
Abstract
Glioblastoma is a Grade 4 primary brain tumor defined by therapy resistance, diffuse infiltration, and near-uniform lethality. The underlying mechanisms are unknown, and no treatment has been curative. Using a recently developed creatine kinase inhibitor (CKi), we explored the role of this inhibitor on GBM biology in vitro. While CKi minimally impacted GBM cell proliferation and viability, it significantly affected migration. In established GBM cell lines and patient-derived xenografts, CKi ablated both the migration and invasion of GBM cells. CKi also hindered radiation-induced migration. RNA-seq revealed a decrease in invasion-related genes, with an unexpected increase in glutathione metabolism and ferroptosis protection genes post-CKi treatment. The effects of CKi could be reversed by the addition of cell-permeable glutathione. Carbon-13 metabolite tracing indicated heightened glutathione biosynthesis post-CKi treatment. Combinatorial CKi blockade and glutathione inhibition or ferroptosis activation abrogated cell survival. Our data demonstrated that CKi perturbs promigratory and anti-ferroptotic roles in GBM, identifying the creatine kinase axis as a druggable target for GBM treatment.
Similar content being viewed by others


Introduction
Creatine metabolism plays a crucial role in normal brain physiology, contributing to various aspects of brain function. Creatine is synthesized primarily from arginine, glycine, and methionine in the liver and kidneys1. It is then transported to the brain and other tissues through the creatine transporter SLC6A82. Creatine is converted to phosphocreatine (PCr) within the brain by the enzyme creatine kinase B (CKB). This conversion allows for the storage and efficient transfer of high-energy phosphate groups, which are essential for cellular energy metabolism. PCr acts as a reservoir of high-energy phosphates in the brain, providing an immediate source of ATP during times of increased energy demand3. By replenishing ATP levels, phosphocreatine helps maintain optimal neuronal function and promotes overall brain health. Additionally, creatine has been found to have neuroprotective effects by mitigating oxidative stress, reducing neuroinflammation, and promoting neuronal survival4. Evidentiary of its critical role in CNS health, inborn deficiency of creatine transport leads to severe mental retardation5. However, in adult tissues, there appears to be an ample therapeutic index for inhibiting creatine energetics, as loss-of-function models and inhibition of creatine energetics have modest effects on organismal health6,7,8,9.
It is therefore unsurprising that creatine metabolism plays a role in brain tumor biology. In our previous work10, we found that tumor-associated myeloid cells produce creatine within the hypoxic niche, which promotes the survival and stem-cell phenotypes of glioblastoma (GBM). The role of creatine metabolism has also been previously studied in other tumor models. Creatine metabolism can promote cancer metastasis through SMAD2/3 activation11 and can be regulated by HIF activity12 and the extracellular milieu13. Other studies have shown that creatine can enhance breast cancer survival by ameliorating oxidative stress in vitro14.
In our previous work, we used a well-established, clinically relevant inhibitor of creatine uptake (RGX-202-0114) to treat murine models of brain tumors. This compound is the only clinically used inhibitor of creatine metabolism and is currently under phase 1a/2 trials for gastrointestinal cancer (ClinicalTrials.gov Identifier: NCT03597581). However, as an antimetabolite, consistently high concentrations are needed to reach the CNS, and its effects are quite limited in our brain tumor models10. To avoid these limitations, in this study, we utilized a newly developed inhibitor of creatine kinase (CKi), which covalently binds to the creatine kinase family of enzymes, potently inhibiting its activity15. This inhibitor binds in the reactive cysteine pocket of all creatine kinase family enzymes (CKB, CKM, CKMT1, CKMT2) and thus potently shuts down the entire creatine kinase system.
Surprisingly, while CKi exerts minimal effects on cellular viability, it completely abrogates tumor migration and invasion in several models of GBM. Furthermore, by performing metabolic and transcriptomic analyses, our data revealed that CKi creates synthetic vulnerability to glutathione inhibition and ferroptosis induction. CKi in combination with glutathione inhibition (or ferroptosis induction) synergizes to kill GBM cells. The results of this work suggest that dual targeting of these pathways may represent an effective way to treat glioblastoma.
Results
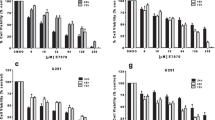
As it is a newly developed creatine kinase B inhibitor, the first experiments aimed to demonstrate its on-target effects. To test this hypothesis, the U251 GBM cell line was treated with a previously established concentration of CKi (20 µM)15, and targeted metabolomics was performed at several time points posttreatment to evaluate creatine and phosphocreatine levels (Fig. 1A). Over the course of 8 h of treatment, the creatine level remained unchanged, while the phosphocreatine (PCr) level decreased in a time-dependent manner (Fig. 1A). Consequently, the Cr/PCr ratios significantly increased over the 8-hour treatment period from 1.78 ± 0.21 without treatment to 4.9 ± 0.27-fold after 8 h (Fig. 1B; p < 0.001). Importantly, all the experiments were performed under hypoxia (1% O2), as CK activity and creatine metabolism in general are regulated by hypoxia12,16,17. We also performed the same assay with two human Patent-derived xenografts, GBM39 and GBM43, and found the same increase in creatine/phosphocreatine ratio as U251 (Fig. 1C).

CKi prevents PCr accumulation and causes several changes in the metabolome. (A) Levels of creatine and phosphocreatine in U251 cells at different time points following CKi treatment. (B) Ratio of creatine to phosphocreatine after CKi inhibition in the U251 cell line. (C), Ratio of creatine to phosphocreatine after CKi inhibition in GBM39, 43. (C) Heatmap showing changes in peripheral versus tumoral myeloid cells, organized according to one-way ANOVA (top 25 shown). (D) Metabolic pathway enrichment analysis comparing vehicle control and CKi-treated cells. (E-F) Basal oxygen consumption rate (OCR) and extracellular acidification rate (ECAR) of U251 cells incubated with CKi for the indicated times. (G) Cell Titer Glo luminescence assay results for several cell lines and patient-derived xenografts (PDXs) treated with 20 µM CKi for 24 h under hypoxia. For (A,B), n = 3 per group; data are representative of 2 independent experiments. For (C), data is n = 2 compiled from 2 independent experiments. For (D,E), n = 3–4 per group. For (G), n = 4–6 per group; data are representative of at least 2 independent experiments. Statistical analysis for (A,B) was performed using one-way ANOVA, followed by Tukey’s post hoc test. For (B,G), significance was calculated using a paired Student’s t-test. *p < 0.05; **p < 0.01; ***p < 0.001.
