
Abstract
Bannayan–Riley–Ruvalcaba syndrome (BRRS) is a rare overgrowth condition caused by a pathogenic variant in the phosphatase and tensin homolog (PTEN) gene and belongs to a group of disorders called PTEN hamartoma tumor syndrome (PHTS). The diagnosis is often complicated by great phenotypic diversity. Furthermore, to this date treatment options are limited. Here we performed a systematic review using PubMed, Cochrane, and Scopus databases to identify cases of pediatric patients diagnosed with BRRS and summarized information about the clinical presentation, treatment, and long-term patient care. A total of 83 pediatric patients with BRRS were identified. The most common clinical findings were macrocephaly (77%) and developmental disorders (63%). Surgical interventions were the treatment of choice, described in 19 articles. Patient surveillance was proposed in 15 case reports and mostly aimed at periodic cancer screening. Recognition of BRRS clinical symptoms and early referral to a geneticist is important for better disease control and overall prognosis. As targeted treatment is still lacking, symptom relief and long-term surveillance remain the main management strategies.
Similar content being viewed by others


Introduction
The phosphatase and tensin homolog (PTEN) gene is a tumor suppressor gene that controls cell proliferation, survival, metabolism, and migration by downregulating the PI3K-AKT-mTOR pathway1 . PTEN pathogenic variants can cause several overgrowth syndromes, together known as PTEN hamartoma tumor syndrome (PHTS). These conditions include Cowden syndrome (CS), Bannayan-Riley-Ruvalcaba syndrome (BRRS), Lhermitte-Duclos disease, autism spectrum disorders with macrocephaly, Proteus, Proteus-like and VATER (Vertebral defects, Anal atresia, Tracheo-esophageal fistula with Esophageal atresia, and Radial and Renal dysplasia) syndromes2 . They share common key features, such as multiple hamartomas, developmental anomalies, and a higher risk of cancer. However, phenotypes of PHTS can vary greatly, especially when comparing pediatric and adult patients due to age-related penetrance. We chose to explore the available literature on BRRS in the pediatric cohort. This article aims to raise awareness among clinicians and patients about this rare syndrome to facilitate earlier diagnosis and choose a prompt management strategy.
BRRS is a rare genetic disorder that is mostly diagnosed among children and is typically characterized by the growth of hamartomatous, lipomatous, or vascular tumors, congenital macrocephaly, developmental delay, and pigmented genital macules3. Prenatal overgrowth and musculoskeletal system disorders such as myopathy of proximal muscles and scoliosis are other clinical features commonly found among individuals with BRRS4. PTEN pathogenic variants in BRRS cases usually demonstrate autosomal dominant inheritance; nevertheless, it is estimated that up to 48% of pathogenic variants can be de novo2,5. Patients with BRRS are considered to share the same lifetime risk of cancer as individuals with Cowden syndrome, however, it has not been confirmed by formal studies4. Additionally, they are susceptible to complications related to gastrointestinal polyps and vascular anomalies. Therefore, early diagnosis and consistent surveillance are important3,6.
There is no established standard treatment for PHTS patients; however, a promising therapy with mammalian target of rapamycin (mTOR) inhibitors is currently under research and can be prescribed as an off-label drug2. mTOR inhibitors have been investigated in patients with vascular anomalies when previous treatments were ineffective7. Here, we present a systematic review that was conducted to summarize the clinical features of pediatric patients with BRRS and to discuss potential treatment and long-term surveillance.
Methods
This systematic review was pursued to summarize the clinical features of pediatric patients with BRRS and to discuss potential treatment and long-term surveillance strategies. The systematic review was conducted following the Preferred Reporting Items for Systematic Review and Meta-Analyses (PRISMA) guidelines8. The study protocol was registered in the International Prospective Register of Systematic Reviews (PROSPERO) (registration number: CRD42022370744) before starting the search. The study protocol was approved by Vilnius University Santaros Clinics research committee.
Eligibility criteria
The inclusion criteria were pediatric patients (0-18 years old) diagnosed with BRRS and patients of any age with BRRS who received treatment with sirolimus. We included full-text, free or available observational studies, case series, and case reports from Vilnius University. Editorials, letters, systematic reviews, literature reviews, comments, guidelines, conference abstracts, expert opinions, and non-English articles were excluded.
