
Abstract
There is an urgent need to pinpoint novel targets for drug discovery in the context of chronic kidney disease (CKD), and the proteome represents a significant pool of potential therapeutic targets. To address this, we performed proteome-wide analyses using Mendelian randomization (MR) and colocalization techniques to uncover potential targets for CKD. We extracted summary-level data from the ARIC study, focusing on 7213 European American (EA) individuals and 4657 plasma proteins. To broaden our analysis, we incorporated genetic association data from Icelandic cohorts, thereby enhancing our investigation into the correlations with chronic kidney disease (CKD), creatinine-based estimated glomerular filtration rate (eGFRcrea), and estimated glomerular filtration rate (eGFR). We utilized genetic association data from the GWAS Catalog, including CKD (765,348, 625,219 European ancestry and 140,129 non-European ancestry), eGFRcrea (1,004,040, European ancestry), and eGFR (567,460, European ancestry). Employing MR analysis, we estimated the associations between proteins and CKD risk. Additionally, we conducted colocalization analysis to evaluate the existence of shared causal variants between the identified proteins and CKD. We detected notable correlations between levels predicted based on genetics of three circulating proteins and CKD, eGFRcrea, and eGFR. Notably, our colocalization analysis provided robust evidence supporting these associations. Specifically, genetically predicted levels of Transcription elongation factor A protein 2 (TCEA2) and Neuregulin-4 (NRG4) exhibited an inverse relationship with CKD risk, while Glucokinase regulatory protein (GCKR) showed an increased risk of CKD. Furthermore, our colocalization analysis also supported the associations of TCEA2, NRG4, and GCKR with the risk of eGFRcrea and eGFR.
Similar content being viewed by others
Introduction
Chronic kidney disease (CKD) is a condition that is widespread worldwide and has a substantial impact on both illness and death rates. Various complications, including cardiovascular disease and kidney failure, are linked to the condition. As a result, CKD imposes significant caregiving strains upon individuals, families at large1,2. A notable indicator of CKD is the reduction in glomerular filtration rate3. The estimated effect on eGFRcrea is of specific worry4. In most instances, the underlying causes of CKD remain unclear, and its intricate biological etiology poses limitations for the creation of novel medications5. Consistently, proteins that circulate and act as crucial controllers of molecular pathways are widely recognized as primary objectives for pharmacological interventions. Prior research has indicated associations between CKD and specific agents such as SGLT-2i, GLP-1 and Urinary CKD-2736,7. Nevertheless, these associations are susceptible to confounding factors or reverse causality. While randomized controlled trials are widely recognized for studying causality, conducting such trials to explore the causal relationship between thousands of proteins and CKD is often cost-prohibitive and impractical.
Utilizing random allocation of alleles, Mendelian randomization (MR) studies offer a reliable method to reduce the impact of confounding biases. Moreover, MR studies are valuable in circumventing reverse causality issues that are often encountered in other types of observational studies8,9. In a prior investigation, over 1.2 million individuals were analyzed to explore genetic variations and genes linked to kidney function10. Genetic studies have offered opportunities to look at circulating protein levels in CKD from a comprehensive perspective. In this study, we conducted MR proteomic analysis along with co-localization techniques, aimed at investigating candidate therapeutic targets for CKD and their defining factors, particularly eGFRcrea and eGFR.
Methods
Study design
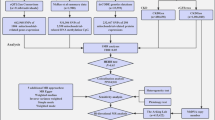
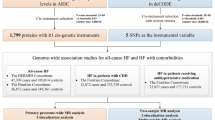
Refer to Fig. 1 for a visual representation of the study design. The present study adhered to the Strengthening the Reporting of Observational Studies in Epidemiology-Mendelian Randomization (STROBE-MR) guidelines (see Supplementary Table 1)11. All GWAS and protein quantitative trait loci (pQTL) data utilized were obtained from publicly accessible databases that have received the necessary ethical approvals. The research relies on data that is accessible to the public from a large-scale genome-wide association study on blood proteome (https://www.decode.com/summarydata/)12, plasma proteome analysis of European descent plasma(nilanjanchatterjeelab.org/pwas/)13 and GWAS Catalog (GWAS Catalog (ebi.ac.uk))14,15,16 (Supplementary Table 2). Our research methodology adopts a two-phase strategy, commencing with the ARIC study as the primary discovery phase. This phase is dedicated to investigating associations between CKD and a comprehensive range of circulating proteins, leveraging data on 4657 proteins obtained from this cohort. Subsequent validation of our findings is conducted in the replication phase, utilizing proteomic data from the Icelandic study, which encompasses 4,907 proteins. This systematic approach ensures the robustness and reproducibility of our conclusions.

