
Abstract
Rad51 maintains genome integrity, whereas Rad52 causes non-canonical homologous recombination leading to gross chromosomal rearrangements (GCRs). Here we find that fission yeast Srr1/Ber1 and Skb1/PRMT5 promote GCRs at centromeres. Genetic and physical analyses show that srr1 and skb1 mutations reduce isochromosome formation mediated by centromere inverted repeats. srr1 increases DNA damage sensitivity in rad51 cells but does not abolish checkpoint response, suggesting that Srr1 promotes Rad51-independent DNA repair. srr1 and rad52 additively, while skb1 and rad52 epistatically reduce GCRs. Unlike srr1 or rad52, skb1 does not increase damage sensitivity. Skb1 regulates cell morphology and cell cycle with Slf1 and Pom1, respectively, but neither Slf1 nor Pom1 causes GCRs. Mutating conserved residues in the arginine methyltransferase domain of Skb1 greatly reduces GCRs. These results suggest that, through arginine methylation, Skb1 forms aberrant DNA structures leading to Rad52-dependent GCRs. This study has uncovered roles for Srr1 and Skb1 in GCRs at centromeres.
Similar content being viewed by others

Introduction
Gross chromosomal rearrangements (GCRs), such as translocations, can occur using repetitive sequences that are abundant and widespread in eukaryotic genomes1. In humans, the total number of repetitive sequences, including satellite repeats and transposable elements, accounts for 54% of the genome2,3. GCRs cause cell death and genetic disorders, including cancer. On the other hand, GCRs can be a driving force of evolution by creating genome diversity4. Therefore, GCRs are not only pathological but also physiological phenomena.
The centromere that ensures proper chromosome segregation contains repetitive DNA sequences in many eukaryotes. Human centromeres (≥ 3 Mb) contain α satellite and other types of satellite repeats, transposable elements, and segmental duplications5. The orientation of the centromere repeats including higher-order repeats of α satellites, switches within a centromere, forming inverted DNA repeats. Despite the important role in chromosome segregation, the centromere is a hotspot for chromosomal breakage and rearrangement6,7,8,9,10. Recombination between repetitive sequences at the centromere forms abnormal chromosomes11,12,13. Robertsonian translocations, a fusion of two acrocentric chromosomes at or around centromeres, are the most frequently observed form of chromosomal abnormality in humans, affecting 1 out of 1000 newborns14. Isochromosomes whose arms are mirror images of each other are commonly found in cancer cells15. Isochromosomes of chr21 and chrX cause Down and Turner syndromes, respectively16,17. Compared to mammalian centromeres, the fission yeast S. pombe centromeres are short (35~110 kb) but contain inverted DNA repeats flanking a non-repetitive core sequence18,19. In this fungus, isochromosomes are produced using inverted DNA repeats in the centromere20,21,22. Less complexity of the centromere DNA sequence makes fission yeast an excellent system to study the mechanism of centromeric GCRs.
Homologous recombination is required to repair detrimental DNA damage such as double-strand breaks23. Rad51 is the key player in canonical homologous recombination and catalyses homology search and DNA strand exchange, forming displacement loops. Mammalian BRCA1 and BRCA2 facilitate Rad51-dependent recombination, and their mutations increase GCRs and predispose the carriers to cancer24,25. Homologous recombination maintains centromere integrity. In mammals, the inactivation of Rad51 increases aberrant recombination at centromeres9,10,26. In fission yeast, loss of Rad51 increases isochromosome formation at centromeres20,21,27. Detailed analysis using fission yeast showed that Rad51 preferentially promotes a conservative way of recombination: non-crossover recombination at centromeres27,28, thereby suppressing isochromosome formation.
Another recombinase Rad52 promotes homology-dependent DNA recombination/repair independent of Rad5129,30. Rad52, on its own, promotes displacement loop formation, single-strand annealing (SSA), and inverse-strand exchange using RNA strands. Yeast Rad52 also facilitates Rad51 loading onto replication protein A (RPA)-coated single-stranded DNA, while human Rad52 does not have the loader activity31. In both mammals and fission yeast, Rad52-dependent non-canonical recombination causes GCRs at centromeres9,32. In fission yeast, Rad52 causes isochromosome formation via crossover recombination with Mus81, a crossover-specific endonuclease27,32,33,34,35. PCNA ubiquitination at lysine 107 and Msh2-Msh3 have been implicated in the Rad52-dependent GCR pathway32,36. The DNA sliding clamp PCNA may form DNA structures leading to Rad52-dependent GCRs because PCNA K107 is dispensable for DNA damage repair36. The rad52 deletion does not eliminate isochromosome formation, suggesting the presence of a Rad52-independent GCR pathway(s). Moreover, the initial event that leads to GCRs remains unclear.
To gain insights into the GCR mechanism, we search for the factors that cause GCRs in the rad51Δ mutant strain and find Srr1 and Skb1. In A. thaliana and mice, the Srr1 homolog affects the transcription of the genes involved in the circadian rhythm37,38,39. Skb1 is involved in a range of pathways, including cell morphology and cell cycle regulation in fission yeast40,41,42,43, and is the homolog of the human protein arginine methyltransferase 5 (PRMT5)44,45. Srr1 and Skb1 specifically promote isochromosome formation. Remarkably, the srr1 mutation increases DNA damage sensitivity and chromosome loss but is not essential for checkpoint response to DNA damage, suggesting that Srr1 promotes DNA damage repair. srr1 and rad52 mutations additively reduced GCR rates, suggesting that Srr1 and Rad52 have overlapping and non-overlapping roles in GCRs. In contrast to srr1, the skb1 deletion does not increase DNA damage sensitivity and, intriguingly, reduces chromosome loss in rad51Δ cells. Loss of Slf140,41 or Pom142,43, which functions with Skb1 in cell morphology and cell cycle regulation, did not reduce GCRs. However, mutating conserved residues in the arginine methyltransferase (RMTase) domain of Skb1 strongly reduced GCRs, suggesting that Skb1 causes isochromosome formation through its RMTase activity. These findings pave new avenues to decipher the mechanism of GCR events at the centromere.
Results
Srr1 and Skb1 cause gross chromosomal rearrangements (GCRs)
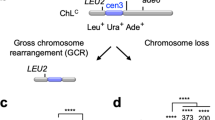
To gain insights into the mechanism of GCRs, we introduced random mutations into rad51Δ cells that show elevated GCR rates and searched for the clones that exhibit reduced levels of GCRs. To detect otherwise lethal GCRs in haploid cells, we used an extra-chromosome ChLC (~530 kb) derived from fission yeast chromosome 3 (chr3) and detected spontaneous GCRs that had lost ura4+ and ade6+ marker genes (Fig. 1a)20,27,46. To assess GCR rates, yeast clones grown on Edinburgh minimum media supplemented with uracil and adenine (EMM+UA) were replicated onto the media containing 5-fluoroorotic acid (5-FOA+UA) that is toxic to ura4+ cells. Of 24,000 clones, three reproducibly exhibited reduced levels of GCRs. Genome sequencing of one of them identified the srr1/ber1-W157R and skb1-A377V mutations in their SRR1-like and arginine methyltransferase (RMTase) domains, respectively (Fig. 1b). The srr1 and skb1 genes are only 51 kb apart from each other on chr2. The replica plating assay shows that, compared to the parental rad51Δ strain, the rad51Δ clone containing the srr1 and skb1 mutations from the reduced number of colonies on the 5-FOA+UA plate (Fig. 1c).

a Depicted is an extra-chromosome ChLC used to detect GCRs in this study. GCRs resulting in Leu+ Ura– Ade– from Leu+ Ura+ Ade+ were detected. b Srr1/Ber1 and Skb1 proteins contain the SRR1-like domain and the arginine methyltransferase (RMTase) domain, respectively. Aligned are the amino acid sequences around the srr1-W157R and skb1-A377V mutations (blue circles) in different species. Similar and identical residues among the different species are highlighted in pale and dark gray, respectively. c The parental rad51∆ strain and the clone additionally containing srr1-W157R and skb1-A377V mutations (TNF5411 and 5954) grown on EMM+UA were replicated onto 5-FOA+UA plates. Leu+ Ura– cells selectively form colonies on 5-FOA+UA plates. d GCR rates of wild-type, srr1∆, skb1∆, rad51∆, srr1∆ rad51∆, skb1∆ rad51∆, and srr1∆ skb1∆ rad51∆ strains (TNF5369, 5774, 5772, 5411, 5904, 5788, and 8432). e GCR rates of wild-type, rad51∆, srr1∆ rad51∆, srr1-W157R rad51∆ (TNF8344), skb1∆ rad51∆, skb1-A377V rad51∆ (TNF8359), and srr1-W157R skb1-A377V rad51∆ (TNF8547). Each dot represents a value obtained from an independent experiment. Black lines show the median. Rates relative to the wild-type are shown on top of each dot cluster. The two-tailed Mann-Whitney test between the wild-type and mutant strains and between the indicated pairs. ns, non-significant; **p < 0.01; ***p < 0.001; ****, p < 0.0001. Numerical data underlying the graphs d and e are provided in Table A in Supplementary Data 1.