Literature search
The PubMed, Scopus, and Cochrane collaboration databases were searched using the following keywords and their combinations: Bannayan Riley Ruvalcaba Syndrome, Bannayan Zonana Syndrome, BRRS, BZS, Myhre Riley Smith syndrome, Riley Smith syndrome, Ruvalcaba Myhre syndrome, Ruvalcaba Myhre Smith syndrome, Bannayan Zonana Syndrome, child, children, and adolescent. We also used the truncation technique (*) with the following words to expand the number of results: pediatri*/, infan*/, newbor*/. An additional search adding the keywords sirolimus, rapamycin, rapamycin, AY 22 989, and 2190A was performed to review the relevant literature on sirolimus efficacy in BRRS. The literature selection was not limited by publication date due to the rarity of the syndrome. Automation search tools were used to include open-access journal articles in the English language. The search was conducted by two reviewers (MK and NS) independently.
Study selection and data extraction
Deduplication of found articles was performed with the citation management software EndNote. First, the titles and abstracts were screened for eligibility, followed by a full-text review of the potentially included publications. The extracted data included authorship, publication year, sample size, demographics of cases, status of PTEN pathogenic variant, family history, clinical presentation, treatment, follow-up care, and outcomes. Two reviewers (MK and NS) independently performed the screening of articles and eligible information selection. Chosen publications and extracted data by both team members were compared, and any disagreements were resolved by discussion.
Evaluating the risk of bias
The quality of the included publications was assessed by each reviewer independently using Joanna Briggs Institute's (JBI) Critical Appraisal Checklist for case reports and case series9,10. The following evaluation criteria were established: low risk of bias was attributed to articles with more than 70% “yes” answers; moderate risk for articles with 50% to 69% “yes” answers; and high risk of bias for studies with less than 49% “yes” answers.
Data analyses
This review consists of case reports and case series; therefore, the heterogeneity of study designs and outcomes made statistical analysis not applicable. Henceforth, a descriptive approach and tabulation were selected to present the results and conclusions.
Results
Of the 2600 publications acquired from the search, 55 duplicates and 2475 original articles were excluded following the inclusion/exclusion criteria and automation tools (Figure 1). After the full-text review of the remaining 70 publications, 37 articles were excluded because they did not meet the inclusion criteria or did not contain relevant information. In total, 33 studies were eligible for the review11,12,13,14,15,16,17,18,19,20,21,22,23,24,25,26,27,28,29,30,31,32,33,34,35,36,37,38,39,40,41,42,43. The methodological quality assessment was performed together with the data extraction (see Supplemental Tables S1 and S2 in Additional File 1).

PRISMA flow chart.
Overall, 83 children with BRRS were included in the review. Supplemental Table S3 summarizes the median age and gender of patients, the status of PTEN pathogenic variant, relevant conditions of parents and siblings, management, surveillance, and outcomes. If the report did not provide the information mentioned above, it was marked in the tables as not available (N/A). A 22-year-old patient was included in the review because the case report mostly described his pediatric history35. The majority of patients were males (60/83), and the median age of the entire cohort was 8 years. In addition, most patients had PTEN pathogenic variants (75/83), whereas only approximately half of the studies reported a positive family history (14/33). Treatment mainly comprised surgical removal of lesions (most commonly lipomas, gastrointestinal polyps, and vascular anomalies); a few cases also mentioned thyroidectomies20,35,41,43, and only one study reported the use of rapamycin25.
In 15 reports, follow-up visits were proposed; however, strategies of surveillance were diverse, mostly aiming at cancer screening. Long-term outcomes were reported in only 6 cases11,18,21,25,35,40. In 3 cases, patients did not develop any new symptoms, while 2 reports described the recurrence of lipomas and gastrointestinal polyps.
Macrocephaly was described in almost all case reports (32/33). Pigmented penile macules were described in the majority of male reports (21/28). Lipomas were mentioned in more than half of the cases (18/33), and the second most common lesion was gastrointestinal polyps (16/33). Another frequent finding was vascular abnormalities, namely, hemangiomas (8/33) and AVMs (6/33). Other reported clinical features were as follows: developmental delay or autism spectrum disorders (25/33), skin manifestations (21/33), and thyroid changes (12/33). A complete summary of the phenotypic findings is presented in Table 1. A more detailed list of the clinical characteristics of all included studies is presented in Supplemental Table S4.