Study design. FDR, false discovery rate; MR, Mendelian Randomization.
Data source for proteomics
The circulating proteins associated with genetic markers were aggregated from ARIC studies13. These proteins exhibit associations with common variations in the cis-region. Compared to trans linking, cis-linking demonstrates higher reproducibility across diverse proteome platforms. Another set of summary-level statistics on circulating protein was derived from Ferkingstad E's study12. The use of genome-wide association studies (GWASs) focusing on circulating protein levels allows for the identification of sequence determinants, known as protein quantitative trait loci (pQTLs), associated with protein levels17,18,19,20. Due to their colocalization with disease variants, pQTLs can play a crucial role as informative markers for directing the exploration of causal genes and disease pathways21,22.
Data source for outcome data
Data summaries for CKD, eGFRcrea and eGFR were meticulously curated from the GWAS Catalog. The CKD dataset encompassed 625,219 European ancestry, defined by an eGFR lower than 60 ml/min^−1 per 1.73 m^2. Notably, Wuttke M’s analysis on eGFRcrea, utilizing the Renal Disease Study equation, included 1,004,040 individuals of European descent. Conversely, the eGFR dataset comprised 567,460 individuals, also of European ancestry. It is imperative to clarify that the CKD dataset served as the primary outcome for our analysis, while the eGFRcrea and eGFR datasets were utilized as secondary outcomes.
MR analysis
In our Mendelian randomization (MR) analysis, it is essential to clarify that we utilized the random allocation of alleles, emphasizing the broader impact of genetic variation rather than focusing solely on the distribution of specific single nucleotide polymorphisms (SNPs). This distinction underscores our methodological approach, which aligns more closely with the principles of Mendelian Randomization, utilizing genetic variants as instrumental variables to infer causal relationships. An effective instrumental variable is grounded in three key assumptions: The instrumental variable (such as genetic variation) must be significantly associated with the exposure. It should be independent of confounding factors, meaning it is not correlated with potential confounders unrelated to the exposure, thereby avoiding interference in causal inference. Additionally, the instrumental variable should affect the outcome only through the exposure, without directly influencing the outcome by any other means23.
To elucidate the connections between circulating proteins and the susceptibility to CKD and its respective indicators, we conducted a two-sample MR analysis using index SNPs for proteins. In this study, we employed a targeted approach by selecting a genomic region of 100 kilobases around the start and end sites of the target genes from the database. Within this region, we identified SNPs that exhibited significant associations (P < 1 × 10−5, linkage disequilibrium [LD] r2 < 0.01) with CKD and its respective indicators. To quantify the associations between the identified proteins and the outcomes, we employed the odds ratios (ORs) along with their corresponding confidence intervals (CIs) using either the Wald ratio or inverse-variance weighted method. In the “MR analysis” section, we performed a false discovery rate (FDR) test to avoid false positive results and ensured the robustness and validity of our instrumental variables by calculating the F-statistic, a crucial metric for assessing instrument quality.