To establish whether Srr1 or Skb1 is required for GCRs, we deleted the genes and determined GCR rates by the fluctuation test (Fig. 1d). In the wild-type background, srr1Δ but not skb1Δ slightly reduced GCR rates, showing that Srr1 is required for GCRs even in the presence of Rad51. To our surprise, not only srr1Δ but also skb1Δ reduced GCR rates in the rad51Δ background, demonstrating that both Srr1 and Skb1 cause GCRs. Remarkably, srr1Δ skb1Δ double mutation further reduced GCRs than the single mutations, suggesting that Srr1 and Skb1 have non-overlapping roles in GCRs (see below). To determine whether the srr1-W157R and skb1-A377V point mutations reduce GCR rates, we introduced each mutation into yeast by transformation (see Methods) and determined GCR rates in the rad51Δ background (Fig. 1e). srr1-W157R reduced GCR rates although less prominent than srr1Δ, suggesting a residual activity of the Srr1 mutant protein. Unlike skb1Δ, skb1-A377V did not significantly reduce GCR rates in srr1+ rad51Δ cells. However, skb1-A377V reduced GCR rates in srr1-W157R rad51Δ cells, suggesting that skb1-A377V partially inactivates the Skb1 function. The additive effect of srr1-W157R and skb1-A377V on GCR rates can explain why our genetic screening identified both mutations in the same clone. These results are consistent with the idea that Srr1 and Skb1 have non-overlapping roles in GCRs.
Budding yeast srr1/ber1Δ (benomyl resistant) cells are hyper-resistant to microtubule-destabilizing benzimidazole compounds: benomyl and nocodazole39. However, fission yeast srr1Δ cells were no more resistant than wild type to thiabendazole (TBZ), the most popular benzimidazole used in this fungus (Supplementary Fig. 1). Based on this, we decided to call this gene srr1 rather than ber1 to avoid confusion.
Srr1 and Skb1 promote isochromosome formation at the centromere
Previous studies showed that rad51Δ cells produce isochromosomes and relatively few chromosomal truncations27,32,36. Isochromosomes are produced using inverted DNA repeats at the centromere, whereas chromosomal truncations are formed by the de novo addition of telomere sequences to new chromosome ends (Fig. 2a). Isochromosomes (300~400 kb) and truncations (< 220 kb) are distinguished from each other by their lengths. To determine which GCRs Srr1 and Skb1 cause, chromosomal DNAs of the parental and independent GCR clones were prepared in agarose plugs, separated by pulse-field gel electrophoresis (PFGE), and stained with ethidium bromide (Fig. 2b). The sizes of GCR products were determined using the parental ChLC (530 kb) and the lambda (λ) DNA ladder as references. rad51Δ cells produced many isochromosomes (300~400 kb) and a small number of truncations (< 220 kb), as observed previously20,27,32,36. Loss of Srr1 or Skb1 reduced the proportion of isochromosomes among GCR products. The total GCR rates were multiplied by the proportion of isochromosomes or truncations (Table 1) to obtain the rates of isochromosomes and truncations, respectively (Fig. 2c). Either srr1Δ or skb1Δ eliminated ~90% of isochromosomes formed in rad51Δ cells, but neither reduced chromosomal truncations. These data indicate that Srr1 and Skb1 are specifically required for isochromosome formation at the centromere.

a Depicted are the non-repetitive (cnt3) and repetitive sequences (the innermost imr3, dg, dh, and the outermost irc3) in cen3. The ura4+ marker gene is placed at 10 kb from cen3. Loss of Rad51 increases isochromosomes and chromosomal truncations. b Chromosomal DNAs prepared from the parental (P) and independent GCR clones of rad51∆, srr1∆ rad51∆, and skb1∆ rad51∆ strains (TNF5411, 5904, and 5788) were separated by PFGE. Sizes of lambda (λ) DNA ladders are indicated on the left of the panels. Sample numbers of isochromosomes and truncations are shown in blue and magenta, respectively. c Rates of isochromosome formation and chromosomal truncation in wild-type (TNF5369), rad51∆, srr1∆ rad51∆, skb1∆ rad51∆, and skb1-F319Y rad51∆ (TNF8391) strains. Rates relative to the rad51∆ strain are indicated on the top of the bars. The two-tailed Fischer’s exact test between the rad51∆ and other mutant strains. **p < 0.01; ****p < 0.0001. Numerical data underlying c are provided in Table B in Supplementary Data 1. Uncropped gel images are shown in Supplementary Fig. 5.
Srr1 has a Rad51-independent role in DNA damage repair and chromosome maintenance
As Srr1 and Skb1 promote isochromosome formation mediated by centromere repeats, they might be involved in the recombinational repair of DNA damage. To test this possibility, we performed a serial dilution assay and determined the sensitivity of srr1 and skb1 mutant strains to DNA-damaging agents (Fig. 3a). Methyl methanesulfonate (MMS) is a DNA alkylating agent; hydroxyurea (HU) depletes dNTP pool; camptothecin (CPT) is a topoisomerase inhibitor. These agents interfere with the progression of replication forks and create DNA breaks. Compared to wild-type, srr1Δ cells exhibited hypersensitivity to all the DNA-damaging agents (Fig. 3a, top panels). Notably, srr1Δ rad51Δ cells were more sensitive than the single mutants, suggesting a role for Srr1 in Rad51-independent DNA damage response. The srr1-W157R mutation that partially reduced GCR rates (Fig. 1e) also partially increased the damage sensitivity. These results suggest that Srr1 facilitates Rad51-independent DNA damage response.

a A serial dilution assay to determine the sensitivity to DNA damaging agents. Log-phase cultures of wild-type, srr1-W157R, srr1Δ, rad51Δ, srr1-W157R rad51Δ, and srr1Δ rad51Δ (TNF3885, 8280, 5847, 5845, 8573, and 5849) prepared in YE3S medium were spotted onto YE3S plates supplemented with the indicated concentrations of MMS, HU, or CPT (top panels). Log-phase cultures of wild-type, skb1Δ, rad51Δ, and skb1Δ rad51Δ (TNF35, 8321, 8107, and 8320) prepared in YE medium were spotted onto YE plates (bottom panels). b Wild-type, srr1Δ, skb1Δ, and chk1Δ cells (TNF35, 5943, 8321, and 3559) in the log phase in EMM were treated with 0.01% MMS. The percentage of cells containing a septum is indicated. > 300 cells are counted at each point. Pearson’s Chi-square test of the septation index between wild-type and other strains at t = 8 h showed that srr1Δ or skb1Δ did not significantly change the septation index (p > 0.05) but chk1Δ increased it. c Chk1 phosphorylation in response to MMS treatment. Before and after 4 h treatment with 0.01% MMS, extracts were prepared from chk1+ (TNF7555) and chk1-HA+ cells of wild-type, srr1Δ, and skb1Δ (TNF8441, 8799, and 8802) and separated by 8% SDS-PAGE. Chk1-HA was detected by Western blotting using anti-HA antibodies (16B12). Whole proteins were stained using Coomassie brilliant blue. Size markers (Takara, 3454 A, CLEARLY stained protein ladder) are shown on the left. wt, wild-type. Uncropped images are shown in Supplementary Fig 6. d Wild-type and srr1Δ (TNF5369 and 5774) cells were plated onto adenine-limited YE plates, on which ade6– cells form red colonies. e Chromosome loss rates of wild-type, srr1Δ, srr1-W157R, skb1Δ, rad51Δ, srr1-W157R rad51Δ, and skb1Δ rad51Δ strains (TNF5369, 5774, 8308, 5772, 5411, 8344, and 5788). The two-tailed Mann-Whitney test. **p < 0.01; ***p < 0.001. Numerical data underlying b, e are provided in Tables C and D, respectively, in Supplementary Data 1.
Cell cycle arrest caused by DNA damage checkpoint gives cells time to repair DNA. To determine whether Srr1 is required to arrest cell cycle progression in response to DNA damage, we determined the percentage of septated cells before and after exposure to MMS (Fig. 3b). In the wild-type, the septation index declined from ~30 to 5% after MMS exposure, demonstrating MMS-induced cell cycle arrest. The checkpoint kinase Chk1 is required for cell cycle arrest47. In the chk1Δ strain, the septation index did not decline to the wild-type level. Unlike chk1Δ, in the srr1Δ strain, the septation index declined to the wild-type level by 6 h after MMS addition, suggesting that Srr1 is dispensable for cell cycle arrest. A delay in the reduction of septated cells can be due to the slow growth of the srr1Δ strain. The doubling time of wild-type and srr1Δ cells grown in EMM media at 30 °C were 2.48 ± 0.11 and 2.73 ± 0.10 h, respectively (p = 0.042, the two-tailed student’s t-test) (Table E in Supplementary Data 1). Like wild-type cells, srr1Δ cells were elongated after MMS exposure (Supplementary Fig. 2a). These data suggest that Srr1 is dispensable for DNA damage-induced cell cycle arrest. Chk1 kinase is phosphorylated and activated in response to DNA damage48,49. To determine whether Srr1 is required for the Chk1 phosphorylation, HA-tagged Chk1-HA was expressed from the original chromosomal locus, separated by SDS-PAGE, and detected using anti-HA antibodies (Fig. 3c). A slow-migrating Chk1-HA band indicative of the phosphorylation48 was observed upon MMS treatment in both wild-type and srr1Δ strains, showing that Srr1 is not essential for DNA damage checkpoint activation.