Discussion
Clinical phenotypes of BRRS
Studies have shown that approximately 80% to 100% of patients with PHTS have marked macrocephaly (defined as head circumference ≥97th percentile)44. A similar prevalence is seen among children in the reviewed studies. However, height deviations are not associated with the syndrome3.
Mucocutaneous manifestations are present in the majority of PTEN-related syndromes. Most of them appear later in life due to age-related penetrance44. One of the pathognomonic findings in BRRS is lentiginous genital macules which can be present in both males and females45. It is frequently reported in pediatric BRRS cases in patients as young as 1 year old3. Nevertheless, genital speckles can also develop later in adolescence; therefore, the absence of lentigines alone is not a plausible reason to exclude BRRS23. Other skin findings in the reviewed cases were diverse and nonspecific for the syndrome, but the most frequently mentioned were café au lait spots, verrucous papules, and subcutaneous hemangiomas. Vascular and lipomatous lesions are key findings in BRRS. In the included studies, the most common tumors were lipomas, which, although described as benign, can reach a significant volume and act as space-occupying lesions21. Complications of lipomas depend on their localization and can manifest as occlusion, torsion, or cosmetic defects29,46. In addition, testicular lipomas are a rare entity in the general population, occur more often in individuals with PTEN pathogenic variants, and can be a significant indicator for genetic testing13,44.
Other important findings in BRRS are vascular anomalies, namely, AVMs and hemangiomas. Similar to lipomas, vascular lesions can develop in any part of the body. If located in more superficial layers, they can be seen as discoloration of the skin. Most commonly, vascular anomalies cause swelling and pain, but some cases also reported aggressive growth on the extremities, resulting in the loss of function or amputation25,39. Notably, AVMs can occur in the CNS and carry a potential risk of bleeding and complications related to mass effects28,40. A significant prevalence of gastrointestinal polyps in PHTS has been observed in several studies47. Multiple polyps are usually found in both the upper and lower gastrointestinal tracts and have mixed histology, especially with ganglioneuromatous features48. BRRS patients with polyps may present with abdominal pain, hematochezia, anemia, invagination, constipation, or diarrhea22,23,24,31. Extensive symptom burden or complications such as invagination or blockage may require bowel resection20,23.
Thyroid pathologies are a very common manifestation of PTEN-related syndromes. Up to 75% of patients develop thyroid changes, and approximately one-third will develop thyroid cancer in their lifetime46. While other malignancies tend to appear in adulthood, thyroid carcinoma also occurs among children, with the youngest described patient being 6 years old46. From the included studies, the majority of outlined thyroid changes were nodules; however, 4 studies reported 5 thyroid tumors, of which 3 were carcinomas13,28,35,43.
Developmental delay and autism spectrum disorders (ASD) are also often associated with PHTS. A meta-analysis by Cummings et al. suggests that up to 25% of patients with PTEN pathogenic variants may express characteristics of ASD49. Motor delay is the most common developmental disorder among patients3. Studies included in this systematic review described developmental disorders of different degrees, ranging from normal psychomotor development and mild motor or speech delay to severe intellectual disability. A few cases of ASD and attention deficit disorder have also been reported13,14,22,32,34.
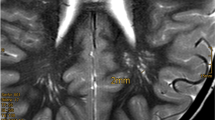
The reviewed studies also outlined less frequent yet notable clinical features of BRRS. Patients are more prone to obesity than the general population due to profound insulin sensitivity50. In addition, some PTEN pathogenic variant carriers can suffer from muscle hypotonia, which is associated with motor delay3. The literature also presents a variety of brain lesions in PHTS patients, mostly located in white matter46. For example, a study by Bhargava et al. assessed brain MRI scans of 7 children with BRRS: almost all patients demonstrated marked macrocephaly, white matter cysts, and abnormal cerebrospinal fluid (CSF) signal intensity (hyperintense on T2 and hypointense on FLAIR)13. Particularly associated with BRRS are musculocutaneous findings such as pectus excavatum, scoliosis, and joint hypermobility51.