To measure the intensity of each plasma protein, the F-statistic is calculated using the following formula: \({\text{F}} = {\text{R}}_{{{\text{combined}}}}^{2} {\text{ (N}} - {\text{k}} - {\text{1)/k (1}} - {\text{R}}_{{{\text{combined}}}}^{2} {\text{)}}\), where \({\text{R}}_{{{\text{combined}}}}^{2}\) represents the sum of the proportions of plasma protein variability explained by each instrument, N denotes the sample size of the GWAS for SNP-plasma protein associations, and k indicates the number of instrumental variables. The formula for calculating R2 is as follows: R2 = 2 × β2 × MAF ×(1 − MAF), where MAF is the minor allele frequency and β is the estimated genetic effect on the plasma protein. The formula for calculating \({\text{R}}_{{{\text{combined}}}}^{2}\) is as follows: \({\text{R}}_{{{\text{combined}}}}^{{\text{2}}} {\text{ = }}\sum\nolimits_{{{\text{i = 1}}}}^{{\text{k}}} {{\text{R}}_{{\text{i}}}^{2} }\). This statistic serves as a key indicator of instrumental variable strength, with higher values denoting stronger instruments and thereby enhancing the reliability of our MR estimates. An F-statistic exceeding 10 is typically regarded as indicative of a robust instrument, thereby mitigating the risk of weak instrument bias in our MR analysis.
Colocalization analysis
We utilized the coloc R package to perform colocalization analysis in order to examine if the observed connections between proteins and CKD as well as its indicators were affected by linkage disequilibrium24. The Bayesian method appraised evidence for five distinct hypotheses concerning each locus: (1) no connection with either trait, (2) exclusive connection with trait 1, (3) a unique link to trait 2, (4) both traits display links, but each has its own unique causal variants, and (5) both traits share a common causal variant, and they are interconnected25. Posterior probabilities were obtained for every hypothesis (H0, H1, H2, H3, and H4) as a result of the analysis. We assigned initial probabilities for specific situations in the following manner: a SNP linked solely to trait 1 (p1) with a probability of 1 × 10−4, a SNP linked solely to trait 2 (p2) with a probability of 1 × 10−4, and a SNP associated with both traits (p12) with a probability of 1 × 10−526.In this study, we adopted a specific approach for selecting SNPs located 250 kilobases above and below the target gene in the Iceland dataset and ARIC study.
Protein-CKD associations with FDR-corrected P value < 0.05 in MR were subsequently classified into three groups. Proteins that exhibit statistically significant p-values in both the discovery and validation cohorts, and have high colocalization support evidence (PH4 > 0.8), are considered tier 1 targets. Proteins that exhibit statistically significant p-values in both the discovery set and the validation set, coupled with medium colocalization support evidence (0.5 < PH4 < 0.8), are considered tier 2 targets. the remaining proteins were considered Tier 3 targets.
All statistical analyses were implemented by the package TwoSampleMR(version 0.5.6)27, MendelianRandomization package (version 0.5.0)28, coloc R package24 and locuscomparer package (version 1.0.0)29.
Results
Proteome-wide MR analysis
The research examined the connections between 1788 proteins, which had accessible index pQTL signals, and the likelihood of experiencing CKD outcomes using MR analysis. The MR analysis results are outlined in Fig. 2. Through this examination, we pinpointed 155 combinations of protein and CKD that showed slight importance (P < 0.05) in the MR analysis. Nevertheless, following the application of the FDR test to address potential pleiotropy, only 10 of these associations remained significant (as shown in Supplementary Table 3). Following the exclusion of associations that did not satisfy the FDR test criteria, we identified seven circulating proteins, namely Transcription elongation factor A protein 2 (TCEA2), Isopentenyl-diphosphate delta-isomerase 2 (IDI2), Microfibril-associated glycoprotein 4 (MFAP4), Lactosylceramide 4-alpha-galactosyltransferase (A4GALT), Leukocyte immunoglobulin-like receptor subfamily A member 5 (LILRA5), N-acetylglucosamine-1-phosphotransferase subunit gamma (GNPTG), and Neuregulin-4 (NRG4), which were inversely associated with CKD risks. On the other hand, higher levels of Glucokinase regulatory protein (GCKR) and Rho GTPase-activating protein 1 (ARHGAP1) were determined to be a contributing factor to an elevated CKD risk (Fig. 2 and Supplementary Table 3).