To analyze how CK inhibition may affect other metabolic pathways, untargeted metabolomics was performed at 0, 1, 4, and 8 h post-CKi treatment, and the top 25 genes were plotted (Fig. 1D). Time-dependent increases in cystine, D-galactonic acid, 6-phosphogluconic acid, and serine were observed, whereas decreases in deoxyadenosine, deoxycytidine, aspartic acid, and orotic acid were observed with CKi treatment (among others). Subsequent unbiased analysis of pathways using metaboanalyst18 was performed, and the control and 8 h CKi treatment groups were compared, as the assay can be performed on only two experimental groups (Fig. 1E). The pathways associated with the greatest changes were the pentose-phosphate pathway (PPP) and pyrimidine, riboflavin, glutathione, and glycine/serine/threonine metabolism, indicating several changes in the overall metabolic health of cells treated with CKi. Considering these changes, we next sought to determine how CKi might affect the overall metabolic health of GBM cells using Seahorse Extracellular flux analysis to measure mitochondrial and glycolytic function (Supplementary Fig. 1). After 8 h of CKi treatment, we found a small but significant reduction in ATP-linked respiration (Supplementary Fig. 1A,B) and glucose-stimulated extra-cellular acidification rate (ECAR) in U251 and GBM43 (Supplementary Fig. 1C,D). There were no significant changes in GBM39 cells. Therefore, the effect of CKi on mitochondrial and glycolytic metabolism is limited, which is consistent with our bulk metabolomics data and the previously reported effects of CKi on metabolism15.
To determine the effects of CKi on the viability of cells, we performed CCK-8 assays with increasing concentrations of CKi at 24, 48, and 72 h (Fig. 1F). While no concentration of CKi achieved ic50 in GBM39 and GBM43, in U251, ic50 could be reached with approximately 5 μm of CKi after 72 h of treatment. Furthermore, we performed Annexin-V/Live-dead assays with 20 μm of CKi on U251, GBM39 and GBM43 (Fig. 1G). Data shows that even though CKi induced significant necrosis on all three lines, (U251-2.9 ± 0.6% in control vs. 17.3 ± 1.0% with 20 μm CKi treatment, p < 0.001; GBM39-5.9 ± 0.3% in control vs. 30.3 ± 3.4% with 20 μm CKi treatment, p < 0.001; GBM43-1.0 ± 0.1% in control vs. 5.9 ± 0.3% with 20 μm CKi treatment; p < 0.001) a majority of cells remained viable. Thus, results demonstrate some level of cell death induced by CKi, although the effects seem limited.
The creatine phosphagen system transfers active phosphate from the mitochondria to the cytoplasm, facilitating processes within that cellular compartment19. This is predominantly performed by the enzyme CKB, which has been shown to be a critical regulator of motility and metastasis in other models of cancer12,13. This suggests that inhibiting the creatine kinase system using CKi may lead to changes in cellular motility. Using the IVYGAP database20, which employed laser capture microdissection to provide gene expression data from different microscopic regions of GBM, we found that CKB expression is enriched at the leading edge and in cellular tumors of GBM (Supplementary Fig. 2A,B), supporting the potential role of CKi in perturbing GBM motility. To test this hypothesis, U251 cells were cultured in the presence of increasing concentrations of CKi, and cellular migration was assessed over time (Fig. 2A,B). Surprisingly, at concentrations that do not inhibit U251 cell survival, CKi caused a dose-dependent decrease in the migration of tumor cells under hypoxia according to the wound healing assay. After 24 h of hypoxia treatment, wound closure was complete in the control group, whereas 20 µM CKi treatment almost completely abolished cellular migration. To extend these findings, we performed these assays in both GBM39 (Fig. 2C,D) and GBM43 (Fig. 2E,F) PDX models in which CKi treatment also dramatically inhibited migration in both GBM39 (pixel width 18.9 ± 12.2 in the control vs. 422.8 ± 14.4 in the CKi-treated group, p < 0.001) and GBM43 (pixel width 41.9 ± 17.4 in the control vs. 318.9 ± 17.7 in the CKi-treated group, p < 0.001).

CKi abrogates the migration of GBM cells in vitro. (A,B) U251 cells were plated overnight in Ibidi chambers (5 × 104 cells per well). After removing the barrier, wells were imaged every 12 h with increasing concentrations of CKi. (C–F) The same migration assay was performed with 20 µM CKi with GBM PDX GBM39 (C,D) and GBM43 (E,F), and chambers were imaged at 12- and 24-hours post-treatment. For all experiments in this figure, n = 3–4 images were obtained from three individual chamber assays, data is representative of 2–3 independent experiment per cell line. Statistics in (B) were calculated via one-way ANOVA, followed by Tukey’s post hoc test to assess individual significance. In (G), significance in (D) and (F) was calculated via paired Student’s t-test. *p < 0.05; **p < 0.01; ***p < 0.001.
While Radiation therapy (RT) is an essential and effective component of standard-of-care surgery for GBM21 it has the unintended side effect of promoting cellular migration and invasion22,23. We therefore wanted to test if CKi could block the enhanced migration endowed by radiation. To test the effects of RT on CKi migration, we performed wound healing assays +/- 4 Gy radiation and +/- CKi (Supplementary Fig. 3). While RT had no effect on U251 cell migration, it significantly increased wound closure at 24 h in both GBM39 and GBM43 cells. Like in the previous assay, U251 cells exhibited abrogated wound closure, and RT had no effect on the efficacy of CKi (Supplementary Fig. 3A,B). This finding was also verified in both GBM39 (Supplementary Fig. 3C,D) and GBM43 (Supplementary Fig. 3E,F) cells. Notably, although CKi prevented RT-induced migration in GBM39 cells, CKi was not more effective as an inhibitor without RT.

CKi inhibits GBM invasion in vitro. In (A–C), U251, GBM39 and GBM43 cells were plated in Matrigel-coated invasion chambers (+/- 20 µM CKi) for 18 h under 1% O2 hypoxia. Some cells were subjected to 4 Gy X-ray radiation 48 h prior to being plated into invasion chambers. Following a 24-hour incubation, the cells were fixed overnight using 4% formaldehyde and stained the following morning with 0.1% crystal violet stain. For all the experiments in this figure, the number of invaded cells was quantified from the images (n = 5 images per experimental group) representative of two independent experiments. Significance was calculated via paired Student’s t test. *p < 0.05; **p < 0.01; ***p < 0.001.
To complement the wound healing assays, we also performed Matrigel invasion assays, which are a well-established method of assessing tissue invasion24 (Fig. 3). The cells were treated with +/- CKi and +/- RT and then subjected to a Matrigel invasion assay under hypoxia; after 24 h, the invaded cells were manually counted. Regardless of RT, CKi almost completely abolished the migration of U251 (Fig. 3A), GBM39 (Fig. 3B), and GBM43 cells (Fig. 3C). These results strongly indicate that creatine kinase is essential for tumor cell migration and invasion and that targeted CKi inhibition is effective at preventing these behaviors.