It is important to note that both CS and BRRS share the same PTEN alterations and are now considered to be different phenotypic expressions of the same disease52. In addition, there are cases of CS/BRRS overlap families, in which members present with clinical features of either one or both syndromes17,23,42. CS is associated with cancer, and BRRS patients can also develop malignancies later due to CS and BRRS allelism and age-dependent penetrance52.
Diagnostic work-up
There are no internationally established guidelines specifically for the diagnosis of BRRS; nevertheless, the National Comprehensive Cancer Network® (NCCN®) has established diagnostic criteria for PHTS, together with surveillance recommendations for PTEN pathogenic variant carriers44. Patients meeting diagnostic criteria are referred to genetic counseling. Additional critical judgment should be used when applying criteria to the pediatric cohort, since children may express clinical phenotypes different from adults44,46. Pediatric criteria proposed by Tan et al. include the most prevalent PHTS clinical findings in children, namely, macrocephaly, gastrointestinal polyps, neurologic disorders (ASD and developmental delay), dermatologic (lipomas, pigmented genital macules, papillomas), and vascular (AVMs, hemangiomas) features53. German pediatric guidelines updated the Tan et al. criteria by adding positive family history, thyroid changes, and enlarged perivascular spaces in brain MRI46.
Treatment options
Currently, therapy for BRRS is mostly limited to symptom relief, prevention, and eradication of complications. In our reviewed cases, the most frequent intervention was the excision of tumors. Lipomas were the most common masses to be removed. Polypectomies of hamartomatous polyps were also frequent, with two cases of proctocolectomy and subtotal colectomy due to severe symptom burden and recurrence20,23. Thyroid resection and thyroidectomies were described in four cases20,35,41,43. Single cases of scoliosis, seizures, intracranial hypertension, and intracranial tumor treatment were also presented11,19,35,37.
Vascular anomalies are a very common manifestation of BRRS. Sclerotherapy, embolization, and surgery are typical therapy approaches. Nevertheless, these interventions do not always bring satisfactory results due to the complexities of malformations, high frequency of recurrence, and periprocedural morbidity25,39. Therefore, the mTOR inhibitor rapamycin (sirolimus) was proposed as an off-label target agent for treatment of vascular anomalies. PTEN pathogenic variants cause excessive activity of the PI3K-AKT-mTOR pathway, which is responsible for cellular proliferation and angiogenesis induction, leading to the formation of vascular anomalies54. As a result, by downregulating the mTOR pathway, sirolimus could potentially reduce vascular lesions in patients with PHTS.
The first human study of sirolimus for CS investigated its efficacy in 18 adult patients with cutaneous and gastrointestinal lesions55. The trial demonstrated the effectiveness of sirolimus in suppressing mTOR signaling and improving cerebellar function as well as adequate tolerability of the drug. To our knowledge, currently, there are no studies designated for the specific pediatric PHTS group; nevertheless, research has been done among children with vascular anomalies of diverse origins7,56. In retrospective study Hammill et al. 6 children (100%) with treatment-resistant vascular anomalies showed a partial or complete response to sirolimus7. Notably, in a review by Geeurickx et al., 334 cases of pediatric vascular anomalies treated with sirolimus were presented56. It was noticed that the drug efficacy partially depends on the type of malformation. Namely, low-flow vascular anomalies demonstrated the best results. Sirolimus efficacy on fast-flow vascular malformations is yet to be investigated by future clinical trials. In addition, AVMs associated with PTEN pathogenic variants were more responsive to sirolimus monotherapy than isolated AVMs. The systematic literature review by Badawy et al. evaluated sirolimus efficacy in 39 patients with isolated limb overgrowth disorders57. Sirolimus was given as first-line treatment or as an alternative option after previously failed therapies. The reviewed studies were highly variable in terms of sirolimus dosage, length of therapy, and outcome measurement; nevertheless, the results showed a partial response to sirolimus therapy and did not report any case of disease progression. Only one case describing treatment with an mTOR inhibitor was included in our systematic review25. A 6-year-old boy received sirolimus therapy due to resistance to embolization and surgery, persistent pain, and progressive loss of left-hand function. In 6 months, the AVM size decreased, and the patient became pain-free and regained hand mobility. The literature suggests that 6 months is the optimal time to see the maximum response to sirolimus therapy56,58.