Manhattan plots for associations of genetically predicted 1778 circulating proteins levels with CKD. The X-axis represents genetic associations on autosomes, with each point above each chromosome representing a circulating protein. Blue points denote plasma proteins in odd chromosome regions, orange points denote plasma proteins in even chromosome regions, and green points represent plasma proteins confirmed as Tier 1 targets. Parallel to the X-axis, the blue line represents the threshold line of p-values after FDR correction, with points above this line indicating these plasma proteins are associated with the outcome. The red line parallel to the X-axis is the genome-wide significance threshold line (P < 5 × 10−8), with points above this line indicating significant associations of these plasma proteins with the outcome. The Y-axis represents the results of the association analysis, depicted as − log10(p-values), where higher points indicate plasma proteins with more significant associations.
For every uptick of one standard deviation in protein levels, the odds ratio (OR) of CKD was 0.614 (95% CI 0.507–0.743) for TCEA2, 0.642 (95% CI 0.527–0.783) for NRG4, and 1.188 (95% CI 1.087–1.281) for GCKR. The same connections were duplicated in the 35,559 individuals from the Icelandic group, showing consistent effects in the same direction during the analysis of replication (Table 1 and Supplementary Table 4). Likewise, Per SD increment of genetically predicted levels of protein, the OR of CKD was 0.742 (95% CI 0.662–0.831) for TCEA2, 0.341 (95% CI 0.211–0.552) for NRG4, and 1.460 (95% CI 1.210–1.762) for GCKR.
By eliminating associations that did not meet the criteria of the FDR test, we discovered 125 proteins that showed a significant connection to eGFRcrea in the two-sample MR analysis, with a P-value less than 0.05 (see Fig. 3 and Supplementary Table 5). Consistently, the validation consistently confirmed the effects of TCEA2, NRG4, and GCKR in the same direction. The levels of TCEA2 (OR 1.020, 95% CI 1.013–1.027) and NRG4 (OR 1.055, 95% CI 1.049–1.061) showed a positive relationship with eGFRcrea, whereas GCKR (OR 0.975, 95% CI 0.972–0.977) exhibited an inverse association with the probability of eGFRcrea. Moreover, the associations for TCEA2, NRG4, and GCKR were replicated in the 35,559 individuals from the Icelandic cohort, with consistent directional effects observed in the replication analysis (Table 1 and Supplementary Table 4). For every increase of 1 standard deviation in genetically estimated protein levels, OR for eGFRcrea showed values of 1.012 (95% CI 1.008–1.016) for TCEA2, 1.139 (95% CI 1.124–1.155) for NRG4, and 0.943 (95% CI 0.936–0.949) for GCKR.

Manhattan plots for associations of genetically predicted 1778 circulating proteins levels with eGFRcrea. The X-axis represents genetic associations on autosomes, with each point above each chromosome representing a circulating protein. Blue points denote plasma proteins in odd chromosome regions, orange points denote plasma proteins in even chromosome regions, and green points represent plasma proteins confirmed as Tier 1 targets. Parallel to the X-axis, the blue line represents the threshold line of p-values after FDR correction, with points above this line indicating these plasma proteins are associated with the outcome. The red line parallel to the X-axis is the genome-wide significance threshold line (P < 5 × 10−8), with points above this line indicating significant associations of these plasma proteins with the outcome. The Y-axis represents the results of the association analysis, depicted as − log10(p-values), where higher points indicate plasma proteins with more significant associations.
Regarding eGFR, following FDR adjustment, higher genetically predicted levels of circulating GCKR were linked to a reduction in eGFR, while higher levels of circulating proteins such as TCEA2 and NRG4 were linked to a heightened likelihood of eGFR. The OR for eGFR was 1.024 (95% CI 1.015–1.032) for TCEA2, 1.049 (95% CI 1.041–1.057) for NRG4, and 0.977 (95% CI 0.974–0.981) for GCKR with each one-standard-deviation increase in protein levels (Fig. 4 and Supplementary Table 6). Furthermore, the findings for TCEA2, NRG4, and GCKR were duplicated in the 35,559 participants of the Icelandic group (Table 1 and Supplementary Table 4). In particular, increased levels of TCEA2 (OR = 1.014, 95% CI 1.009–1.019) and NRG4 (OR = 1.123, 95% CI 1.102–1.144) exhibited a connection with a heightened probability of eGFR, while elevated GCKR (OR = 0.949, 95% CI 0.941–0.956) were connected to a decreased likelihood of eGFR.