Repair of spontaneous DNA damage is vital to maintaining chromosomes. To determine whether Srr1 is required to maintain chromosomes, we determined spontaneous loss rates of ChLC by the fluctuation test. A colony formed on YE3S plates was suspended in sterilized water, and the cells were plated onto adenine-limited YE plates, on which cells lacking ade6+ formed red colonies50 (Fig. 3d). The red colonies were further inspected for Leu and Ura auxotrophy to obtain the rate of chromosome loss (i.e., Leu– Ura– Ade–) (Fig. 3e). We found that srr1Δ and srr1-W157R increased the rate of chromosome loss. rad51Δ also increases chromosome loss, as observed previously32. Notably, srr1-W157R and rad51Δ synergistically increased chromosome loss, demonstrating that Srr1 and Rad51 have different roles in maintaining chromosomes.
In contrast to the srr1 mutations, skb1Δ did not increase the sensitivity to DNA-damaging agents either in the presence or absence of Rad51 (Fig. 3a, bottom panels), showing that Skb1 is dispensable to repair DNA damage induced by MMS, HU, or CPT. Skb1 was also dispensable for cell cycle arrest and Chk1 phosphorylation induced by MMS treatment (Fig. 3b, c). Intriguingly, however, skb1Δ strongly reduced chromosome loss in rad51Δ cells (Fig. 3e), showing that Skb1 causes isochromosome formation and chromosome loss in rad51Δ cells.
Srr1 and Rad52 have overlapping and non-overlapping roles in GCRs
Rad52 promotes isochromosome formation in rad51Δ cells. The rad52-R45K mutation in the N-terminal DNA-binding domain specifically impairs in vitro single-strand annealing (SSA) activity and reduces isochromosome formation to the same extent as rad52Δ32, suggesting a role of Rad52-mediated SSA in isochromosome formation. The rad52-R45K, rad52Δ, and srr1Δ mutations eliminate ~90% of isochromosomes in rad51Δ cells (Fig. 2c and ref. 32), indicating that both Rad52 and Srr1 are essential for the major pathway of isochromosome formation. To define the relationship between Srr1 and Rad52, we created the srr1 rad52 double mutants and determined GCR rates (Fig. 4a). Notably, srr1Δ and rad52-R45K additively reduced GCR rates in the wild-type background, suggesting that Srr1 and Rad52 also have non-overlapping roles in GCRs. Consistent with this idea, srr1-W157R and rad52-R45K additively reduced GCR rates in the rad51Δ background. Ubiquitination of PCNA (encoded by the pcn1 gene) at lysine 107 (K107) plays a role in Rad52-dependent GCRs36. As expected, srr1-W157R and pcn1-K107R also additively reduced GCR rates in rad51Δ cells (Fig. 4a). These data show that Srr1 and Rad52 have both overlapping and non-overlapping roles in GCRs.

a GCR rates of wild-type, srr1∆, rad52-R45K, srr1∆ rad52-R45K, rad51∆, srr1-W157R rad51∆, rad52-R45K rad51∆, srr1-W157R rad52-R45K rad51∆, pcn1-K107R rad51∆, and srr1-W157R pcn1-K107R rad51∆ strains (TNF5369, 5774, 6599, 8281, 5411, 8344, 7122, 8663, 6761, and 8601). The two-tailed Mann-Whitney test. b Tetrad analysis of srr1∆ and rad52∆. srr1::kanR and rad52::hygR haploids (TNF5943 and 7988) were crossed, and the resulting tetrads were dissected on YE plates under a microscope. Images of three sets of three-spore viable tetrads in which the srr1::kanR rad52::hygR progenies did not form colonies are shown. c Depletion of Rad52 by the AID system impairs the growth of srr1∆ cells. rad52-AID, srr1∆ rad52-AID, OsTIRF74A, srr1∆ OsTIRF74A, rad52-AID OsTIRF74A, and srr1∆ rad52-AID OsTIRF74A (TNF8614, 8621, 8616, 8623, 8617, and 8627) were spotted on YE plates supplemented with 200 nM 5’a-IAA which induces Rad52 depletion. d Rpa2-mCherry foci (arrowhead) were observed by fluorescence microscopy in wild-type cells (TNF5492). Fluorescence and DIC images are overlayed. DIC, differential interference contrast. A bar shown below the image indicates 10 µm. The bar graph shows percentages of nuclei containing at least one Rpa2-mCherry focus in wild-type and srr1∆ (TNF8803) strains. The bars represent the mean of three independent experiments. The two-tailed student’s t-test. e Rad52-GFP foci (arrowhead) were observed in wild-type cells (TNF4442). The bar graph shows percentages of nuclei containing at least one Rad52-GFP focus in wild-type and srr1∆ (TNF6130) strains. Numerical data underlying a are provided in Table A, and those underlying d and e are in Table F in Supplementary Data 1.
We crossed srr1Δ and rad52Δ haploid strains and dissected the tetrads but failed to obtain srr1Δ rad52Δ progenies (Fig. 4b), suggesting synthetic growth defects of the srr1 rad52 double deletion. To confirm this, we depleted Rad52 in srr1Δ cells using the auxin-induced degron 2 (AID2) system51. In the presence of the F-box protein OsTIR1F74A, an auxin analog 5’-adamantyl-IAA (5’a-IAA) induces polyubiquitin-dependent degradation of the AID-tagged Rad52 protein, Rad52-AID. The addition of 5’a-IAA to the media significantly impaired cell growth of the srr1Δ rad52-AID compared to the rad52-AID strain (Fig. 4c, last two rows). These results show non-overlapping roles for Srr1 and Rad52 in cell growth. Given the hypersensitivity of srr1Δ cells to DNA-damaging agents (Fig. 3a), Srr1 may promote the repair of spontaneous DNA damage. Single-stranded DNA is formed during replication, transcription, and DNA damage repair/recombination. The replication protein A (RPA) complex binds single-stranded DNA with high affinity52. We detected the spontaneous focus formation of Rpa2-mCherry expressed from the original chromosomal locus32 and found that srr1Δ increased the fraction of cells containing Rpa2-mCherry focus (Fig. 4d), suggesting the accumulation of single-stranded DNA in srr1Δ cells. Rad52 forms nuclear foci at spontaneous DNA damage sites27,53. srr1Δ also increased the cells containing Rad52-GFP focus (Fig. 4e), showing that Srr1 suppresses the Rad52 focus formation.
A role of the SRR1-like domain in GCRs and DNA damage response
Srr1 has an evolutionally conserved domain called the SRR1-like domain (Fig. 5a). The Srr1 protein structure predicted by AlphaFold methods54,55 consists of the SRR1-like domain containing β-sheets sandwiched by α-helixes with intrinsically disordered extensions56 at both N- and C-termini (Fig. 5b, c). The srr1-W157R mutation site is present in the SRR1-like domain. To extend this, we changed conserved residues in the SRR1-like domain to alanine: srr1-D111A,P112A and srr1-H148A (Fig. 5a, b). Both srr1-D111A,P112A and srr1-H148A mutations reduced GCR rates (Fig. 5d). srr1-D111A,P112A and srr1-H148A also increased DNA damage sensitivity (Fig. 5e). These results demonstrate that the SRR1-like domain plays an essential role in GCRs and DNA damage repair. As expected from the prolonged doubling time (Table E in Supplementary Data 1), srr1Δ cells formed small colonies on plate media compared to wild-type cells (Supplementary Fig. 2b). Like srr1Δ cells, srr1-W157R, srr1-D111A,P112A, and srr1-H184A cells produced small colonies (Supplementary Fig. 2b), consistent with the role of the SRR1-like domain even in the absence of exogenous DNA damage.

a Positions of the fission yeast srr1-D111A,P112A, -H148A, and -W157R mutation sites are indicated by blue circles. Similar and identical residues among the different species are highlighted in pale and dark gray, respectively. b A ribbon model of the Srr1 structure predicted by AlphaFold methods. Positions of the mutation sites are indicated. c A surface model of the Srr1 structure. Positively and negatively charged residues are shown in blue and red, respectively. d GCR rates of wild-type, rad51∆, srr1∆ rad51∆, srr1-W157R rad51∆, srr1-D111A,P112A rad51∆, and srr1-H148A rad51∆ (TNF5369, 5411, 5904, 8344, 8686, and 8387). The two-tailed Mann-Whitney test. Numerical data are provided in Table A in Supplementary Data 1. e Mutating the conserved residues in the SRR1-like domain increases the sensitivity to MMS, HU, and CPT. Wild-type, srr1∆, srr1-W157R, srr1-H148A, and srr1-D111A,P112A (TNF3885, 5847, 8280, 8275, and 8274) cells were spotted onto YE3S supplemented with the indicated concentrations of MMS, HU, or CPT.