We next sought to understand how GBM cells respond to CKi. We treated U251 cells for 8 h with CKi under (1% O2) and performed bulk RNA sequencing (Fig. 4). In response to CKi, 668 genes were found to be differentially regulated, with 411 downregulated and 257 upregulated (Fig. 4A). The most significantly upregulated genes were directly associated with glutathione metabolism and redox homeostasis (Fig. 4B). To assess whether the expression of the genes associated with these pathways increased in response to CKi treatment, GSEA was performed. The data clearly showed that CKi treatment upregulated the expression of genes associated with ferroptosis (Fig. 4C) and glutathione metabolism (Fig. 4D). Upon more detailed analysis, the expression of critical redox/reactive oxygen species (ROS)-related genes was dramatically upregulated (Fig. 4E). For example, thioredoxin reductase 1 (TXNRD1) is a key player in oxidative stress control and is essential for survival under glutathione deprivation25. Another gene, heme oxygenase 1 (HMOX1), is essential for the response to ROS-induced stress26. Importantly, several genes involved in both glutathione biosynthesis (GCLM, GCLC) and the uptake of glutathione precursors (SLC7A11, SLC38A2, SLC7A5) were upregulated. SLC7A11 is essential for glutamate uptake and cysteine export, and SLC38A2, which is typically associated with glutamine import to cells, was also upregulated (Fpkm values of 47.7 ± 1.6 in control cells vs. 126.8 ± 1.8 in CKi-treated cells, p < 0.001). These results suggest that cells respond to CKi treatment by upregulating glutathione biosynthesis. Importantly, we re-ran these assays using qPCR in U251, GBM39, and GBM43 and found a significant upregulation of TNXRD1, HMOX1, and SLC7A11 across the models (Fig. 4F).

CKi treatment induces transcriptional upregulation of genes involved in glutathione/ferroptosis regulation. U251 cells were treated with vehicle (1% DMSO or 20 µM CKi), and after 8 h under hypoxia (1% O2), RNA was isolated and sent to Novogene for bulk RNA sequencing. (A), Number of genes dysregulated by CKi treatment; (B), Volcano plot of genes differentially expressed in response to CKi treatment. (C,D) Gene-set enrichment analysis (GSEA) of genes associated with ferroptosis and glutathione, respectively. In (E), FKPM expression was upregulated in 8 genes involved in redox stress and glutathione/ferroptosis regulation. In (F), U251 GBM39 and GBM43 were cultured with CKi for 8 h, and qPCR was performed on all the indicated genes. The results are from n = 3 independent experiments. (E,F) Significance was calculated via paired Student’s t test. *=p < 0.05; **p < 0.01; ***p < 0.001.
This in turn suggests that CKi puts GBM cells under REDOX stress. To determine this we first performed a CellRox assay which with increasing intracellular ROS levels there is an increase in green fluorescence27. We treated U251 with CKi (and H2O2 as a positive control) and found that beginning at one hour, CKi induced a significant increase in intracellular ROS (Supplementary Fig. 4A,B). Furthermore, we performed DCFDA assays and found that addition of N-acetylcysteine could protect GBM cells from ROS production by CKi (Supplementary Fig. 4C). Therefore, these data support that CKi induces redox stress in GBM cells. Considering that the transporters and enzymes responsible for glutathione biosynthesis were upregulated by CKi, we next sought to determine whether de novo glutathione biosynthesis is indeed upregulated by CKi treatment (Fig. 5). Several inputs are needed for glutathione biosynthesis, but glutamine is an essential building block (Fig. 5A). We performed a series of 13C-glutamine tracing experiments for 4 h of CKi treatment in U251, GBM39 and GBM43 cells (Fig. 5B,C). After 4 h of tracing, the labeled glutamine and glutamate amounts were essentially unchanged, indicating that these metabolites had reached a steady state (Fig. 5B). The only exception was in GBM43, where CKi increased the percentage enrichment of M + 5 glutamate. In contrast, the enrichment of de novo-synthesized glutathione and glutathione disulfide (as measured by M + 5 labeling) was dramatically upregulated (Fig. 5C). There was a significant enrichment of M + 5 glutathione disulfide in U251 cells (29.3 ± 1.0% in the control vs. 65.7 ± 2.7% in the CKi treatment; p < 0.001), GBM39 (6.6 ± 0.9% in the control vs. 11.6 ± 0.5% in the CKi treatment; p < 0.01), and GBM43 (0.02 ± 0.0% in the control vs. 7.5 ± 2.3% in the CKi treatment; p < 0.05). There was also significant upregulation of glutathione in U251 and GBM39 cells. In GBM43 cells, there was a trend toward increased M + 5 labeling, but this difference was not significant.

CKi treatment rewires cells to produce GSH de novo. In (A), a schematic showing inputs for glutathione biosynthesis from glutamine and from glucose (green is what was observed in the data). In (B-C), U251, GBM43, and GBM39 cells were cultured +/- 20 µM CKi in the presence of uniformly labeled13C-glutamine (2 mM) for 4 h before the metabolites were isolated, and LC‒MS/MS was used to determine the enrichment of each metabolite in glutathione biosynthesis. In (D), the same assay was performed with glucose-free DMEM supplemented with 10 mM uniformly labeled 13C-glucose. The percentage enrichment for the most common isotopomers is shown in the figure. n = 3 per group for each experiment, the data are representative of two independent experiments. Significance was calculated via paired Student’s t test. *=p < 0.05; **p < 0.01; ***p < 0.001.
Previous work has shown that TCA metabolites can be shunted into glutathione biosynthesis under metabolic stress28, and we wanted to determine whether this could contribute to the reduced mitochondrial activity observed with CKi. Therefore, we subsequently performed 13C-glucose tracing in U251, GBM39 and GBM43 cells (Fig. 5D). As expected, the fractional enrichment of glutathione disulfide (as measured by M + 2 labeling) was significantly upregulated in U251, GBM39, and GBM43 cells. The results clearly demonstrate that CKi induces oxidative stress, and tumor cells attempt to alleviate this stress by synthesizing more glutathione.
These results indicate that CKi perturbs redox homeostasis and that targeting either glutathione biosynthesis or ferroptosis induction can potentiate antitumor responses against GBM. To determine whether CKi indeed perturbs redox homeostasis, we performed a series of migration experiments in which we supplemented CKi-treated cells with a cell-permeable version of glutathione (glutathione ethyl-ester; GSHee29) concurrently or 24 h after CKi addition (Fig. 6; Supplementary Fig. 5). When GSHee was added at the same time as CKi (CKi + GSHee 0 h), there was a significant rescue of the migration of all three models tested. The most dramatic rescue occurred in the GBM43 model, in which complete wound closure was observed at 48 and 72 h, whereas CKi treatment completely prevented wound closure (p < 0.001; Fig. 6A,B). Similar effects were detected in the U251 and GBM39 tumor models (Fig. 6C,D; Supplementary Fig. 5). Interestingly, the addition of GSHee after 24 h of treatment (CKi + GSHee for 24 h) significantly rescued the migration of GBM43 cells at 48 (stats) and 72 h (stats) (Fig. 6A,B). Rescue after 24 h of CKi treatment was not significant in U251 or GBM39 cells (Fig. 6C,D; Supplementary Fig. 5). The results of this work indicate that the effects of CKi on cellular migration occur through the inhibition of redox homeostasis.