Manhattan plots for associations of genetically predicted 1775 circulating proteins levels with eGFR. The X-axis represents genetic associations on autosomes, with each point above each chromosome representing a circulating protein. Blue points denote plasma proteins in odd chromosome regions, orange points denote plasma proteins in even chromosome regions, and green points represent plasma proteins confirmed as Tier 1 targets. Parallel to the X-axis, the blue line represents the threshold line of p-values after FDR correction, with points above this line indicating these plasma proteins are associated with the outcome. The red line parallel to the X-axis is the genome-wide significance threshold line (P < 5 × 10−8), with points above this line indicating significant associations of these plasma proteins with the outcome. The Y-axis represents the results of the association analysis, depicted as − log10(p-values), where higher points indicate plasma proteins with more significant associations.
Colocalization analysis
We performed colocalization analyses to explore the connection between circulating proteins and CKD as well as its indicators. Initially, we investigated if the connections between the circulating protein and CKD, as well as its markers, were influenced by common causal variants (Table 2, Supplementary Figs. 1–9). Notably, we discovered compelling evidence of colocalization between three proteins (TCEA2, NRG4, and GCKR) and CKD, categorizing them as tier 1 targets. Furthermore, three proteins exhibited high support for colocalization with eGFRcrea and eGFR associations, also classified as tier 1 targets. Unfortunately, 7 plasma proteins were classified as Tier 3 targets due to failure to pass the relevant validation, and the number of Tier 2 targets was zero in Tables 1 and 2.
In an effort to establish the credibility of our results, we performed colocalization analyses on the dataset of 35,559 individuals from Iceland to examine the co-occurrence of circulating proteins. Specifically, we observed strong indication of colocalization between CKD and three proteins: TCEA2, NRG4, and GCKR. Similarly, these three circulating proteins exhibited high support for colocalization with eGFRcrea and eGFR (Table 1, Supplementary Figs. 10–18).
Discussion
In order to reveal the possible causal effects of over 4000 circulating proteins on CKD and its indicators, we conducted a thorough analysis of the proteome using MR and colocalization techniques. The aim was to offer worthy preclinical information for the development of drugs. Through the MR analysis, we spotted three proteins that were associated with CKD, and these associations were further confirmed in their respective indicators. Subsequently, by employing colocalization analysis to account for the impacts of linkage disequilibrium, we gathered compelling evidence supporting the inverse association between higher levels of TCEA2 and NRG4 with CKD risks. Furthermore, it was found that elevated circulating GCKR are linked to an increased vulnerability to CKD. In addition, it was noted that increased levels of TCEA2 and NRG4 showed a positive correlation with eGFRcrea and eGFR. We also observed that, after FDR adjustment, higher genetically predicted circulating levels of IDI2, MFAP4, A4GALT, LILRA5, and GNPTG were tied to decreased CKD risk, whereas elevated levels of ARHGAP1 were associated with heightened CKD risk. Regrettably, due to not passing the detection in subsequent validation, we did not classify them as Tier 1 targets.
In our study, we discovered strong evidence supporting the potential targeting of TCEA2, GCKR, and NRG4 for CKD. GCKR, a member of the Sugar ISomerase (S IS) protein family, encodes a protein with regulatory function that suppresses glucokinase activity. This inhibition is achieved by forming a non-covalent complex with the enzyme, rendering it inactive. Previous research has indicated an association between the GCKR gene and CKD, as well as eGFRcrea and eGFR14,30,31,32. Notably, evidence suggests that GCKR is a potential genetic predisposition site for Non-alcoholic fatty liver disease (NAFLD)33 and individuals with elevated fibrosis levels in NAFLD are at an Intensified likelihood of developing CKD34. Furthermore, by concentrating our examination on individuals with European descent, we reduced possible prejudices arising from population stratification, thereby enhancing the credibility and applicability of our findings.