Skb1 promotes Rad52-dependent isochromosome formation via its arginine methyltransferase activity
Skb1 has been implicated in a wide range of pathways, including cell morphology and cell cycle regulation. Skb1 interacts with Slf1 and localizes to cell cortical nodes depending on Slf1, promoting rod-like cell morphology of fission yeast40,41. The DYRK-family kinase Pom1 negatively regulates cell cycle progression to ensure that cells grow to a certain size before entering mitosis42. Genetic evidence shows that Skb1 acts in the Pom1 pathway to regulate the cell cycle independently of its methyltransferase activity43. To ask whether Skb1 promotes isochromosome formation through these pathways, we disrupted slf1 or pom1 genes and determined the GCR rates of the mutant strains (Fig. 6a). Unlike skb1Δ, slf1Δ and pom1Δ slightly increased GCR rates in the wild-type background and neither reduced GCR rates in the rad51Δ background, showing that Skb1 promotes isochromosome formation through the function independent of Slf1 or Pom1.

a GCR rates of wild-type, skb1∆, slf1∆, pom1∆, rad51∆, skb1∆ rad51∆, slf1∆ rad51∆, and pom1∆ rad51∆ strains (TNF5369, 5772, 8811, 8813, 5411, 5788, 8834, and 8838). b Shown are the structure of the arginine methyltransferase domain of the fission yeast S. pombe Skb1 predicted by AlphaFold methods and the crystal structure of the arginine methyltransferase domain of C. elegans PRMT5 (PDB code 3UA3) with SAH, a SAM analog. Positions of the phenylalanine (F) and glutamic acid (E) residues essential for arginine methyltransferase activity are indicated. c GCR rates of wild-type, rad51∆, skb1∆ rad51∆, skb1-A377V rad51∆, skb1-F319Y rad51∆, and skb1-E422A,E431A rad51∆ strains (TNF5369, 5411, 5788, 8359, 8391, and 8474). d PFGE separated GCR products of the skb1-F319Y rad51∆ strain. Sample numbers of isochromosomes and truncations are shown in blue and magenta, respectively. e GCR rates of wild-type, skb1∆, rad52-R45K, skb1∆ rad52-R45K, rad51∆, skb1∆ rad51∆, rad52-R45K rad51∆, and skb1∆ rad52-R45K rad51∆ strains (TNF5369, 5772, 6599, 8324, 5411, 5788, 7122, and 8345). The two-tailed Mann-Whitney test. Numerical data underlying a, c, and e are provided in Table A in Supplementary Data 1. Uncropped gel images are shown in Supplementary Fig. 5.
Skb1 is the homolog of PRMT5 arginine methyltransferase (RMTase)44,45. The RMTase domain structure of S. pombe Skb1 predicted by AlphaFold methods54,55 is very similar to the crystal structure of C. elegans PRMT5 RMTase domain (Fig. 6b) and to the predicted structures of H. sapiens, A. thaliana, and S. cerevisiae Skb1/PRMT5 homologs (Supplementary Fig. 3). The skb1-A377V mutation that we isolated is present in the RMTase domain (Figs. 1b, 6b), but the mutant phenotype was not as prominent as skb1Δ (Fig. 6c). To determine whether the RMTase activity is essential for Skb1 to promote isochromosome formation, we mutated the Skb1 residues F319, E422, and E431 equivalent to those important for the in vitro RMTase activity of C. elegans PRMT557. We found that the skb1-F319Y and skb1-E422A,E431A mutations strongly reduced GCR rates in rad51Δ cells (Fig. 6c). Importantly, the same skb1-E422A,E431A mutation does not interfere with its role in cell cycle regulation43. PFGE analysis showed that, like skb1Δ, skb1-F319Y reduced isochromosomes but not chromosomal truncations in rad51Δ cells (Figs. 2c, 6d, and Table 1). We also examined the relationship between Skb1 and Rad52 and found that skb1Δ did not significantly reduce GCR rates in rad52-R45K or rad52-R45K rad51Δ cells (Fig. 6e). Together, these data indicate that Skb1 promotes Rad52-dependent isochromosome formation via its RMTase activity.
Discussion
GCRs occur using repetitive sequences present in the eukaryote genome. However, the mechanism of repeat-mediated GCRs is largely unknown. Here, we found that the evolutionally conserved Srr1 and Skb1 promote isochromosome formation using inverted DNA repeats at the centromere. Remarkably, srr1 increased DNA damage sensitivity in rad51Δ cells but did not abolish checkpoint response, suggesting that Srr1 promotes Rad51-independent DNA repair prone to GCRs. The srr1 and rad52 mutations additively, whereas skb1 and rad52 epistatically reduced GCR rates. Contrary to srr1 mutations, skb1Δ did not increase DNA damage sensitivity and reduced chromosome loss in rad51Δ cells. Through RMTase activity, Skb1 might form DNA structures leading to Rad52-dependent GCRs.
Srr1 and Skb1 promote isochromosome formation at the centromere. We found the srr1-W157R and skb1-A377V mutations in the same clone that exhibits reduced GCR levels (Fig. 1c). Either srr1 or skb1 deletion reduced GCR rates in rad51Δ cells (Fig. 1d), indicating that both Srr1 and Skb1 promote GCRs. Srr1 seems more integral to GCRs than Skb1 because srr1Δ but not skb1Δ reduced GCR rates even in the presence of Rad51 (Fig. 1d). Physical analysis of GCR products showed that Srr1 and Skb1 are responsible for ~90% of isochromosomes produced in rad51Δ cells but dispensable for chromosomal truncations (Fig. 2c). Given the fact that breakpoints of the isochromosomes are present in the centromere repeat20, these results indicate that Srr1 and Skb1 promote repeat-mediated GCRs at the centromere. Whether Srr1 or Skb1 causes GCRs outside the centromere remains to be elucidated. The srr1 and skb1 mutations additively reduced GCR rates in rad51Δ cells (Fig. 1d, e), suggesting that Srr1 and Skb1 have non-overlapping roles in GCRs (see below).
How does Srr1 promote isochromosome formation? We propose that Srr1 promotes DNA damage repair prone to GCRs. In the wild type, Rad51 primally repairs detrimental DNA damage, such as DNA double-strand breaks, and safeguards chromosome integrity. However, in the absence of Rad51, DNA damage left unrepaired will be channelled into other repair pathways, such as the Rad52-dependent recombination pathway29,30. Srr1 is involved in Rad51-independent DNA damage response, as the srr1 and rad51 mutations additively increased the sensitivity to MMS, HU, or CPT (Figs. 3a, 5e). Srr1 was not essential for MMS-induced cell cycle arrest and Chk1 phosphorylation (Fig. 3b, c), suggesting that Srr1 promotes DNA damage repair rather than checkpoint response. This is consistent with the fact that the checkpoint kinases Chk1 and Rad3 suppress but do not promote GCRs20,58. Like rad52Δ and rad52-R45K32, srr1Δ eliminated ~90% of isochromosomes in rad51Δ cells (Fig. 2c), showing that Srr1 and Rad52 are essential for the major pathway of isochromosome formation. However, Srr1 and Rad52 may also have non-overlapping roles in GCRs, as srr1 and rad52 mutations additively reduced GCR rates (Fig. 4a). This is supported by the additive effect of srr1 and skb1 (Fig. 1d, e) and the epistatic effect of skb1 and rad52 on GCR rates (Fig. 6e). srr1Δ caused synthetic growth defects with rad52Δ and accumulated spontaneous foci of Rpa2 and Rad52 (Fig. 4b–e). srr1Δ increased the doubling time and produced small colonies compared to the wild type (Table E in Supplementary Data 1; Supplementary Fig. 2b). Srr1 may promote the repair of spontaneous DNA damage. The hypersensitivity of srr1 cells to DNA-damaging agents supports this, but we do not exclude the possibility that Srr1 suppresses the formation of spontaneous DNA damage. Although its biochemical function remains unknown, we found the SRR1-like domain critical for GCRs and DNA damage repair. The srr1-D111A,P112A and srr1-H148A mutations altering evolutionally conserved residues in the SRR1-like domain strongly reduced GCR rates and increased sensitivity to the DNA-damaging agents, which interfere with the fork progression and eventually create DNA breaks (Fig. 5). The N-terminal extension of Srr1 is predicted intrinsically disordered in ref. 56 and contains positively charged residues in fission yeast and other organisms including humans, suggesting its role in the interaction with other molecules. Interestingly, protein structure comparison using Dali server59 suggests that the SRR1-like domain has structural similarity to a set of proteins, including methyltransferases (Supplementary Data 2). The knockout of the Srr1 homolog SRRD in mouse cells impairs DNA replication37, suggesting that mammalian Srr1 also plays a role in the repair of spontaneous DNA damage created during DNA replication. In plants and mammals, SRR1/SRRD affects the transcription of the genes involved in the circadian rhythm37,38,60, raising the possibility that Srr1 promotes GCRs and DNA damage response through transcriptional regulation. Interestingly, the srr1/ber1 mutation in budding yeast causes synthetic growth defects with mutations in the centromere proteins, including the centromere-specific histone H3 variant Cse4/CENP-A39. Although, unlike budding yeast srr1Δ/ber1Δ cells, fission yeast srr1Δ cells were not hyper-resistant to a microtubule-destabilizing drug, it is still possible that Srr1 promotes isochromosome formation by affecting the centromere structure. Srr1 has been localized to both the nucleus and cytosol in fission yeast61, but whether Srr1 localizes to the centromere remains unknown. Future works are required to address how Srr1 promotes isochromosome formation and DNA damage response.