The addition of glutathione rescues the inhibitory effects of CKi on migration. In (A–D), GBM43, GBM39, or U251 cells were plated overnight into Ibidi (company) chambers (50,000 cells per well), and the following day, the barrier was removed, and the wells were imaged every 24 h under 20 µM CKi and 20 µM Cki + GSHee. For all the experiments in this figure, n = 3–4 images were obtained from three individual chamber assays, representative of three independent experiments. Statistics in (B–D) were calculated via one-way ANOVA, followed by Tukey’s post hoc test to assess individual significance. *p < 0.05; **p < 0.01; ***p < 0.001.
These results led us to determine whether blocking glutathione biosynthesis (Fig. 7A–C) or promoting ferroptosis (Fig. 7D–F) could work in concert with CKi to promote GBM cell death. We utilized the well-known inhibitor of glutathione biosynthesis, Buthionine sulfoximine (BSO), to determine whether CKi treatment could promote cell death. While no concentration of BSO elicited cell death at any concentration, the introduction of CKi induced cell death in U251, GBM39 and GBM43 cells. In U251 cells, the combination of CKi and BSO had an IC50 of 84 nM (Fig. 7A); in GBM39 cells, the IC50 was 12 nM (Fig. 7B); and in GBM43 cells, the IC50 was ≈ 1.5 µM (Fig. 7C). The ferroptosis inducer RSL3 was able to induce cell death at higher concentrations, but CKi had an additive effect on cell death. In U251 cells, the combination of CKi and RSL3 had an IC50 of 283 nM (Fig. 7D); in GBM39 cells, the IC50 was 62 nM (Fig. 7E); and in GBM43 cells, the IC50 was 12 nM (Fig. 7F). The results of these experiments clearly demonstrated that CKi treatment exacerbates redox stress and promotes ferroptosis.

Glutathione inhibition or ferroptosis activation cooperate with CKi to promote cell death in GBM. In (A–C), U251, GBM43, and GBM39 cells were cultured with increasing concentrations of the glutathione inhibitor BSO alone or with increasing concentrations of CKi +/- 20 µM. After 24 hours, Cell-Titer Glo© was used to examine cellular viability. In (D–F), U251, GBM43, and GBM39 cells were cultured with increasing concentrations of the ferroptosis activator RSL3 alone or with increasing concentrations of CKi (approximately +/- 20 µM). After 24 hours, cell-titer glass was used to examine cellular viability. After the data were obtained, a nonlinear regression was performed using [Inhibitor] vs. response (three parameters) with least squares fit. This was subsequently used to determine the IC50 of either the single agent BSO/RSL3 or BSO/RSL3 in the presence of CKi. “To test for significance in curves, a two-way ANOVA was performed, and Sidak’s multiple comparison post hoc test was used to test for individual significance. *p < 0.05; **p < 0.01; ***p < 0.001.
Discussion
The results of this study clearly reveal that creatine metabolism is an essential player in GBM biology. In the field of GBM, creatine is typically used as a denominator for magnetic resonance spectroscopy (MRS) imaging, as it is supposed to be constant in tissues30,31. However, as this study showed, the effects of creatine are beyond those of simple steady-state metabolites. This has not been the first attempt to inhibit creatine metabolism in tumors, as several previous studies have used two distinct pharmacologic approaches. The first compound is cyclocreatine, a cell-permeable and highly electrophilic metabolite. Cyclocreatine has been tested for cancer treatment in several publications32,33,34,35, and it has been shown to be effective in several studies. The other is the guandinoproprionic acid (β-GPA) analog RGX-202-0136,37. This creatine uptake inhibitor is currently being tested in clinical trials for GI cancer treatment. While these compounds have shown great promise, several limitations are associated with metabolite analogs, such as the need for constant dosing and off-target effects. In this study, we demonstrated that a covalent inhibitor of CK enzymes is a potentially actionable therapy for brain tumors. This compound has its limitations: this first-generation compound lacks stability in plasma and a short half-life, which limits its use in vivo. Furthermore, while CKi is on-target to CK enzymes, we cannot rule out the possibility of other targets of CKi. For example, our previous work has identified that CKi may also target GSTO1 and GSTCD15, which also involves redox homeostasis38 and may influence cellular adhesion39. Therefore, future work will have to determine exactly to what extent CKi specifically targets the CK family of enzymes.
Despite this limitation, this publication lays out a roadmap for understanding how CKi influences GBM biology and provides insight into which therapies may be combined to promote antitumor responses. First, we demonstrated that the inhibitor causes a reduction in Cr/PCr ratios, demonstrating an inhibition of the creatine phosphagen system. However, the effects of CKi caused significant changes in the metabolome, including a general reduction in both OXPHOS and glycolysis. As the creatine shuttle can quickly translocate phosphate from newly generated mitochondrial ATP19, it is logical that the inhibition of CK activity has downstream effects on mitochondrial metabolism. This phenomenon has been previously shown in studies of oligodendrocyte function during remyelination40 and in adipose tissue during futile cycling41. Another potential explanation is that our data show that a substantial amount of carbon from glucose is redirected toward de novo glutathione metabolism in response to CKi treatment. This stress-responsive “shift” in metabolism may be responsible for the decrease in mitochondrial output (there are reductions in several mitochondrial genes with CKi treatment) and, potentially, glycolysis, but future studies are needed to understand this phenomenon more directly.
Second, inhibition of CK did not, by itself, induce significant cell death, even under hypoxic stress. However, what did change was an almost complete ablation of cellular migration and invasion. These findings are consistent with previous studies demonstrating that creatine is involved in migration and metastasis11,12,13. The necessity of CK for cellular motility is an interesting observation, as these two papers are the only others, to our knowledge, to show a link between cellular motility and CK activity. There may be several reasons for this. As the role of the creatine phosphate shuttle is to transmit active phosphate to the cytoplasm, CKis may restrict cytoplasmic ATP levels, which are essential for cellular migration42. Another possibility is the downregulation of several collagen and invasion-related genes in response to CKi inhibition. This difference may be due to stress induction, as several genes associated with the ISR were upregulated in response to CKi treatment. However, upregulation of the ISR is typically associated with increased migration/metastasis43,44. This finding suggested that the effects of CKi are most likely associated with interfering with cellular bioenergetics.