NRG4 belongs to the neuregulin family, recognized for its ability to trigger the receptors for growth factor type-1. Previous research has provided evidence that NRG4 attenuates tubulointerstitial fibrosis through the regulation of TNF-R1 signaling35. Significantly, the administration of exogenous NRG4 during colitis has been documented to decrease the population of colon macrophages and ameliorate inflammation36. The disrupted gut-kidney axis establishes a detrimental cycle that ultimately accelerates the advancement of CKD37. The findings regarding NRG4’s effects on CKD in this study align with previous research, thereby reinforcing the accuracy of the results. TCEA2 encodes a protein predominantly localized in the nucleus, serving as an SII class transcription elongation factor. Previous research has highlighted varied expression levels of TCEA2 in the kidney38,39. While the associations of NRG4 and GCKR with CKD have been extensively investigated, the relationship between TCEA2 and CKD remains relatively understudied. However, considering the consistent findings of GCKR and NRG4 effects on CKD in this study and previous research, the potential effects of TCEA2 on CKD hold significant reference value.
Our study possesses several notable advantages. Firstly, by employing a MR design, we were able to emulate a randomized controlled trial. Randomized control features are highly recognized for establishing causality; however, they are often expensive and impractical to implement. In contrast, MR studies effectively mitigate confounding biases by leveraging the random allocation of SNPs during conception. Furthermore, MR studies offer the advantage of addressing reverse causation, which is a common concern in other observational studies. Secondly, we utilized colocalization analysis, a powerful tool for elucidating pleiotropic effects of specific loci on multiple traits. By employing this analysis, we gained valuable insights into the interconnectedness of various traits and their shared genetic underpinnings. Furthermore, our study leveraged GWASs featuring extensive sample sizes. In our analysis, we exclusively employed robust instrumental variables (IVs) that exhibited F statistics greater than 10. This meticulous approach substantially heightened our capacity to identify even the most subtle associations. Additionally, we conducted analyses across multiple datasets, reinforcing the robustness and consistency of our findings. Moreover, by focusing our analysis on individuals of European ancestry, we minimized potential biases stemming from population stratification, thus increasing the validity and generalizability of our results.
Nonetheless, it is imperative to acknowledge various constraints in our research. To begin with, during the execution of sensitivity analyses in MR, encompassing weighted median, MR-PRESSO, and MR-Egger intercept tests, we encountered specific limitations. Our analysis predominantly relied on a single SNP as the instrumental variable, which may have constrained the detection of intricate causal relationships and led to an underestimation of the risk of horizontal pleiotropy. While weighted median and MR-Egger tests provide supplementary evidence regarding the robustness of causal effects, their efficacy is contingent upon sample size and the strength of instrumental variables, with MR-Egger being particularly sensitive to instrument imbalance. Additionally, while colocalization analysis helped mitigate bias resulting from linkage disequilibrium, the presence of horizontal pleiotropy could not be completely eliminated, which may introduce confounding effects. Nevertheless, our study sought to overcome this limitation by conducting a thorough colocalization analysis of circulating proteins. This approach enabled us to identify potential candidate genes that could potentially have a causal effect on CKD. It is important to note that performing colocalization analyses can sometimes result in apparent conflicts with the results obtained through Mendelian randomization. However, exercising caution and considering the implications of such discrepancies is often warranted and appropriate. Lastly, in our study, we acknowledged the inclusion of samples from non-European ancestries within the broader datasets. This recognition underscores the necessity of examining the European ancestry proportion to ensure the applicability and relevance of our findings across different populations. In addition, future clinical trials are crucial for confirming its therapeutic potential.
Conclusion
Our study presented evidence that TCEA2, NRG4, and GCKR emerged as promising candidates for targeted drug interventions in CKD. Nevertheless, further validation through future clinical trials is warranted to ascertain their therapeutic potential.
Data availability
The study is based on publicly available data from large-scale genome-wide association study on blood proteome (https://www.decode.com/summarydata/) and Plasma proteome analysis of individuals of European ancestry (nilanjanchatterjeelab.org/pwas/). Summary-level data for chronic kidney disease (CKD), estimated glomerular filtration rate using serum creatinine (eGFRcrea), and estimated glomerular filtration rate (eGFR) were sourced from the GWAS Catalog (GWAS Catalog (ebi.ac.uk)). Specifically, “GCST008064” for CKD, “GCST90103634” for eGFRcrea, “GCST008059” for eGFR.