It has been shown that a mutation in the Fbh1 helicase suppresses growth and recombination defects of rad52Δ cells62, raising the possibility that spontaneous fbh1 mutations have been introduced into our rad52Δ strain and affect GCRs. However, we previously showed no fbh1 mutations in our rad52Δ rad51Δ strain36 used to determine GCR rates. We also confirmed no fbh1 mutations in all the rad52 mutant strains and the srr1Δ rad51Δ strain used in this study (Supplementary Data 3). Furthermore, the fbh1 deletion did not significantly affect isochromosome formation in rad51Δ cells (Supplementary Fig. 4).
Skb1 has been implicated in many pathways. However, our data show that Skb1 promotes isochromosome formation independent of its role in cell morphology and cell cycle regulation mediated by Slf1 and Pom1, respectively (Fig. 6a). Skb1 and Slf1 bind each other and are mutually required for their localization to cell cortical nodes40. The cortical node localization is not essential for Skb1 to promote isochromosome formation, as slf1Δ did not reduce GCR rates. In rad51Δ cells, rad52-R45K reduces isochromosome formation and increases DNA damage sensitivity32. The pcn1-K107R mutation in a DNA sliding clamp PCNA and rad52-R45K epistatically reduce isochromosome formation, but pcn1-K107R does not increase DNA damage sensitivity36. Like pcn1-K107R, skb1Δ impairs Rad52-dependent isochromosome formation (Fig. 6e) but does not increase DNA damage sensitivity (Fig. 3a). In concert with PCNA, Skb1 might form DNA structures leading to Rad52-dependent GCRs. rad51Δ cells showed elevated levels of chromosome loss and GCRs, probably due to the inability to repair spontaneous DNA damage. As in the case of GCRs (Fig. 1d), skb1Δ reduces chromosome loss in rad51Δ but not in wild-type cells (Fig. 3e), showing that Skb1 causes GCRs and chromosome loss in rad51Δ cells. Skb1 is the fission yeast homolog of PRMT5 arginine methyltransferase (RMTase)44,63. PRMT5 is overexpressed in many malignant tumours, including breast cancer, and plays a role in the development of cancer64. Interestingly, the proteins affecting chromatin structure and replication, such as histones, Fen1 DNA-flap endonuclease, and p53, have been found as PRMT5 substrates65,66,67,68. Skb1 also interacts with Shk1, a p21-activated kinase (PAK) that negatively regulates cell cycle progression69. Mutating the residues essential for the RMTase activity:57 skb1-F319Y and skb1-E422A,E431A strongly reduced GCR rates, suggesting that Skb1 promotes GCRs through the RMTase activity. It is crucial to identify the key substrate(s) of Skb1 RMTase that induces chromosome instability in the future. Nonetheless, our findings paved a new avenue for elucidating the mechanism of GCRs at the centromere.
Methods
Genetic procedures and yeast strains
The fission yeast strains used in this study are listed in Supplementary Table 1 and are available from the corresponding author upon request. DNA primers used in this study are listed in Supplementary Table 2. Cells were grown in yeast extract (YE) medium or Edinburgh minimal medium 2 (EMM)70 at 30 °C unless otherwise indicated. Amino acids or bases were added at a final concentration of 225 mg L−1. Yeast nitrogen base (YNB) medium contained 7 g L−1 of yeast nitrogen base (BD Difco, BD 291940) and 20 g L−1 glucose. YNB medium is supplemented with 1 g L−1 5-fluoroorotic acid (Apollo Scientific, PC4054) and 56 mg L−1 uracil (Nacalai Tesque, 35824–82) to prepare 5-FOA media. Solid media contained 1.5% agarose (Nacalai Tesque, 01028–85). Yeast transformation was performed by the lithium acetate/PEG method71. Yeast cells were grown in YE or YE3S medium until log phase (1–2 × 107 cells mL−1) and harvested by centrifugation. Cells were washed once with sterilized water and twice with 1 mL LiAc/TE buffer (0.1 M lithium acetate, 10 mM Tris-HCl (pH 7.5), 1 mM EDTA). Cells were suspended in LiAc/TE buffer at > 2 × 109 cells mL−1. 100 μL of the cell suspension was mixed with 5 µL of salmon sperm DNA (10 mg mL−1) and the introducing DNA and incubated at room temperature for 10 min. After adding 260 μL of PEG/LiAc/TE (40% PEG4000, 0.1 M lithium acetate, 10 mM Tris-HCl (pH 7.5), 1 mM EDTA), the tube was further incubated for 30 min with rotation. After adding 43 μL of dimethyl sulfoxide (DMSO), the tube was incubated at 42 °C for 5 min. Cells were harvested by centrifugation at 503 × g for 30 s, suspended in YE or YE3S media, and plated on non-selective media. After one day of incubation, the cells were replica plated onto a medium supplemented with G418 (Nacalai Tesque, 09380–86) or hygromycin B (Nacalai Tesque, 09287–87) at a final concentration of 100 µg mL−1 or clonNAT (Werner BioAgents, 5.001.000) at 50 µg mL−1 to select the transformants. We did not pick up exceptionally large colonies of rad52 mutants because they can contain an fbh1 mutation62.
To search for the genes causing GCRs in rad51Δ cells, we introduced random mutations into yeast essentially, as described previously in ref. 32. Nitrous acid was used as the mutagen because it efficiently introduces mutations in DNA-repair deficient cells, as in wild-type cells72. rad51Δ cells containing ChLC (TNF5411) grown in EMM were collected at the log phase (5 × 106 cells mL−1), suspended in water, and kept overnight at 4 °C. After centrifugation, cells were suspended in 0.8 mL of 0.01 M nitrous acid solution prepared before use by dissolving sodium nitrate (Wako, 195–20562) in 0.5 M sodium acetate (pH 4.8) and incubated at room temperature for 20 min. After adding an equal volume of the stop buffer (3.6% Na2HPO4·12H20 and 1% yeast extract) to the cell suspension, cells were plated on EMM+UA plates. The plating efficiency of the mutagenized cells determined using EMM plates was around 10%. 24,000 independent clones were incubated as patches on EMM+UA plates for 2–3 d at 30 °C and then transferred onto 5-FOA+UA plates to semi-quantitatively determine the rate of uracil auxotroph, resulting from GCR or a point mutation in the ura4 gene. 80 clones produced reduced numbers of colonies on 5-FOA+UA plates. PFGE analysis showed that six contained the aberrant sizes of the parental ChLC. The remaining 74 clones were crossed with wild-type cells containing ChLC. Three clones reproducibly exhibited reduced GCR rates. Deep sequencing of genomic DNA was carried out using MiSeq (Illumina, San Diego, CA), and the mutations were identified by pooled-linkage analysis73,74. Nucleotide sequence data of the parental strain and a pool of nine mutant segregants obtained by backcrossing one of the three clones are available in the DDBJ Sequenced Read Archive under the accession numbers DRX042095 and DRX042098, respectively.
The srr1::kanMX6 strain was created by two rounds of polymerase chain reaction, PCR75. srr1-kan3 and srr1-kan5 primers were designed such that the 3’ side was complementary to srr1 and the 5’ side was complementary to the kanMX6 gene on pFA6a-kanMX6 plasmid76. In the first round of PCR, the srr1 flanking regions of 0.5 kb were amplified, using srr1-kan3/srr1-3 or srr1-kan5/srr1-1 primer pairs and fission yeast genomic DNA as a template. The second round of PCR was carried out in the presence of the two PCR fragments, pFA6a-kanMX6, and srr1-1/srr1-3 primers, to produce the DNA fragment containing the srr1::kanMX6 construct. The 2.5 kb PCR fragment was introduced into yeast.
The skb1::kanMX6 (or skb1::hphMX6) strain was created similarly as described above. In the first round of PCR, skb1 flanking regions of 0.5 kb each were amplified, using skb1-1/skb1-kan5 or skb1-kan3/skb1-2 primer pairs. The second round of PCR was carried out in the presence of the two PCR fragments, pFA6a-kanMX6 (or pFA6a-hphMX6), and skb1-1/skb1-2 primers, to produce the DNA fragment containing the skb1::kanMX6 or skb1::hphMX6 construct. The 2.5 or 2.7 kb PCR fragment was introduced into yeast.
To create the srr1-W157R mutant strain, the ura4+ gene was introduced into the srr1 gene in ura4-D18 cells, making ura4+:srr1 cells. In the first round of PCR, 0.5 kb regions were amplified using srr1-F1/srr1-ura4AN5 and srr1-ura4AN3/srr1-R1 primer pairs. The second round of PCR was carried out in the presence of the two PCR fragments, a plasmid containing the ura4+ genomic fragment, and srr1-F1/srr1-R1 primers. The 3 kb PCR fragment was transformed into yeast cells, and the ura4+ transformants were selected on EMM media. Next, the genomic region containing the srr1-W157R mutation was amplified by PCR using srr1-1/srr1-R1 primers and the template DNA prepared from the srr1-W157R skb1-A377V mutant isolated in the screening. The 1.5 kb PCR product was introduced into the ura4+:srr1 strain, and ura4– transformants were selected on 5-FOA plates. DNA sequencing confirmed the integration of the srr1-W157R mutation and no additional mutations. Following the construction of the ura4+:skb1 strain, the skb1-A377V strain was created in the same way, except skb1-F1/skb1-2 primers were used.