In another critical observation, we investigated the impact of creatine kinase inhibition (CKi) on glioblastoma multiforme (GBM) cell migration and its relationship with redox homeostasis. CKi treatment significantly reduced cellular migration in U251, GBM39, and GBM43 cells, even at concentrations that did not induce substantial cell death, as demonstrated by wound healing assays under hypoxic conditions. Importantly, concurrent supplementation with the cell-permeable glutathione derivative (GSHee) rescued the migratory capacity of CKi-treated cells, with the most pronounced effect observed in the GBM43 model. This highlights the intricate connection between CK inhibition, redox imbalance, and cellular migration in GBM, suggesting potential therapeutic strategies involving the modulation of redox pathways in conjunction with CK inhibition to influence tumor cell behavior.
This work’s last observation was identifying glutathione/ferroptosis as a combinatorial target for GBM cells. This observation is fascinating because of the findings of a recent study that CK directly phosphorylates GPX4, promoting protection from ferroptosis/redox stress45. Therefore, CKi may directly regulate the activity of the glutathione/ferroptosis machinery. Indeed, our data showed that CKi inhibition causes the selective upregulation of both glutathione biosynthesis and the uptake of precursor metabolites needed to synthesize it. These exciting findings suggest that the inhibition of CK activity may sensitize tumor cells to glutathione inhibition. Although the results of the combinatorial treatment are quite dramatic, the absence of IC50 measurements for both the CKi and BSO prevented us from conducting a synergy assay to evaluate synergism. Despite this, the combination of glutathione/ferroptosis inducers and CKi lead to significant cell-death greater than any single agent.
It is important to understand the intricate connection between glutathione metabolism and ferroptosis, as it complicates the dissection of these processes and the role CKi plays in them. SLC7A11, a key component of System xc-, facilitates the exchange of extracellular cystine and intracellular glutamate. This cystine is essential for the synthesis of glutathione which feeds into the ferroptosis protection machinery46. Therefore, our data regarding Buthionine Sulfoximine (BSO) potentiating CKi therapy might still have a more direct effect on ferroptosis, and thus future studies are warranted to understand the complex role of CKi in promoting redox dysregulation in GBM.
Our future studies are aimed at developing an in vivo formulation of this compound to determine whether this approach is effective in preclinical practice. In summary, CK activity is essential for the biology of GBM, and its inhibition may be a promising approach for treating brain tumors.
Experimental procedures
Cell culture
CT2A cells were obtained from Millipore (Sigma Aldrich); U87 and U251 human-derived glioma cell lines were obtained from ATCC; the GL261 murine glioma cell line was obtained from the National Cancer Institute (NCI); and GBM39 and GBM43 patient-derived xenograft (PDX) glioma cell lines were obtained from Dr. David James at Northwestern University. All cell lines were maintained in DMEM (Corning) supplemented with 10% fetal bovine serum (FBS, HyClone) and 1% penicillin‒streptomycin (Gibco). Dissociations were performed enzymatically using 0.25% trypsin solution with 2.21 mM EDTA (Corning). All the cells were kept at 5% CO2 and 37 °C.
Cell X-ray radiation
All cell lines were plated in T-75 flasks and radiated at 4 Gy using a Rad Source RS 2000 X-ray Radiator.
Seahorse extracellular flux analysis
The cartridge from the Seahorse XFp FluxPak (Agilent; 103022-100) was hydrated overnight with XF Calibrant solution in a non-CO2 incubator. 5 × 104 cells were seeded in 80uL per well in a 96 well XF96 plate. The following morning, cells were incubated for 8 h in 20 μM CKi in complete DMEM. The plate was washed once with Agilent Seahorse XF Base Medium (Agilent; 103334-100) containing 2mM glutamine (Agilent 103579-100) prior to addition of 150uL Agilent Base Medium. The Seahorse was then run with a first injection of 25mM glucose followed by the Seahorse XF Cell Mito Stress Test Kit (Agilent; 103015-100) per manufacturer’s instructions.
CCK8 and annexin-V/Live dead analysis
For CCK8 assays, GBM cells were plated at a density of 5 × 103 cells/well in a flat bottom 96-well plate treated with indicated concentrations of CKi for periods of time indicated within the figures under 1% O2 hypoxia. At each time point, cells were analyzed using Cell Counting Kit – 8 (Sigma) and absorbance was measured at 450 nm using a Biotek Cytation 5 (Agilent). Viability was then normalized to untreated cells at each time point.
For Live dead assays, GBM cells were plated at a density of 250,000 cells/well and treated with 20 μM CKi under 1% O2 hypoxia for 24 h. After 24 h, cells were washed and first stained with live/dead Efluor780 1:1000 in PBS for 20 min on ice. Cells were then washed, stained with annexin-V FITC (Biolegend, 1:20 dilution) using Annexin-V binding buffer (Becton Dickinson), and incubated for 15 min at room temperature. Cells were then washed with and finally resuspended in binding buffer, and samples were analyzed using a BD symphony flow cytometer (Becton Dickinson). Data analysis was performed using FlowJo software (Becton Dickinson).
IVYGAP/GLIOVIS data acquisition
To determine the spatial expression of CKB in GBM we accessed the IVYGAP database (https://glioblastoma.alleninstitute.org/rnaseq/search/index.html) which allows for the search of genes across microscopic niches in GBM20. We also utilized the Gliovis portal to provide quantitative interpretation of the data from GlioVis plaform (http://gliovis.bioinfo.cnio.es/)47. Data shown as acquired from the platforms.
Creatine kinase and glutathione biosynthesis inhibition and ferroptosis activation survival assays
Cells were seeded in a white 96-well flat bottom plate at a density of 5 × 104 cells in 100 µl of growth medium supplemented with 10% fetal bovine serum (FBS, HyClone) and 1% penicillin‒streptomycin antibiotic (Gibco). Cells were assessed 24 h after seeding for successful adherence before beginning treatment. 100 µl of medium was added to experimental wells supplemented with CKi (20 µM), BSO (10− 2-104 nM), or RSL3 (10− 2-104 nM), or BSO in combination with CKi or RSL3 combined with CKi at these concentrations for 12–24 h. Then, 100 µl of medium was removed, and 100 µl of CellTiter-Glo 2.0 reagent (Promega, Madison, WI, USA) was added to each well. After incubating at room temperature for 10 min on an orbital shaker, cell viability was measured according to luminescence signals by a BioTek Cytation 5 plate reader, and signals were measured via the following settings: Luminescence, Read Type: Endpoint/Kinetic, Optics Type: Luminescence Fiber, Gain: 135, Integration Time: 0:01.00 (MM: SS.ss), Read Height: 1.00 mm. The percentage of surviving cells treated with each compound was calculated based on the percentage of vehicle (DMSO)-treated cells.