In all studies evaluating sirolimus efficacy, adverse reactions were reported, the most common being infections, mucositis, bone marrow suppression, hypercholesterolemia, and elevated liver enzymes7,56,57. Most adverse reactions were not severe and resolved after the dose adjustment7,56. However, awareness of possible drug toxicities and consistent laboratory blood analysis is essential54. Importantly, recurrent symptoms and regrowth of vascular anomalies were observed after discontinuing the treatment56. In addition, it was noticed that sirolimus reduces the volume of vascular anomalies but does not lead to the disappearance of the lesions56,58. Therefore, sirolimus could be used as a polytherapy agent before radical interventions of large complicated vascular anomalies56,58.
It appears that mTOR inhibitors can be an alternative treatment strategy for PHTS patients on individual basis, although current studies are limited by small sample sizes and the absence of randomization. Consequently, more research is required to confirm the potency of sirolimus in treating PTEN-related syndromes.
Long-term surveillance strategies
PHTS is known to be a malignancy-causing syndrome; therefore, the major aspect of follow-up is cancer screening6. Targeted malignancy surveillance in pediatric patients is mostly limited to the thyroid since early-onset changes such as nodules or thyroiditis are common50. Expanded malignancy surveillance and awareness should start in adulthood46,59. The reviewed cases showed similar follow-up strategies and mostly aimed at cancer screening by performing thyroid ultrasound. Dermatological consultation, occult blood tests, and abdominal ultrasound were used in a few cases. As a multisystem disease, PHTS requires a broader follow-up approach, particularly surveillance of nonmalignant manifestations. German pediatric guidelines suggest a screening strategy for children and adolescents which are presented in Supplemental Table S546. They recommend annual physical examinations, including dermatological inspection, neurological and psychomotor assessment, and abdominal ultrasound. Additionally, it is advised to perform thyroid ultrasound after the diagnosis and repeat it periodically if any pathological changes are present. Furthermore, symptom inquiry is important, especially for anemia-related manifestations or gastrointestinal complaints such as abdominal pain, rectal bleeding, obstipation, or diarrhea. Parent education about sun protection, seizures, and concerning symptoms is also encouraged3.
Limitations
The results of our review must be interpreted together with its limitations. Due to the rarity of BRRS, all included studies were case reports and case series. These studies have a high risk of bias. Moreover, study types, heterogeneity, and sometimes incomplete data made generalization impossible. Additionally, we incorporated a number of historical reports that may have interpreted cases with less accuracy than they would with the current knowledge.
Conclusions
Bannayan-Riley-Ruvalcaba syndrome is a rare entity that can manifest in a very broad and diverse spectrum of phenotypes, ranging from mild and subtle symptoms to severe and refractory conditions. Therefore, it is important for clinicians to consider a PTEN pathogenic variant and refer a patient to a geneticist on time. Due to a higher risk of cancer and aggressive growth of hamartomatous anomalies, patients with BRRS require periodic multidisciplinary care that should be individualized to fit every patient’s needs. Currently, surgical interventions are the most common treatment strategy in patients with complicated vascular anomalies; however, the use of mTOR inhibitors could be offered to the patients when other treatment options exhausted. Nevertheless, more research is still needed to fully evaluate their potency in BRRS management.
Data availability
The dataset supporting the conclusions of this article is included within the article and its additional file.
References:
Pulido, R. PTEN: A yin-yang master regulator protein in health and disease. Methods San Diego Calif. 77–78, 3–10 (2015).
Yehia, L., Keel, E. & Eng, C. The clinical spectrum of PTEN mutations. Annu. Rev. Med. 27(71), 103–116 (2020).
Macken, W. L., Tischkowitz, M. & Lachlan, K. L. PTEN hamartoma tumor syndrome in childhood: A review of the clinical literature. Am. J. Med. Genet. C Semin. Med. Genet. 181(4), 591–610 (2019).
Gorlin, R. J., Cohen, M. M., Condon, L. M. & Burke, B. A. Bannayan-Riley-Ruvalcaba syndrome. Am. J. Med. Genet. 44(3), 307–314 (1992).
Mester, J. & Eng, C. Estimate of de novo mutation frequency in probands with PTEN hamartoma tumor syndrome. Genet. Med. 14(9), 819–822 (2012).
Tan, M. H. et al. Lifetime cancer risks in individuals with germline PTEN mutations. Clin. Cancer Res. 18(2), 400–407 (2012).