The srr1-H148A mutant fragment was also created in two rounds of PCR. First, 0.6 and 0.5 kb fragments were amplified using srr1-H148A-F/srr1-R1 and srr1-H148A-R/srr1-F1 primer pairs, respectively, and fission yeast genomic DNA as a template. Both the srr1-H148A-F and srr1-H148A-R primers contain the srr1-H148A mutation. The two overlapping PCR fragments were joined in the second round of PCR in the presence of srr1-F1/ srr1-R1 primers. The 1.1 kb product was introduced into the ura4+:srr1 strain, and ura4– transformants were selected on 5-FOA plates. The srr1-D111A,P112A mutant was created in a similar manner. In the first round of PCR, 0.7 and 0.5 kb fragments were amplified using srr1-DPAA-F/srr1-R1 and srr1-DPAA-R/srr1-F1 primer pairs, respectively. The two PCR fragments were joined in the second PCR reaction containing srr1-F1/srr1-R1 primers. The 1.1 kb product was introduced into the ura4+:srr1 strain. skb1-F319Y and skb1-E422A,E431A mutants were generated in the same way using skb1-F1/skb1-F319Y-R and skb1-F319Y-F/skb1-R1 primer pairs, and skb1-F1/skb1-doubleE-R and skb1-doubleE-F/skb1-R1 primer pairs, respectively. Correct integration of the point mutations, but no additional mutations were confirmed by Sanger sequencing.
GCR rates
GCR rates were determined by the fluctuation test as described previously in ref. 36. Yeast cells were incubated on EMM+UA plates for 6–8 days. Single colonies were used to inoculate 10 mL EMM+UA liquid medium. After 1–2 days of incubation, cells were plated onto 5-FOA+UA and YNB+UA plates. After 5–9 days of incubation, colonies formed on 5-FOA+UA and YNB+UA were counted to determine the number of Leu+ Ura– and Leu+ cells, respectively. About six colonies from each 5-FOA+UA plate were streaked onto EMM+UA to examine the colony size and then transferred to EMM+U plates to inspect adenine auxotrophy. The number of Leu+ Ura– Ade– cells, indicative of GCR, was obtained by subtracting the number of Leu+ Ura– Ade+ cells from that of Leu+ Ura– cells. The GCR rates per cell division were determined as described in ref. 77. When we started yeast cultures, we randomly picked up colonies of different sizes. We recovered both large and small colonies on 5-FOA+UA plates for PFGE analyses, according to the ratio of their appearance.
Chromosome loss rates
Chromosome loss rates were determined by the fluctuation test as described previously in ref. 27. A single colony formed on YE3S plates after 3–4 days incubation was suspended in sterilized water, and the cells were plated on YE plates. After 4–6 days incubation, white and red colonies were counted. The red colonies, indicative of ade6 loss, were transferred to YE3S and EMM+UA plates to inspect leucine auxotrophy. Colonies grown on YE3S plates were replica plated on EMM+UL and EMM+AL plates to test adenine and uracil auxotrophy, respectively. The number of Leu– Ura– Ade– cells, indicative of ChLC chromosome loss, and the total number of colony-forming cells were used to obtain the chromosome loss rate per cell division77.
PFGE analysis of GCR products
Each GCR clone was obtained from an independent culture to avoid multiple clones from the same parent. Cells were grown in YE3S medium at 25 °C for 12–24 h. 1 × 108 cells were harvested, suspended in 2.5 mL ice-cold 50 mM EDTA, and stored at 4 °C. The cells were centrifuged and resuspended in 1 mL CSE buffer (20 mM citrate phosphate, 1 M sorbitol, 50 mM EDTA (pH 5.6)). To prepare spheroplasts, 5 μL Zymolyase 20 T (Seikagaku, Tokyo, Japan, 25 mg mL−1) and 5 μL lyzing enzyme (Sigma, St. Louis, Missouri, 25 mg mL−1) were added to the cell suspension and incubated at 30 °C for 20 to 50 min. Spheroplasts were harvested by centrifugation at 33 × g for 10 min at 4 °C, and the pellet was suspended in 140 μL CSE buffer. An equal volume of 1.6% low melting agarose gel (Nacalai Tesque, 01161–12) pre-heated at 50 °C was added to the cell suspension and distributed into molds. The agarose plugs were incubated at 4 °C for 20 min. The plugs were incubated in SDS-EDTA solution (1% SDS, 0.25 M EDTA) at 60 °C for 2 h and then in ESP solution (0.5 M EDTA, 1% N-lauroyl sarcosine, 1.5 mM calcium acetate) supplemented with 0.5 mg mL−1 proteinase K (Nacalai Tesque, 39450–01–6) at 50 °C overnight. The plugs were transferred into another ESP solution supplemented with 0.5 mg mL−1 proteinase K and incubated at 50 °C for an additional 8 h. The plugs were stored in TE buffer (10 mM Tris-HCl (pH 8.0), 1 mM EDTA) at 4 °C. Chromosomal DNAs were resolved using a CHEF-DRII pulsed-field electrophoresis system (Bio-Rad, Hercules, California). PFGE ran at 4.2 V cm−1 with a pulse time of 40 to 70 s for 24 h, at 4 °C in 0.5× TBE buffer (89 mM Tris-borate, 2 mM EDTA) using 0.55% Certified Megabase agarose gel. DNA was stained with 0.2 µg mL−1 ethidium bromide (EtBr) (Nacalai Tesque, 14631–94) and detected using a Typhoon FLA9000 gel imaging scanner (GE Healthcare, Chicago, Illinois) or GelDoc Go imaging system (Bio-Rad, Hercules, CA). Gel images were processed using ImageJ2 2.9.0 (NIH, United States) or Adobe Photoshop Elements 2020 (Adobe, San Jose, CA).
Serial dilution assay
A single colony formed on YE (or YE3S) plates after 3–4 days incubation was used to inoculate 2 mL YE (or YE3S) liquid media. The 2 mL overnight culture was used to prepare 10 mL log-phase cultures. Five-fold serial dilutions of the indicated strains were prepared with sterilized water. 6 µL from each dilution was spotted onto YE (or YE3S) plates supplemented with the indicated concentration of MMS, HU, CPT, or TBZ. The plates were incubated for 3−5 days at 30 °C. Images were taken using GT-X800 (Epson, Nagano, Japan) and processed using Adobe Photoshop Elements 2020 (Adobe, San Jose, CA).
Western blot
Cell extracts were prepared using an alkaline lysis method78. 1 × 108 cells from log-phase YE cultures were collected, washed with 1 ml H2O, and suspended in 300 µl H2O. After adding 300 µl of 0.6 M NaOH, the cell suspension was incubated at 30 °C for 5 min with the tube rotating. After centrifugation at 6000 rpm for 3 min (TOMY, MX-201), the alkali-treated cells were suspended with 140 µ of SDS sample buffer (60 mM Tris-HCl (pH6.8), 5% glycerol, 4% sodium dodecyl sulfate, 4% β-mercaptoethanol, 0.005% bromophenol blue) and incubated at 95 °C for 3 min. Cell extracts were recovered from the supernatant after centrifugation at 15,000 rpm for 1 min (TOMY, Kitman), separated by 8% sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) (acrylamide to bisacrylamide ratio, 37.5:1), and transferred onto PolyScreen PVDF Transfer Membrane (Perkin Elmer, NEF1002001PK). To detect Chk1-HA, a mouse monoclonal antibody against the HA tag (16B12, Abcam, Cambridge, MA) (1:2000) and peroxidase AffiniPure goat anti-mouse IgG (heavy+light) (Jackson ImmunoResearch Laboratories, 115–035−146) (1:10,000) were used as the primary and secondary antibodies, respectively. The blots were developed using Supersignal West Femto substrate (ThermoScientific, 34095). Images were captured using ImageQuant LAS 500 (GE Healthcare).
Septation index
MMS was added to log-phase EMM cultures to a final concentration of 0.01%. At 0, 2, 4, 6, and 8 h after adding MMS, cells were harvested, suspended in 70% ethanol, and stored at 4 °C. Cells were suspended in PEMS buffer (100 mM PIPES (pH6.9), 1 mM EGTA, 1 mM MgSO4, 1 M sorbitol), containing 2 µg mL−1 4’,6-diamidino-2-phenylindole (DAPI) and 4 µg mL−1 calcofluor. The cell suspension was mixed with a mounting medium (90% glycerol, 1 mg mL−1 n-propyl gallate, 1 mg mL−1 1,4-phenylenediamine dihydrochloride, 0.1× phosphate buffered saline) on poly-L-lysine coated coverslip. Using a fluorescence microscope (BX51, Olympus, Tokyo, Japan) with a 100× objective (NA = 1.40, Olympus, Tokyo, Japan), nuclei and septa were visualized with DAPI and calcofluor, respectively. Images were obtained using a charge-coupled device camera (DP72, Olympus, Tokyo, Japan) and processed using cellSens Standard 2.3 (Olympus) and Adobe Photoshop Elements 2020 (Adobe, San Jose, CA).