Wound healing and migration assay
The cells were seeded at 5 × 104 cells per well in the culture insert chamber (Ibidi, Fitchburg, WI, USA). The cells were incubated overnight at 37 °C and 5% CO2. The cells were checked twelve hours after seeding for successful adherence. At the first timepoint, the cells were washed with Dulbecco’s phosphate-buffered saline (DPBS; Corning), and the silicone insert was carefully lifted using sterile forceps. The medium was switched to glucose-free DMEM supplemented with 10% fetal bovine serum (FBS, HyClone) and 2 mM glucose. CKi was added to experimental groups and cells were incubated in a hypoxic chamber at 37 °C and 1% O2 for the duration of treatment. At the experimental timepoints, images were obtained via bright field microscopy via a Leica microscope until the gap was visible. The distance or area between cells was measured via pixels on ImageJ and compared between treatment groups over time intervals.
Glutathione rescue assays
Human GBM cells were seeded in white 96-well plates at a density of 5 × 104 cells/ml in 100 µl of growth medium, resulting in approximately 5 × 103 cells per well. Two experimental groups (control and 20 µM CKi treatment) were established, each with a corresponding duplicate, and were allowed to grow overnight. The following day, the medium was replaced with fresh medium containing various concentrations of GSHee for titration purposes, concurrently with creatine kinase inhibitor (CKi) treatment. Cell viability was assessed 24 h posttreatment using the CellTiter-Glo 2.0 Assay, aiming to identify the concentration at which GSHee best mitigated the effects of CKi.
Subsequently, the cells were seeded in Ibidi migration chambers and allowed to adhere and grow overnight. The next day, the cells were washed with PBS, and the silicone inserts were gently removed. The growth medium was then replaced with medium containing 20 µM CKi. GSHee was subsequently added at a concentration of 2 mM at various time points. The cells were incubated in a hypoxic chamber at 37 °C and 1% O2 for the duration of treatment, and migration was monitored under a Leica microscope at 24-hour intervals. Cell migration distances were quantified using ImageJ software, which measures the pixel distance between the edges of the cell-free space as an indicator of migration capability.
Corning matrigel invasion assay
Cells were seeded at 5 × 103 cells per transwell (Corning) in blank DMEM. DMEM supplemented with 10% fetal bovine serum was used as a chemoattractant in 24-well plates. The cells were incubated for 24 h at 37 °C in 5% CO2. Following incubation, the media was aspirated, and the cells were fixed overnight in Matrigel supplemented with 4% formaldehyde. The next morning, the cells were stained with 0.1% crystal violet stain, and once the cells were dried, the Matrigel layer was removed, and the cells were mounted on slides for imaging. Five images were taken per well, and the cells were manually counted via ImageJ.
Metabolomics analysis
Untargeted bulk metabolomics
U251, GBM39, and GBM43 samples were analyzed via untargeted bulk metabolomics. Cells were seeded overnight in a 6-well plate at a density of 3.5 × 105 cells in 3 mL of growth medium supplemented with 10% fetal bovine serum (FBS, HyClone) and 1% penicillin‒streptomycin antibiotic (Gibco). The cells were checked twelve hours after seeding for successful adherence. At the first timepoint, the cells were washed and reconstituted in blank DMEM supplemented with 10% dialyzed fetal bovine serum (FBS; HyClone), 10 mM glucose (Fisher), and 2 mM glutamine (Gibco). The experimental wells contained 13C media supplemented with CKi (20 µM). The cells were placed in a hypoxic chamber at 37 °C and 1% O2 for the duration of treatment. At the 4-hour time point, the cells were lifted and washed twice with chilled sterile 0.9% saline. The cell pellets were resuspended in 80% methanol/20% sterile H2O and lysed via 3 freeze‒thaw cycles. The samples were spun at 21,000 × g for 10 min, after which the supernatant was collected and analyzed.
Glucose labeled flux data
U251, GBM39, and GBM43 cells were analyzed via a glucose flux assay. Cells were seeded overnight in a 6-well plate at a density of 3.5 × 105 cells in 3 ml of growth medium supplemented with 10% fetal bovine serum (FBS, HyClone) and 1% penicillin‒streptomycin antibiotic (Gibco). The cells were checked twelve hours after seeding for successful adherence. At the first timepoint, the cells were washed and reconstituted in blank DMEM supplemented with 10% dialyzed fetal bovine serum (FBS, HyClone), glutamine (2 mM), and U13C-D-glucose (10 mM; Cambridge Isotope Laboratories). The experimental wells contained 13C media supplemented with CKi (20 µM). The cells were placed in a hypoxic chamber at 37 °C and 1% O2 for the duration of treatment. At the 4-hour time point, the cells were lifted and washed twice with chilled sterile 0.9% saline. The cell pellets were resuspended in 80% methanol/20% sterile H2O and lysed via 3 freeze‒thaw cycles. The samples were spun at 21,000 × g for 10 min, after which the supernatant was collected and analyzed.
Glutamine labeled flux data
U251, GBM39, and GBM43 cells were analyzed via a glucose flux assay. Cells were seeded overnight in a 6-well plate at a density of 3.5 × 105 cells in 3 mL of growth medium supplemented with 10% fetal bovine serum (FBS, HyClone) and 1% penicillin-streptomycin antibiotic (Gibco). The cells were checked twelve hours after seeding for successful adherence. At the first timepoint, the cells were washed and reconstituted in blank DMEM supplemented with 10% dialyzed fetal bovine serum (FBS; HyClone), 10 mM glucose, and U13C-D-glutamine (2 mM; Cambridge Isotope Laboratories). The experimental wells contained 13C media supplemented with CKi (20 µM). The cells were placed in a hypoxic chamber at 37 °C and 1% O2 for the duration of treatment. At the 4-hour time point, the cells were lifted and washed twice with chilled sterile 0.9% saline. The cell pellets were resuspended in 80% methanol/20% sterile H2O and lysed via 3 freeze-thaw cycles. The samples were spun at 21,000 × g for 10 min, after which the supernatant was collected and analyzed.
Method for sample reconstitution after extraction
Extracted cell samples were dried using a SpeedVac. Afterwards, 60% acetonitrile was used to reconstitute the pellet, followed by an overtaxing period of 30 s. The samples were then centrifuged at 20,000 × g for 30 min at 4 °C. After preparation, the samples were analyzed via high-performance liquid chromatography (HLP) and high-resolution mass spectrometry (HRMS) and tandem mass spectrometry (TMS).
RNA sequencing
Samples were lysed with RLT Plus buffer (Qiagen, catalog no. 74134) and homogenized with an insulin syringe. RNA was extracted using the RNeasy Plus Mini Kit (Qiagen, catalog no. 74134) according to the manufacturer’s protocol. RNA sample quality control, library preparation (polyA selection, nonstranded), sequencing of 20 M paired-end reads, and analysis were performed by Novogene.
qPCR analysis
RNA was isolated as described above and then synthesized using iScript reverse transcriptase supermix (Bio-Rad) using manufacturers’ protocols. Quantitative PCR was then performed on the cDNA using SYBR® Green master mix (Bio-Rad) and amplified using CFX96 Touch Real-Time PCR qPCR System (Bio-Rad). The following primers used for analysis are all 5’-3’ and synthesized by IDT. ACTB-F: CCTGGCACCCAGCACAAT, ACTB-R: GCTGATCCACATCTGCTGGAA. HMOX1-F: CCAGGCAGAGAATGCTGAGTTC, HMOX1-R: AAGACTGGGCTCTCCTTGTTGC. SLC7A11-F: TCTCCAAAGGAGGTTACCTGC, SLC7A11-R: AGACTCCCCTCAGTAAAGTGAC. TXNRD1-F: ACACAAAGCTTCAGCATGTCA, TXNRD1-R: CAATTCCGAGAGCGTTCC.
