
Abstract
Bone is a highly dynamic tissue undergoing continuous formation and resorption. Here, we investigated differential but complementary roles of hypoxia-inducible factor (HIF)-1α and HIF-2α in regulating bone remodeling. Using RNA-seq analysis, we identified that specific genes involved in regulating osteoblast differentiation were similarly but slightly differently governed by HIF-1α and HIF-2α. We found that increased HIF-1α expression inhibited osteoblast differentiation via inhibiting RUNX2 function by upregulation of Twist2, confirmed using Hif1a conditional knockout (KO) mouse. Ectopic expression of HIF-1α via adenovirus transduction resulted in the increased expression and activity of RANKL, while knockdown of Hif1a expression via siRNA or osteoblast-specific depletion of Hif1a in conditional KO mice had no discernible effect on osteoblast-mediated osteoclast activation. The unexpected outcome was elucidated by the upregulation of HIF-2α upon Hif1a overexpression, providing evidence that Hif2a is a transcriptional target of HIF-1α in regulating RANKL expression, verified through an experiment of HIF-2α knockdown after HIF-1α overexpression. The above results were validated in an ovariectomized- and aging-induced osteoporosis model using Hif1a conditional KO mice. Our findings conclude that HIF-1α plays an important role in regulating bone homeostasis by controlling osteoblast differentiation, and in influencing osteoclast formation through the regulation of RANKL secretion via HIF-2α modulation.
Similar content being viewed by others

Introduction
Bone is a dynamic tissue characterized by old matrix resorption by osteoclasts and new matrix formation by osteoblasts. The balance between bone formation and bone resorption is essential for bone homeostasis. Osteoblasts, originating from mesenchymal stem cells, are controlled by osteogenic transcription factors, runt-related transcription factor (RUNX2)1, osterix2, and TCF/LEF3. Osteoclasts are derived from hematopoietic progenitors of the monocyte-macrophage lineage4. The initial proliferation of pre-osteoclasts is controlled by macrophage colony-stimulating factor (M-CSF), and osteoclast differentiation and maturation are governed by receptor activator of nuclear factors (NF)-κB ligand (RANKL)5. RANKL is expressed in the osteoblast-lineage cells and stimulates RANK, the receptor for RANKL, on osteoclast precursors. Interaction of RANKL on osteoblasts and RANK on osteoclasts results in the activation of signal transduction for the differentiation and maturation of osteoclasts, such as NF-κB, c-Fos, and nuclear factor of activated T cells 1 (NFATc1)6,7. Osteoprotegerin (OPG) produced by osteoblasts is a soluble decoy receptor for RANKL and regulates RANKL-mediated osteoclastogenesis8.
Hypoxia-inducible factor (HIF) is a heterodimeric transcription factor consisting of an inducible α subunit and constitutive β subunit. HIF-α is oxygen labile, and HIF-α stability is crucial for the activity of HIF as a transcriptional regulator9. Under normoxia, the proline residues of the HIF-α subunit are hydroxylated by prolyl-4-hydrolyase (PHD) following interaction with von Hippel-Lindau (VHL), specific E3 ligase, and then polyubiquitination and proteasomal degradation10. However, in hypoxia, proline hydroxylation is prohibited by the deprivation of oxygen, allowing the accumulation of HIF-α and the formation of an active heterodimeric complex with HIF-1β11. Remarkably, HIF-α can be stabilized even in normoxia by inflammatory factors, such as IL-1β, INF-γ, IGF-1 and TNF-α, in a pathophysiologic microenvironment12,13, and normoxic stabilization of HIF-1α induces osteoclastogenesis and pathological bone resorption14.
Of the three identified isotypes of HIF-α, studies have focused on HIF-1α and HIF-2α. They retain highly conserved sequences, have similar domain structures, and bind to the same hypoxia-responsive element (HRE), 5′-(A/G)CGTG-3′ sequence, in promoters of specific target genes15,16. Studies show that the effects of HIF-1α and HIF-2α in regulating skeletal development, bone remodeling, and pathological bone diseases are different. Some papers suggest the anabolic role of HIF-α. Accumulation of HIF-1α and HIF-2α in osteoblasts via specific deletion of VHL promotes endochondral ossification and long bone formation by promoting vascularization17,18. On the contrary, other studies support the catabolic effects of HIF-1α. Deletion of HIF-1α in osteoblasts of mature bone resulted in the increased accumulation of bone19. Overexpression of HIF-1α negatively regulates mechanical load-induced bone formation20 and inhibits the proliferation and growth of osteoblasts by synergistic inhibition of the Wnt pathway and osterix21. Bone is a hypoxic tissue with different regions, such as cortical bone, bone marrow, cancellous bone, and endosteum, characterized by different hypoxia levels depending on the distribution of large or small blood vessels22. The effects of hypoxia on skeletal development and bone remodeling are complex and differ with the early or late stage of differentiation and diverse oxygen levels22. Therefore, the effects of HIF-1α and HIF-2α involved in skeletal development, bone remodeling, and pathologic bone disease would differ.