Fluorescent microscopy detecting Rpa2-mCherry and Rad52-GFP foci
Exponentially growing cells in EMM (Rpa2-mCherry) or YE (Rad52-GFP) medium were collected, seeded on glass-bottom dishes (Matsunami Glass, Osaka, Japan, D11130H), and observed using a DeltaVision Personal fluorescence microscopy system (GE Healthcare), which is based on an Olympus wide-field IX71 fluorescence microscope equipped with a CoolSNAP HQ2 CCD camera (Photometrics, Tucson, Arizona) and an oil-immersion objective lens (UAPO 40×; NA = 1.35; Olympus, Tokyo, Japan). The percentage of nuclei with Rpa2-mCherry and Rad52-GFP foci was obtained by counting the nuclei containing at least one Rpa2-mCherry and Rad52-GFP focus, respectively, using ImageJ2 2.9.0 (NIH, United States). > 300 nuclei were counted in each experiment. Three independent experimental values and their means were shown in the graph using GraphPad Prism 9 for MacOS (GraphPad Software, San Diego, CA). Images were processed using ImageJ2 2.9.0 or Adobe Photoshop Elements 2020.
Rad52-AID2 depletion assay
Exponentially growing cells of the indicated strains were prepared and five-fold serially diluted with sterilized water. 6 µL from each dilution was spotted onto YE plates supplemented with DMSO or 200 nM 5’a-IAA (Tokyo Chemical Industry, A3390)51. The plates were incubated for 2 days. Images were taken using GT-X800 (Epson, Nagano, Japan) and processed using Adobe Photoshop Elements 2020 (Adobe, San Jose, CA).
Statistics and reproducibility
The two-tailed Mann-Whitney and the two-tailed Fischer’s exact tests were performed using GraphPad Prism 9 for MacOS. The two-tailed student’s t-test and Pearson’s Chi-square test were performed using Microsoft Excel for Mac 16.72. The sample size used to derive each statistic was provided in the figure legend or in the supplementary information.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
The data supporting the findings of this study are included in the paper, Supplementary Information (Supplementary Figs. 1–6 and Supplementary Tables 1-2), and Supplementary Data 1-3. DNA sequence data of the parental strain and a pool of the mutant segregants are available in the DDBJ Sequenced Read Archive under the accession numbers DRX042095 and DRX042098, respectively, at the following URL: https://ddbj.nig.ac.jp/resource/bioproject/PRJDB4206. All raw datasets are available from the corresponding author upon reasonable request.
Change history
05 June 2023
In this Article, the legend to Figure 1 was misplaced so that it overlapped part of the figure. This has now been corrected.
References
Pascarella, G. et al. Recombination of repeat elements generates somatic complexity in human genomes. Cell 185, 3025–3040 (2022).
Carvalho, C. M. & Lupski, J. R. Mechanisms underlying structural variant formation in genomic disorders. Nat. Rev. Genet. 17, 224–238 (2016).
Nurk, S. et al. The complete sequence of a human genome. Science 376, 44–53 (2022).
Koonin, E. V. Evolution of genome architecture. Int J. Biochem Cell Biol. 41, 298–306 (2009).
Altemose, N. et al. Complete genomic and epigenetic maps of human centromeres. Science 376, eabl4178 (2022).
Padilla-Nash, H. M. et al. Jumping translocations are common in solid tumor cell lines and result in recurrent fusions of whole chromosome arms. Genes Chromosomes Cancer 30, 349–363 (2001).
Martinez, A. C. & van Wely, K. H. Centromere fission, not telomere erosion, triggers chromosomal instability in human carcinomas. Carcinogenesis 32, 796–803 (2011).
Rozenzhak, S. et al. Rad3 decorates critical chromosomal domains with γH2A to protect genome integrity during S-Phase in fission yeast. PLoS Genet. 6, e1001032 (2010).
Yilmaz, D. et al. Activation of homologous recombination in G1 preserves centromeric integrity. Nature 600, 748–753 (2021).
Saayman, X., Graham, E., Nathan, W. J., Nussenzweig, A. & Esashi, F. Centromeres as universal hotspots of DNA breakage, driving RAD51-mediated recombination during quiescence. Mol. Cell 83, 523–538 (2023).
Barra, V. & Fachinetti, D. The dark side of centromeres: types, causes and consequences of structural abnormalities implicating centromeric DNA. Nat. Commun. 9, 4340 (2018).
Nakagawa, T. & Okita, A. K. Transcriptional silencing of centromere repeats by heterochromatin safeguards chromosome integrity. Curr. Genet. 65, 1089–1098 (2019).
Black, E. M. & Giunta, S. Repetitive fragile sites: Centromere satellite DNA as a source of genome instability in human diseases. Genes (Basel) 9, 615 (2018).
Therman, E., Susman, B. & Denniston, C. The nonrandom participation of human acrocentric chromosomes in Robertsonian translocations. Ann. Hum. Genet. 53, 49–65 (1989).
Tuna, M., Amos, C. I. & Mills, G. B. Whole-chromosome arm acquired uniparental disomy in cancer development is a consequence of isochromosome formation. Neoplasia 25, 9–17 (2022).
Huijsdens-van Amsterdam, K. et al. Isochromosome 21q is overrepresented among false-negative cell-free DNA prenatal screening results involving Down syndrome. Eur. J. Hum. Genet 26, 1490–1496 (2018).
Cechova, M. & Miga, K. H. Satellite DNAs and human sex chromosome variation. Semin Cell Dev. Biol. 128, 15–25 (2022).
Allshire, R. C. & Ekwall, K. Epigenetic Regulation of Chromatin States. Schizosaccharomyces pombe. Cold Spring Harb. Perspect. Biol. 7, a018770 (2015).
McKinley, K. L. & Cheeseman, I. M. The molecular basis for centromere identity and function. Nat. Rev. Mol. Cell Biol. 17, 16–29 (2016).
Nakamura, K. et al. Rad51 suppresses gross chromosomal rearrangement at centromere. Schizosaccharomyces pombe. EMBO J. 27, 3036–3046 (2008).
Tinline-Purvis, H. et al. Failed gene conversion leads to extensive end processing and chromosomal rearrangements in fission yeast. EMBO J. 28, 3400–3412 (2009).
Li, P. C. et al. Replication fork stability is essential for the maintenance of centromere integrity in the absence of heterochromatin. Cell Rep. 3, 638–645 (2013).
Ranjha, L., Howard, S. M. & Cejka, P. Main steps in DNA double-strand break repair: An introduction to homologous recombination and related processes. Chromosoma 127, 187–214 (2018).
Nalepa, G. & Clapp, D. W. Fanconi anaemia and cancer: an intricate relationship. Nat. Rev. Cancer 18, 168–185 (2018).
Nielsen, F. C., van Overeem Hansen, T. & Sorensen, C. S. Hereditary breast and ovarian cancer: new genes in confined pathways. Nat. Rev. Cancer 16, 599–612 (2016).
Wassing, I. E. et al. The RAD51 recombinase protects mitotic chromatin in human cells. Nat. Commun. 12, 5380 (2021).
Onaka, A. T. et al. Rad51 and Rad54 promote noncrossover recombination between centromere repeats on the same chromatid to prevent isochromosome formation. Nucleic Acids Res. 44, 10744–10757 (2016).
Zafar, F. et al. Regulation of mitotic recombination between DNA repeats in centromeres. Nucleic Acids Res. 45, 11222–11235 (2017).
Rossi, M. J., DiDomenico, S. F., Patel, M. & Mazin, A. V. RAD52: Paradigm of synthetic lethality and new developments. Front. Genet. 12, 780293 (2021).
Jalan, M., Olsen, K. S. & Powell, S. N. Emerging Roles of RAD52 in Genome Maintenance. Cancers (Basel) 11, 1038 (2019).
Kowalczykowski, S. C. An overview of the molecular mechanisms of recombinational DNA repair. Cold Spring Harb. Perspect. Biol. 7, a016410 (2015).
Onaka, A. T. et al. DNA replication machinery prevents Rad52-dependent single-strand annealing that leads to gross chromosomal rearrangements at centromeres. Commun. Biol. 3, 202 (2020).
Boddy, M. N. et al. Mus81-Eme1 are essential components of a Holliday junction resolvase. Cell 107, 537–548 (2001).
Osman, F., Dixon, J., Doe, C. L. & Whitby, M. C. Generating crossovers by resolution of nicked Holliday junctions: a role for Mus81-Eme1 in meiosis. Mol. Cell 12, 761–774 (2003).
Smith, G. R., Boddy, M. N., Shanahan, P. & Russell, P. Fission yeast Mus81.Eme1 Holliday junction resolvase is required for meiotic crossing over but not for gene conversion. Genetics 165, 2289–2293 (2003).
Su, J., Xu, R., Mongia, P., Toyofuku, N. & Nakagawa, T. Fission yeast Rad8/HLTF facilitates Rad52-dependent chromosomal rearrangements through PCNA lysine 107 ubiquitination. PLoS Genet 17, e1009671 (2021).