References
James, M. T., Hemmelgarn, B. R. & Tonelli, M. Early recognition and prevention of chronic kidney disease. Lancet. 375(9722), 1296–1309 (2010).
Global,. National age-sex specific mortality for 264 causes of death, 1980–2016: A systematic analysis for the global burden of Disease Study 2016. Lancet390(10100), 1151–1210 (2017).
Levey, A. S. et al. Nomenclature for kidney function and disease: Executive summary from a KDIGO consensus conference. Nat. Rev. Nephrol.16(8), 427–428 (2020).
Arpegård, J. et al. Comparison of heritability of Cystatin C- and creatinine-based estimates of kidney function and their relation to heritability of cardiovascular disease. J. Am. Heart Assoc.4(1), e001467 (2015).
Levin, A. et al. Global kidney health 2017 and beyond: A roadmap for closing gaps in care, research, and policy. Lancet390(10105), 1888–1917 (2017).
Xie, Y. et al. Comparative effectiveness of SGLT2 inhibitors, GLP-1 receptor agonists, DPP-4 inhibitors, and sulfonylureas on risk of kidney outcomes: Emulation of a target trial using Health Care databases. Diabetes Care. 43(11), 2859–2869 (2020).
Jung, C. Y. & Yoo, T. H. Pathophysiologic mechanisms and potential biomarkers in Diabetic kidney disease. Diabetes Metab. J.46(2), 181–197 (2022).
Smith, G. D. & Ebrahim, S. Mendelian randomization’: Can genetic epidemiology contribute to understanding environmental determinants of disease. Int. J. Epidemiol.32(1), 1–22 (2003).
Smith, G. D. & Ebrahim, S. Mendelian randomization: Prospects, potentials, and limitations. Int. J. Epidemiol.33(1), 30–42 (2004).
Stanzick, K. J. et al. Discovery and prioritization of variants and genes for kidney function in > 1.2 million individuals. Nat. Commun.12(1), 4350 (2021).
Skrivankova, V. W. et al. Strengthening the reporting of observational studies in epidemiology using mendelian randomization: The STROBE-MR statement. JAMA326(16), 1614–1621 (2021).
Ferkingstad, E. et al. Large-scale integration of the plasma proteome with genetics and disease. Nat. Genet.53(12), 1712–1721 (2021).
Zhang, J. et al. Plasma proteome analyses in individuals of European and African ancestry identify cis-pQTLs and models for proteome-wide association studies. Nat. Genet.54(5), 593–602 (2022).
Köttgen, A. et al. New loci associated with kidney function and chronic kidney disease. Nat. Genet.42(5), 376–384 (2010).
Wuttke, M. et al. A catalog of genetic loci associated with kidney function from analyses of a million individuals. Nat. Genet.51(6), 957–972 (2019).
Böger, C. A. et al. Association of eGFR-related loci identified by GWAS with incident CKD and ESRD. PLoS Genet.7(9), e1002292 (2011).
Sun, B. B. et al. Genomic atlas of the human plasma proteome. Nature. 558(7708), 73–79 (2018).
Suhre, K. et al. Connecting genetic risk to disease end points through the human blood plasma proteome. Nat. Commun.8, 14357 (2017).
Emilsson, V. et al. Co-regulatory networks of human serum proteins link genetics to disease. Science. 361(6404), 769–773 (2018).
Yao, C. et al. Genome-wide mapping of plasma protein QTLs identifies putatively causal genes and pathways for cardiovascular disease. Nat. Commun.9(1), 3268 (2018).
Zheng, J. et al. Phenome-wide mendelian randomization mapping the influence of the plasma proteome on complex diseases. Nat. Genet.52(10), 1122–1131 (2020).
Folkersen, L. et al. Genomic and drug target evaluation of 90 cardiovascular proteins in 30,931 individuals. Nat. Metab.2(10), 1135–1148 (2020).
Burgess, S., Daniel, R. M., Butterworth, A. S. & Thompson, S. G. Network mendelian randomization: Using genetic variants as instrumental variables to investigate mediation in causal pathways. Int. J. Epidemiol.44(2), 484–495 (2015).