Data availability
Any data is available at the request of the corresponding author, Jason Miska (Jason.miska@northwestern.edu). RNA-sequencing data can be accessed via BioProject accession number PRJNA1124134 (https://www.ncbi.nlm.nih.gov/bioproject/1124134). Metabolomics Data can be accessed on Metabolomics Workbench Study ID: ST003262 (https://www.metabolomicsworkbench.org/data/DRCCMetadata.php? Mode=Study&StudyID=ST003262).
References
Wyss, M. & Kaddurah-Daouk, R. Creatine and creatinine metabolism. Physiol. Rev.80, 1107–1213. https://doi.org/10.1152/physrev.2000.80.3.1107 (2000).
Ohtsuki, S. et al. The blood-brain barrier creatine transporter is a major pathway for supplying creatine to the brain. J. Cereb. Blood Flow. Metab.22, 1327–1335. https://doi.org/10.1097/01.WCB.0000033966.83623.7D (2002).
Kazak, L. & Cohen, P. Creatine metabolism: energy homeostasis, immunity and cancer biology. Nat. Rev. Endocrinol.16, 421–436. https://doi.org/10.1038/s41574-020-0365-5 (2020).
Beal, M. F. Neuroprotective effects of creatine. Amino Acids40, 1305–1313. https://doi.org/10.1007/s00726-011-0851-0 (2011).
Salomons, G. S. et al. X-linked creatine-transporter gene (SLC6A8) defect: a new creatine-deficiency syndrome. Am. J. Hum. Genet.68, 1497–1500. https://doi.org/10.1086/320595 (2001).
Streijger, F. et al. Mice lacking brain-type creatine kinase activity show defective thermoregulation. Physiol. Behav.97, 76–86. https://doi.org/10.1016/j.physbeh.2009.02.003 (2009).
Streijger, F. et al. Structural and behavioural consequences of double deficiency for creatine kinases BCK and UbCKmit. Behav. Brain Res.157, 219–234. https://doi.org/10.1016/j.bbr.2004.07.002 (2005).
Van der Zee, C. E. E. M. Hypothalamic plasticity of neuropeptide Y is lacking in brain-type creatine kinase double knockout mice with defective thermoregulation. Eur. J. Pharmacol.719, 137–144. https://doi.org/10.1016/j.ejphar.2013.07.027 (2013).
Steeghs, K. et al. Cytoarchitectural and metabolic adaptations in muscles with mitochondrial and cytosolic creatine kinase deficiencies. Mol. Cell. Biochem.184, 183–194 (1998).
Rashidi, A. et al. Myeloid cell-derived creatine in the hypoxic niche promotes glioblastoma growth. Cell Metab.36, 62–77 e68 (2024). https://doi.org/10.1016/j.cmet.2023.11.013
Zhang, L. et al. Creatine promotes cancer metastasis through activation of Smad2/3. Cell. Metab.https://doi.org/10.1016/j.cmet.2021.03.009 (2021).
Krutilina, R. I. et al. HIF-Dependent CKB expression promotes breast cancer metastasis, whereas cyclocreatine therapy impairs cellular invasion and improves chemotherapy efficacy. Cancers (Basel)14. https://doi.org/10.3390/cancers14010027 (2021).
Papalazarou, V. et al. The creatine-phosphagen system is mechanoresponsive in pancreatic adenocarcinoma and fuels invasion and metastasis. Nat. Metab.2, 62–80. https://doi.org/10.1038/s42255-019-0159-z (2020).
Li, Q. et al. SLC6A8-mediated intracellular creatine accumulation enhances hypoxic breast cancer cell survival via ameliorating oxidative stress. J. Exp. Clin. Cancer Res.40, 168. https://doi.org/10.1186/s13046-021-01933-7 (2021).
Darabedian, N. et al. Depletion of creatine phosphagen energetics with a covalent creatine kinase inhibitor. Nat. Chem. Biol.https://doi.org/10.1038/s41589-023-01273-x (2023).
Loo, J. M. et al. Extracellular metabolic energetics can promote cancer progression. Cell160, 393–406. https://doi.org/10.1016/j.cell.2014.12.018 (2015).
Glover, L. E. et al. Control of creatine metabolism by HIF is an endogenous mechanism of barrier regulation in colitis. Proc. Natl. Acad. Sci. U. S. A.110, 19820–19825. https://doi.org/10.1073/pnas.1302840110 (2013).
Chong, J., Wishart, D. S., Xia, J. & Using metaboAnalyst 4.0 for comprehensive and integrative metabolomics data analysis. Curr. Protoc. Bioinf.68, e86. https://doi.org/10.1002/cpbi.86 (2019).
Bessman, S. P. & Carpenter, C. L. The creatine-creatine phosphate energy shuttle. Annu. Rev. Biochem.54, 831–862. https://doi.org/10.1146/annurev.bi.54.070185.004151 (1985).
Puchalski, R. B. et al. An anatomic transcriptional atlas of human glioblastoma. Science360, 660–663. https://doi.org/10.1126/science.aaf2666 (2018).
Stupp, R. et al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N. Engl. J. Med.352, 987–996. https://doi.org/10.1056/NEJMoa043330 (2005).
Shankar, A. et al. Subcurative radiation significantly increases cell proliferation, invasion, and migration of primary glioblastoma multiforme in vivo. Chin. J. Cancer33, 148–158. https://doi.org/10.5732/cjc.013.10095 (2014).
Zhai, G. G. et al. Radiation enhances the invasive potential of primary glioblastoma cells via activation of the rho signaling pathway. J. Neurooncol.76, 227–237. https://doi.org/10.1007/s11060-005-6499-4 (2006).
Benton, G., Arnaoutova, I., George, J., Kleinman, H. K. & Koblinski, J. Matrigel: from discovery and ECM mimicry to assays and models for cancer research. Adv. Drug Deliv. Rev.79–80, 3–18. https://doi.org/10.1016/j.addr.2014.06.005 (2014).
Mandal, P. K. et al. Loss of thioredoxin reductase 1 renders tumors highly susceptible to pharmacologic glutathione deprivation. Cancer Res.70, 9505–9514. https://doi.org/10.1158/0008-5472.CAN-10-1509 (2010).