Adachi, Y. et al. The novel heme-dependent inducible protein, SRRD regulates heme biosynthesis and circadian rhythms. Arch. Biochem Biophys. 631, 19–29 (2017).
Staiger, D. et al. The Arabidopsis SRR1 gene mediates phyB signaling and is required for normal circadian clock function. Genes Dev. 17, 256–268 (2003).
Fiechter, V. et al. The evolutionary conserved BER1 gene is involved in microtubule stability in yeast. Curr. Genet. 53, 107–115 (2008).
Deng, L., Kabeche, R., Wang, N., Wu, J. Q. & Moseley, J. B. Megadalton-node assembly by binding of Skb1 to the membrane anchor Slf1. Mol. Biol. Cell 25, 2660–2668 (2014).
Miller, K. E., Magliozzi, J. O., Picard, N. A. & Moseley, J. B. Sequestration of the exocytic SNARE Psy1 into multiprotein nodes reinforces polarized morphogenesis in fission yeast. Mol. Biol. Cell 32, ar7 (2021).
Martin, S. G. & Berthelot-Grosjean, M. Polar gradients of the DYRK-family kinase Pom1 couple cell length with the cell cycle. Nature 459, 852–856 (2009).
Deng, L. & Moseley, J. B. Compartmentalized nodes control mitotic entry signaling in fission yeast. Mol. Biol. Cell 24, 1872–1881 (2013).
Xu, J. & Richard, S. Cellular pathways influenced by protein arginine methylation: Implications for cancer. Mol. Cell 81, 4357–4368 (2021).
Pollack, B. P. et al. The human homologue of the yeast proteins Skb1 and Hsl7p interacts with Jak kinases and contains protein methyltransferase activity. J. Biol. Chem. 274, 31531–31542 (1999).
Niwa, O., Matsumoto, T. & Yanagida, M. Construction of a mini-chromosome by deletion and its mitotic and meiotic behaviour in fission yeast. Mol. Gen. Genet 203, 397–405 (1986).
Walworth, N., Davey, S. & Beach, D. Fission yeast chk1 protein kinase links the rad checkpoint pathway to cdc2. Nature 363, 368–371 (1993).
Lopez-Girona, A. et al. Serine-345 is required for Rad3-dependent phosphorylation and function of checkpoint kinase Chk1 in fission yeast. Proc. Natl Acad. Sci. USA. 98, 11289–11294 (2001).
Capasso, H. et al. Phosphorylation activates Chk1 and is required for checkpoint-mediated cell cycle arrest. J. Cell Sci. 115, 4555–4564 (2002).
Chaudhuri, B., Ingavale, S. & Bachhawat, A. K. apd1+, a gene required for red pigment formation in ade6 mutants of Schizosaccharomyces pombe, encodes an enzyme required for glutathione biosynthesis: A role for glutathione and a glutathione-conjugate pump. Genetics 145, 75–83 (1997).
Watson, A. T., Hassell-Hart, S., Spencer, J. & Carr, A. M. Rice (Oryza sativa) TIR1 and 5’adamantyl-IAA significantly improve the auxin-inducible degron system. Schizosaccharomyces pombe. Genes (Basel) 12, 882 (2021).
Chen, R. & Wold, M. S. Replication protein A: single-stranded DNA’s first responder: dynamic DNA-interactions allow replication protein A to direct single-strand DNA intermediates into different pathways for synthesis or repair. Bioessays 36, 1156–1161 (2014).
Oshidari, R. et al. DNA repair by Rad52 liquid droplets. Nat. Commun. 11, 695 (2020).
Jumper, J. et al. Highly accurate protein structure prediction with AlphaFold. Nature 596, 583–589 (2021).
Varadi, M. et al. AlphaFold Protein Structure Database: Massively expanding the structural coverage of protein-sequence space with high-accuracy models. Nucleic Acids Res. 50, D439–D444 (2022).
Hu, G. et al. flDPnn: Accurate intrinsic disorder prediction with putative propensities of disorder functions. Nat. Commun. 12, 4438 (2021).
Sun, L. et al. Structural insights into protein arginine symmetric dimethylation by PRMT5. Proc. Natl Acad. Sci. USA 108, 20538–20543 (2011).
Blaikley, E. J. et al. The DNA damage checkpoint pathway promotes extensive resection and nucleotide synthesis to facilitate homologous recombination repair and genome stability in fission yeast. Nucleic Acids Res. 42, 5644–5656 (2014).
Holm, L. Dali server: structural unification of protein families. Nucleic Acids Res. 50, W210–W215 (2022).
Johansson, M. & Staiger, D. SRR1 is essential to repress flowering in non-inductive conditions in Arabidopsis thaliana. J. Exp. Bot. 65, 5811–5822 (2014).
Matsuyama, A. et al. ORFeome cloning and global analysis of protein localization in the fission yeast Schizosaccharomyces pombe. Nat. Biotechnol. 24, 841–847 (2006).
Osman, F., Dixon, J., Barr, A. R. & Whitby, M. C. The F-Box DNA helicase Fbh1 prevents Rhp51-dependent recombination without mediator proteins. Mol. Cell Biol. 25, 8084–8096 (2005).
Branscombe, T. L. et al. PRMT5 (Janus kinase-binding protein 1) catalyzes the formation of symmetric dimethylarginine residues in proteins. J. Biol. Chem. 276, 32971–32976 (2001).
Xiao, W. et al. Role of protein arginine methyltransferase 5 in human cancers. Biomed. Pharmacother. 114, 108790 (2019).
Zhao, Q. et al. PRMT5-mediated methylation of histone H4R3 recruits DNMT3A, coupling histone and DNA methylation in gene silencing. Nat. Struct. Mol. Biol. 16, 304–311 (2009).
Liu, F. et al. PRMT5-mediated histone arginine methylation antagonizes transcriptional repression by polycomb complex PRC2. Nucleic Acids Res. 48, 2956–2968 (2020).
Guo, Z. et al. Methylation of FEN1 suppresses nearby phosphorylation and facilitates PCNA binding. Nat. Chem. Biol. 6, 766–773 (2010).
Berger, S. L. Out of the jaws of death: PRMT5 steers p53. Nat. Cell Biol. 10, 1389–1390 (2008).
Gilbreth, M. et al. Negative regulation of mitosis in fission yeast by the Shk1 interacting protein Skb1 and its human homolog, Skb1Hs. Proc. Natl Acad. Sci. USA 95, 14781–14786 (1998).
Forsburg, S. L. & Rhind, N. Basic methods for fission yeast. Yeast 23, 173–183 (2006).
Sabatinos, S. A. & Forsburg, S. L. Molecular genetics of Schizosaccharomyces pombe. Methods Enzymol. 470, 759–795 (2010).
Prakash, L. Lack of chemically induced mutation in repair-deficient mutants of yeast. Genetics 78, 1101–1118 (1974).
Birkeland, S. R. et al. Discovery of mutations in Saccharomyces cerevisiae by pooled linkage analysis and whole-genome sequencing. Genetics 186, 1127–1137 (2010).
Iida, N., Yamao, F., Nakamura, Y. & Iida, T. Mudi, a web tool for identifying mutations by bioinformatics analysis of whole-genome sequence. Genes Cells 19, 517–527 (2014).
Petracek, M. E. & Longtine, M. S. PCR-based engineering of yeast genome. Methods Enzymol. 350, 445–469 (2002).
Hentges, P., Van Driessche, B., Tafforeau, L., Vandenhaute, J. & Carr, A. M. Three novel antibiotic marker cassettes for gene disruption and marker switching. Schizosaccharomyces pombe. Yeast 22, 1013–1019 (2005).
Lin, M., Chang, C. J. & Green, N. S. A new method for estimating high mutation rates in cultured cells. Mutat. Res. 351, 105–116 (1996).
Matsuo, Y., Asakawa, K., Toda, T. & Katayama, S. A rapid method for protein extraction from fission yeast. Biosci. Biotechnol. Biochem 70, 1992–1994 (2006).
Acknowledgements
We thank Akiko Okita, Jie Su, and Yukiko Kubota for their critical comments on the manuscript and Hirofumi Ohmori and Keiko Kayahara for their technical assistance. We also thank Adam T. Watson and Antony M. Carr for sharing the AID2 system. This work was supported by JSPS KAKENHI Grant Numbers 221S0002, JP23570212, JP26114711, 18K06060, 21H02402, and the Uehara Memorial Foundation Grant Number 202120462 to TN.
Author information
Authors and Affiliations
Contributions
P.M., N.T., and T.N. conceived the study. P.M., N.T., and Z.P. performed most experiments with technical help from R.X., Y.K., K.O., and T.N. Deep sequencing was performed by H.T., Y.O., and T.H. The manuscript was written by T.N. and P.M. and approved by all the authors.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Communications Biology thanks Gerben Vader, and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. Primary Handling Editor: Manuel Breuer. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Mongia, P., Toyofuku, N., Pan, Z. et al. Fission yeast Srr1 and Skb1 promote isochromosome formation at the centromere. Commun Biol 6, 551 (2023). https://doi.org/10.1038/s42003-023-04925-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s42003-023-04925-9
- Springer Nature Limited