Hammill, A. M. et al. Sirolimus for the treatment of complicated vascular anomalies in children. Pediatr. Blood Cancer 57(6), 1018–1024 (2011).
Page, M. J. et al. The PRISMA 2020 statement: An updated guideline for reporting systematic reviews. BMJ 29(372), n71 (2021).
Moola S MPF. Chapter 7: Systematic reviews of etiology and risk. In: JBI Manual for Evidence Synthesis [Internet]. JBI; 2020 [cited 2024 Jun 26]. Available from: https://jbi-global-wiki.refined.site/space/MANUAL/355863557/Previous+versions?attachment=/download/attachments/355863557/JBI_Reviewers_Manual_2020June.pdf&type=application/pdf&filename=JBI_Reviewers_Manual_2020June.pdf#page=217
Munn, Z. et al. Methodological quality of case series studies: An introduction to the JBI critical appraisal tool. JBI Database Syst. Rev. Implement Rep. https://doi.org/10.11124/JBISRIR-D-19-00099 (2019).
Ahmed, S. F. et al. Balanced translocation of 10q and 13q, including the PTENGene, in a boy with a human chorionic gonadotropin-secreting tumor and the Bannayan-riley-Ruvalcaba syndrome. J. Clin. Endocrinol. Metab. 84(12), 4665–4670 (1999).
Arch, E. M. et al. Deletion of PTEN in a patient with Bannayan-Riley-Ruvalcaba syndrome suggests allelism with cowden disease. Am. J. Med. Genet. 71(4), 489–493 (1997).
Bhargava, R. Bannayan-Riley-Ruvalcaba syndrome: MRI neuroimaging features in a series of 7 patients. Am. J. Neuroradiol. 35, 402 (2014).
Bishop, P. R., Nowicki, M. J. & Parker, P. H. What syndrome is this? Ruvalcaba-Myhre-Smith syndrome. Pediatr. Dermatol. 17(4), 319–321 (2000).
Blum, R. R., Rahimizadeh, A., Kardon, N., Lebwohl, M. & Wei, H. Genital Lentigines in a 6-year-old boy with a family history of cowden’s disease: Clinical and genetic evidence of the linkage between Bannayan–Riley–Ruvacalba syndrome and Cowden’s disease. J. Cutan Med. Surg. 5(3), 228–230 (2001).
Buisson, P. et al. Cutaneous lipoma in children: 5 cases with Bannayan-Riley-Ruvalcaba syndrome. J. Pediatr. Surg. 41(9), 1601–1603 (2006).
Celebi, J. T. et al. Phenotypic findings of Cowden syndrome and Bannayan-Zonana syndrome in a family associated with a single germline mutation in PTEN. J. Med. Genet. 36(5), 360–364 (1999).
Erkek, E. et al. Clinical and histopathological findings in Bannayan-Riley-Ruvalcaba syndrome. J. Am. Acad. Dermatol. 53(4), 639–643 (2005).
Ghusayni, R., Sachdev, M., Gallentine, W., Mikati, M. A. & McDonald, M. T. Hemimegalencephaly with Bannayan-Riley-Ruvalcaba syndrome. Epileptic Disord. 20(1), 30–34 (2018).
Golas, M. M. et al. Looking for the hidden mutation: Bannayan–Riley–Ruvalcaba syndrome caused by constitutional and mosaic 10q23 microdeletions involving PTEN and BMPR1A. Am. J. Med. Genet. A 179(7), 1383–1389 (2019).
Gontijo, G. M. A. et al. Bannayan-Riley-Ruvalcaba syndrome with deforming lipomatous hamartomas in infant—Case report. An. Bras. Dermatol. 88(6), 982–985 (2013).
Hansen-Kiss, E. et al. A retrospective chart review of the features of PTEN hamartoma tumour syndrome in children. J. Med. Genet. 54(7), 471–478 (2017).
Hendriks, Y. M. C. et al. Bannayan-Riley-Ruvalcaba syndrome: Further delineation of the phenotype and management of PTEN mutation-positive cases. Fam. Cancer 2(2), 79–85 (2003).
Hızarcıoğlu-Gülşen, H. et al. Polyposis deserves a perfect physical examination for final diagnosis: Bannayan-Riley-Ruvalcaba syndrome. Turk. J. Pediatr. 59(1), 80–83 (2017).