Gozzelino, R., Jeney, V. & Soares, M. P. Mechanisms of cell protection by heme oxygenase-1. Annu. Rev. Pharmacol. Toxicol.50, 323–354. https://doi.org/10.1146/annurev.pharmtox.010909.105600 (2010).
Yokoyama, C. et al. Induction of oxidative stress by anticancer drugs in the presence and absence of cells. Oncol. Lett.14, 6066–6070. https://doi.org/10.3892/ol.2017.6931 (2017).
Mitsuishi, Y. et al. Nrf2 redirects glucose and glutamine into anabolic pathways in metabolic reprogramming. Cancer Cell.22, 66–79. https://doi.org/10.1016/j.ccr.2012.05.016 (2012).
Zeevalk, G. D., Manzino, L., Sonsalla, P. K. & Bernard, L. P. Characterization of intracellular elevation of glutathione (GSH) with glutathione monoethyl ester and GSH in brain and neuronal cultures: relevance to Parkinson’s disease. Exp. Neurol.203, 512–520. https://doi.org/10.1016/j.expneurol.2006.09.004 (2007).
Law, M. et al. Glioma grading: sensitivity, specificity, and predictive values of perfusion MR imaging and proton MR spectroscopic imaging compared with conventional MR imaging. AJNR Am. J. Neuroradiol.24, 1989–1998 (2003).
Bulik, M., Jancalek, R., Vanicek, J., Skoch, A. & Mechl, M. Potential of MR spectroscopy for assessment of glioma grading. Clin. Neurol. Neurosurg.115, 146–153. https://doi.org/10.1016/j.clineuro.2012.11.002 (2013).
Kurmi, K. et al. Tyrosine phosphorylation of mitochondrial creatine kinase 1 enhances a druggable tumor energy shuttle pathway. Cell Metab. 28, 833–847 e838 https://doi.org/10.1016/j.cmet.2018.08.008 (2018).
Miller, E. E., Evans, A. E. & Cohn, M. Inhibition of rate of tumor growth by creatine and cyclocreatine. Proc. Natl. Acad. Sci. U. S. A.90, 3304–3308 (1993).
Adamski, V. et al. The chemokine receptor CXCR6 evokes reverse signaling via the transmembrane chemokine CXCL16. Int. J. Mol. Sci.18https://doi.org/10.3390/ijms18071468 (2017).
Patel, R. et al. Cyclocreatine suppresses creatine metabolism and impairs prostate cancer progression. Cancer Res.82, 2565–2575. https://doi.org/10.1158/0008-5472.CAN-21-1301 (2022).
Kurth, I. et al. RGX-202, a first-in-class small-molecule inhibitor of the creatine transporter SLC6a8, is a robust suppressor of cancer growth and metastatic progression. Cancer Res.78https://doi.org/10.1158/1538-7445.Am2018-5863 (2018).
Bendell, J. C. et al. Phase I monotherapy dose escalation of RGX-202, a first-in-class oral inhibitor of the SLC6a8/CKB pathway, in patients with advanced gastrointestinal (GI) solid tumors. J. Clin. Oncol.38 (2020).
Chai, Y. C. & Mieyal, J. J. Glutathione and glutaredoxin-key players in cellular redox homeostasis and signaling. Antioxid (Basel) 12https://doi.org/10.3390/antiox12081553 (2023).
Xu, Y. et al. Deletion of glutathione S-Transferase Omega 1 activates type I interferon genes and downregulates tissue factor. Cancer Res.80, 3692–3705. https://doi.org/10.1158/0008-5472.CAN-20-0530 (2020).
Chamberlain, K. A., Chapey, K. S., Nanescu, S. E. & Huang, J. K. Creatine enhances mitochondrial-mediated oligodendrocyte survival after demyelinating Injury. J. Neurosci.37, 1479–1492. https://doi.org/10.1523/JNEUROSCI.1941-16.2016 (2017).
Kazak, L. et al. A creatine-driven substrate cycle enhances energy expenditure and thermogenesis in beige fat. Cell163, 643–655. https://doi.org/10.1016/j.cell.2015.09.035 (2015).
Zanotelli, M. R. et al. Regulation of ATP utilization during metastatic cell migration by collagen architecture. Mol. Biol. Cell.29, 1–9. https://doi.org/10.1091/mbc.E17-01-0041 (2018).
Lu, S. et al. Transcriptional control of metastasis by integrated stress response signaling. Front. Oncol.11, 770843. https://doi.org/10.3389/fonc.2021.770843 (2021).
Nagelkerke, A. et al. Hypoxia stimulates migration of breast cancer cells via the PERK/ATF4/LAMP3-arm of the unfolded protein response. Breast Cancer Res.15https://doi.org/10.1186/bcr3373 (2013).
Wu, K. et al. Creatine kinase B suppresses ferroptosis by phosphorylating GPX4 through a moonlighting function. Nat. Cell. Biol.25, 714–725. https://doi.org/10.1038/s41556-023-01133-9 (2023).
Sato, M. et al. The ferroptosis inducer erastin irreversibly inhibits system x(c)- and synergizes with cisplatin to increase cisplatin’s cytotoxicity in cancer cells. Sci. Rep.8, 968. https://doi.org/10.1038/s41598-018-19213-4 (2018).
Bowman, R. L., Wang, Q. H., Carro, A., Verhaak, R. G. W. & Squatrito, M. GlioVis data portal for visualization and analysis of brain tumor expression datasets. Neuro Oncol.19, 139–141. https://doi.org/10.1093/neuonc/now247 (2017).
Acknowledgements
We would like to thank Prof. Ben-Sahra (Northwestern) for his assistance in the development and analyses of the 13 C-flux studies. We would like to thank Navdeep Chandel (Northwestern) for his advice and collaboration on this work.
Funding
This was funded by National Institutes of Health/NCI grant (JM) − 1R01CA279686-01, National Institutes of Health/NCI grant P50CA221747 (MSL, JM) SPORE subaward, National Institutes of Health/R01NS096376-06A1 1R01CA223547-01.
Author information
Authors and Affiliations
Contributions
Author Credit Statement Conceptualization: J.M., E.C. Methodology: J.L.K., Y.G., J.S., S.L.D. N.S.S. ,G.M., L.K.B., H.W., C.S., C.W., L.K.B., T.C., S.W., H.W., D.H., G.I.C, Z.H.S., T.A. Formal analysis: J.M., J.L.K., Y.G., C.W.Supervision: JM, E.C., C.L.C., A.U.A., P.Z., D.M.T. Writing (original draft): JMM, E.C., J.L.K. Writing (review and editing): all authors. Funding acquisition: J.M., E.C.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Katz, J.L., Geng, Y., Billingham, L.K. et al. A covalent creatine kinase inhibitor ablates glioblastoma migration and sensitizes tumors to oxidative stress. Sci Rep 14, 21959 (2024). https://doi.org/10.1038/s41598-024-73051-1
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-024-73051-1
- Springer Nature Limited