 Springer Berlin, Heidelberg, (2013).
Friedman, J. H. Greedy function approximation: A gradient boosting machine. Ann. Stat. 29, 1189–1232. https://doi.org/10.1214/aos/1013203451 (2001).
Chen, T. & Guestrin, C. Xgboost: A scalable tree boosting system. In Proceedings of the 22nd ACM SIGKDD International Conference on Knowledge Discovery and Data Mining, 785–794, https://doi.org/10.1145/2939672.2939785 (2016).
Breiman, L. Bagging predictors. Mach. Learn. 24, 123–140. https://doi.org/10.1007/BF00058655 (1996).
Hopfield, J. J. Neural networks and physical systems with emergent collective computational abilities. Proc. Natl. Acad. Sci. 79, 2554–2558. https://doi.org/10.1073/pnas.79.8.2554 (1982).
Lundberg, S. M. et al. Explainable machine-learning predictions for the prevention of hypoxaemia during surgery. Nat. Biomed. Eng. 2, 749–760. https://doi.org/10.1038/s41551-018-0304-0 (2018).
Acknowledgements
We thank Swapnesh Panigrahi, Leon Espinosa, Tâm Mignot and Renaud Piarroux for their feedback and suggestions in the analysis plan. This work was directly supported by Hôpital Européen Marseille and SESSTIM, and by a PhD Grant Awarded by the ISSPAM, Aix Marseille Université.
Author information
Authors and Affiliations
Contributions
SR developed the study idea. SR, RU, JCD and JG contributed to the analysis plan. SR and RU performed the literature review. SR and SC did the data investigation and management. SR, YB, LV and RU participated in the data management and analysis. SR and RU wrote the first draft. All authors had full access to the data, contributed to the interpretation of the findings, reviewed the analysis, wrote the manuscript, approved the final manuscript, and shared the final responsibility for the decision to submit for publication.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Ethical approval
This study utilised solely anonymised bacterial ecology data generated by the bacteriology laboratory at the Hôpital Européen Marseille. It is part of a broadest project focused on predicting antimicrobial resistance using hospital electronic health records, which received authorisation DR-2020-047 from the French national commission for data protection (CNIL, Commission nationale de l’informatique et des libertés).
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Urena, R., Camiade, S., Baalla, Y. et al. Proof of concept study on early forecasting of antimicrobial resistance in hospitalized patients using machine learning and simple bacterial ecology data. Sci Rep 14, 22683 (2024). https://doi.org/10.1038/s41598-024-71757-w
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-024-71757-w
- Springer Nature Limited