Iacobas, I. et al. Oral rapamycin in the treatment of patients with hamartoma syndromes and PTEN mutation. Pediatr. Blood Cancer 57(2), 321–323 (2011).
Iskandarli, M., Yaman, B. & Aslan, A. A case of B annayan– R iley– R uvalcaba syndrome. A new clinical finding and brief review. Int. J. Dermatol. 55(9), 1040–1043 (2016).
Israel, J., Lessick, M., Szego, K. & Wong, P. Translocation 19;Y in a child with Bannayan-Zonana phenotype. J. Med. Genet. 28(6), 427–428 (1991).
Kurek, K. C. et al. PTEN hamartoma of soft tissue: A distinctive lesion in PTEN syndromes. Am. J. Surg. Pathol. 36(5), 671–687 (2012).
Laguna, B. A., Iyer, R. S., Rudzinski, E. R., Roybal, J. L. & Stanescu, A. L. Torsion of a giant mesocolic lipoma in a child with Bannayan-Riley-Ruvalcaba syndrome. Pediatr. Radiol. 45(3), 449–452 (2015).
Longy, M. et al. Mutations of PTEN in patients with Bannayan-Riley-Ruvalcaba phenotype. J. Med. Genet. 35(11), 886–889 (1998).
Lowichik, A. et al. Bannayan-Riley-Ruvalcaba syndrome: Spectrum of intestinal pathology including juvenile polyps. Pediatr. Dev. Pathol. 3(2), 155–161 (2000).
Lynch, N. E., Lynch, S. A., McMenamin, J. & Webb, D. Bannayan-Riley-Ruvalcaba syndrome: A cause of extreme macrocephaly and neurodevelopmental delay. Arch. Dis. Child. 94(7), 553–554 (2009).
Niklinska, E. B., Lyons, E. M., Hicks, A., Zwerner, J. P. & Albers, S. E. Bannayan-Riley-Ruvalcaba syndrome with gingival hyperpigmentation and facial papules. Pediatr. Dermatol. 38(5), 1351–1353 (2021).
O’Rourke, D. J., Twomey, E., Lynch, S. A. & King, M. D. Cortical dysplasia associated with the PTEN mutation in Bannayan Riley Ruvalcaba syndrome: A rare finding. Clin. Dysmorphol. 21(2), 91–92 (2012).
Valentina, P. et al. Thyroid involvement in two patients with Bannayan-Riley-Ruvalcaba syndrome. J. Clin. Res. Pediatr. Endocrinol. 5(4), 261–265 (2013).
Piccione, M. et al. PTEN hamartoma tumor syndromes in childhood: Description of two cases and a proposal for follow-up protocol. Am. J. Med. Genet. A 161(11), 2902–2908 (2013).
Prat, D., Ben Bassat Mizrachi, I. & Vishnevskia-Dai, V. Intermediate uveitis in a child with phosphatase and tensin homolog gene mutation and Bannayan-Riley-Ruvalcaba syndrome. BMJ Case Rep. https://doi.org/10.1136/bcr-2017-224079 (2019).
Schwab, J. G., Pena, L., Waggoner, D. & Pytel, P. Two children with macrocephaly, developmental delay, and PTEN mutation. Clin. Pediatr. (Phila) 48(1), 89–92 (2009).
Soysal, Y., Acun, T., Lourenço, C., Marques, W. & Yakıcıer, M. Muscle Hemangiomatosis presenting as a severe feature in a patient with the Pten mutation: Expanding the phenotype of vascular malformations in Bannayan-Riley-ruvalcaba syndrome. Balk. J. Med. Genet. 15(1), 45–50 (2012).
Srinivasa, R. N. & Burrows, P. E. Dural arteriovenous malformation in a child with Bannayan-Riley-Ruvalcaba syndrome. AJNR Am. J. Neuroradiol. 27(9), 1927–1929 (2006).
Vincenzi, G. et al. Case report—Multinodular goiter in a patient with CONGENITAL HYPOTHYRoidism and Bannayan-Riley-Ruvalcaba syndrome: The possible synergic role of TPO and PTEN mutation. Front. Endocrinol. 8(14), 1205785 (2023).