Giambartolomei, C. et al. Bayesian test for colocalisation between pairs of genetic association studies using summary statistics. PLoS Genet.10(5), e1004383 (2014).
Foley, C. N. et al. A fast and efficient colocalization algorithm for identifying shared genetic risk factors across multiple traits. Nat. Commun.12(1), 764 (2021).
Zuber, V. et al. Combining evidence from mendelian randomization and colocalization: Review and comparison of approaches. Am. J. Hum. Genet.109(5), 767–782 (2022).
Hemani, G. et al. The MR-Base platform supports systematic causal inference across the human phenome. Elife. 7, e34408 (2018).
Yavorska, O. O. & Burgess, S. MendelianRandomization: An R package for performing mendelian randomization analyses using summarized data. Int. J. Epidemiol.46(6), 1734–1739 (2017).
Liu, B., Gloudemans, M. J., Rao, A. S., Ingelsson, E. & Montgomery, S. B. Abundant associations with gene expression complicate GWAS follow-up. Nat. Genet.51(5), 768–769 (2019).
Mahajan, A. et al. Trans-ethnic fine mapping highlights kidney-function genes linked to salt sensitivity. Am. J. Hum. Genet.99(3), 636–646 (2016).
Pattaro, C. et al. Genetic associations at 53 loci highlight cell types and biological pathways relevant for kidney function. Nat. Commun.7, 10023 (2016).
Pattaro, C. et al. Genome-wide association and functional follow-up reveals new loci for kidney function. PLoS Genet.8(3), e1002584 (2012).
Ghodsian, N. et al. Electronic health record-based genome-wide meta-analysis provides insights on the genetic architecture of non-alcoholic fatty liver disease. Cell. Rep. Med.2(11), 100437 (2021).
Du, R. et al. PDPN positive CAFs contribute to HER2 positive breast cancer resistance to trastuzumab by inhibiting antibody-dependent NK cell-mediated cytotoxicity. Drug Resist. Updates68, 100947 (2023).
Shi, J., Xu, W., Zheng, R., Miao, H. & Hu, Q. Neuregulin 4 attenuate tubulointerstitial fibrosis and advanced glycosylation end products accumulation in diabetic nephropathy rats via regulating TNF-R1 signaling. Am. J. Transl. Res.11(9), 5501–5513 (2019).
Schumacher, M. A. et al. ErbB4 signaling stimulates pro-inflammatory macrophage apoptosis and limits colonic inflammation. Cell. Death Dis.8(2), e2622 (2017).
Huang, Y. et al. Indoxyl sulfate induces intestinal barrier injury through IRF1-DRP1 axis-mediated mitophagy impairment. Theranostics. 10(16), 7384–7400 (2020).
Duff, M. O., Olson, S., & Wei, X., et al.Genome-wide identification of zero nucleotide recursive splicing in Drosophila. Nature521(7552), 376–379 (2015).
Fagerberg, L. et al. Analysis of the human tissue-specific expression by genome-wide integration of transcriptomics and antibody-based proteomics. Mol. Cell. Proteom.13(2), 397–406 (2014).
Acknowledgements
We are deeply thankful to Zhang J, Ferkingstad E. et al. and the investigators in the GWAS Catalog.
Funding
Financial support for this study was granted under the Key Projects Jointly Constructed by Heman Province and Ministry of Medical Science and Technology (Project Code: SBGJ202102140).
Author information
Authors and Affiliations
Contributions
P.Z. and Z.L.: writing and original draft preparation. J.C. and S.X.: engaged in data analysis and assisted in revising the manuscript. Y.Z.: visualization. Z.Z.: supervision. X.Z.: funding acquisition.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
This analysis of publicly available data does not call for ethical approval.
Consent for publication
Permission for the publication has been granted by all authors.
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Zhao, P., Li, Z., Xue, S. et al. Proteome-wide mendelian randomization identifies novel therapeutic targets for chronic kidney disease. Sci Rep 14, 22114 (2024). https://doi.org/10.1038/s41598-024-72970-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-024-72970-3
- Springer Nature Limited