Wanner, M., Elebi, J. T. & Peacocke, M. Identification of a PTEN mutation in a family with Cowden syndrome and Bannayan-Zonana syndrome. J. Am. Acad. Dermatol. 44(2), 183–187 (2001).
Zambrano, E. et al. Abnormal distribution and hyperplasia of thyroid C-cells in PTEN-associated tumor syndromes. Endocr. Pathol. 15(1), 55–64 (2004).
Pilarski, R. et al. Cowden Syndrome and the PTEN hamartoma tumor syndrome: Systematic review and revised diagnostic criteria. JNCI J. Natl. Cancer Inst. 105(21), 1607–1616 (2013).
Pilarski, R. PTEN hamartoma Tumor syndrome: A clinical overview. Cancers 11(6), 844 (2019).
Plamper, M., Gohlke, B. & Woelfle, J. PTEN hamartoma tumor syndrome in childhood and adolescence—a comprehensive review and presentation of the German pediatric guideline. Mol. Cell. Pediatr. 9(1), 3 (2022).
Stanich, P. P. Colonic manifestations of PTEN hamartoma tumor syndrome: Case series and systematic review. World J. Gastroenterol. 20(7), 1833 (2014).
D’Ermo, G. & Genuardi, M. Gastrointestinal manifestations in PTEN hamartoma tumor syndrome. Best Pract. Res. Clin. Gastroenterol. 58–59, 101792 (2022).
Cummings, K., Watkins, A., Jones, C., Dias, R. & Welham, A. Behavioural and psychological features of PTEN mutations: a systematic review of the literature and meta-analysis of the prevalence of autism spectrum disorder characteristics. J Neurodev Disord. 14(1), 1 (2022).
Ngeow, J., Sesock, K. & Eng, C. Clinical implications for germline PTEN spectrum disorders. Endocrinol. Metab. Clin. North Am. 46(2), 503–517 (2017).
Hobert, J. A. & Eng, C. PTEN hamartoma tumor syndrome: An overview. Genet. Med. 11(10), 687–694 (2009).
Blumenthal, G. M. & Dennis, P. A. PTEN hamartoma tumor syndromes. Eur. J. Hum. Genet. 16(11), 1289–1300 (2008).
Tan, M. H. et al. A clinical scoring system for selection of patients for PTEN Mutation testing is proposed on the basis of a prospective study of 3042 probands. Am. J. Hum. Genet. 88(1), 42–56 (2011).
Adams, D. M. & Ricci, K. W. Vascular anomalies. Hematol. Oncol. Clin. North Am. 33(3), 455–470 (2019).
Komiya, T. et al. A pilot study of Sirolimus in subjects with cowden syndrome or other syndromes characterized by germline mutations in PTEN. Oncologist 24(12), 1510-e1265 (2019).
Geeurickx, M. & Labarque, V. A narrative review of the role of sirolimus in the treatment of congenital vascular malformations. J. Vasc. Surg. Venous Lymphat. Disord. 9(5), 1321–1333 (2021).
Badawy, M., Ma, Y., Baldrighi, C., Oestreich, K. & Jester, A. Efficacy of mTOR inhibitors (sirolimus) in isolated limb overgrowth: A systematic review. J. Hand Surg. Eur. 47(7), 698–704 (2022).
Seront, E., Van Damme, A., Boon, L. M. & Vikkula, M. Rapamycin and treatment of venous malformations. Curr. Opin. Hematol. 26(3), 185–192 (2019).
Tischkowitz, M. et al. Cancer surveillance guideline for individuals with pten hamartoma tumour syndrome. Eur. J. Hum. Genet. 28(10), 1387–1393 (2020).
Author information
Authors and Affiliations
Contributions
Conceptualization, J.R., B.V.; acquisition of data, M.K., N.S., I.A.; writing—original draft preparation, M.K.; writing—review and editing, N.S., I.A., J.R., B.V.; supervision, J.R.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Ethics Approval and Consent for Publication
Ethics approval was acquired from the Vilnius University Santaros Clinics research committee.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Kapačinskaitė, M., Stratica, N., Adomaitienė, I. et al. A systematic review of Bannayan – Riley – Ruvalcaba syndrome. Sci Rep 14, 21119 (2024). https://doi.org/10.1038/s41598-024-71991-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-024-71991-2
- Springer Nature Limited