Our previous study observed the catabolic functions of HIF-2α in maintaining bone homeostasis and normoxic stabilization of HIF-1α and HIF-2α when cultured in differentiation-inducing conditions23. Here, we demonstrate the role of normoxic stabilization of HIF-1α in regulating bone remodeling, and by comparing with HIF-2α function, we suggest the differential but complementary regulation of HIF-1α and HIF-2α.
Results
Transcriptional profiles regulated by HIF-1α and HIF-2α during osteoblast differentiation
Our previous study observed the accumulation of HIF-1α and HIF-2α during in vitro osteoblast differentiation under normoxia when pre-osteoblasts were cultured in differentiation media (DM)23. To determine the distinct roles of HIF-1α in regulating bone homeostasis, at first, we confirmed the expression and normoxic stabilization of HIF-1α and HIF-2α during osteoblast differentiation. Expression of Hif1a was significantly increased in the early stage of osteoblast differentiation before the increase of Ocn, Alp, and Runx2 expression, the markers of osteoblasts, which is a similar pattern of Hif2a expression (Fig. 1a, Supplementary Fig. 1a). Increased accumulation and nuclear localization of HIF-1α and HIF-2α were observed on day 6 with the media containing differentiation-inducing agents, such as L-AA and β-Gp (Fig. 1b). Expression of HIF-1α in the nucleus persisted from day 6 to day 12 of differentiation, while HIF-2α increased until day 15 (Supplementary Fig. 1b). RNA-seq analysis showed similar transcriptional profiles in primary calvarial pre-osteoblasts by overexpression of HIF-1α or HIF-2α (Fig. 1c). As a result of RNA-seq analysis, 838 (62.7%) differentially expressed genes (DEGs) (out of 40,213 genes) were shared, and 146 (10.9%) and 353 (26.4%) DEGs were unique to HIF-1α and HIF-2α, respectively (Fig. 1d). To comprehend the complex cellular processes and functions associated with HIF transcriptional regulation, we clustered the DEGs based on fold enrichment and selected the most informative Gene Ontology (GO) terms for each set of HIF DEGs. Fig. 1e shows the top enriched cellular processes, including extracellular signal-regulated kinase (ERK)-1/-2 cascade, immune response, and positive regulation of cell migration strongly shared between HIF-1α and HIF-2α. Of them, 3905 genes involved in cell differentiation were analyzed, and we found that most were similarly regulated by HIF-1α and HIF-2α (Fig. 1f). Representatively, well-known effectors such as Ptgs2, Fas, Il6, Rankl, Twist2, Vegfa, and Vegfc were upregulated and Bglap1, Bglap2, Bmp2, Fgfr2, Fgfr3, Sp7, and Pparg were down-regulated (Fig. 1f). Thus, we determined that about 62.7% of filtered genes were commonly regulated; however, some genes were distinctly regulated by HIF-1α and HIF-2α during osteoblast differentiation under normoxia.

a Primary calvarial pre-osteoblasts from WT mice were cultured in the medium containing 50 μg/ml L-AA and 5 mM β-Gp for up to 6 days for osteogenic differentiation. Relative transcript levels of Hif1a, Hif2a, Ocn, Alp, and Runx2 on the indicated culture days were determined using qRT-PCR (n ≥ 4). b At day 6 differentiation, nuclear translocation of HIF-1α and HIF-2α in osteogenic differentiation medium (DM) was observed using immunocytochemistry with anti-HIF-1α antibody, anti-HIF-2α antibody and Alex-594 (red) (n = 4). Nuclei were marked with DAPI (blue) staining. CM, control media. Scale bar, 25 μm. a, b Values are presented as the mean ± SEM. **P < 0.01, and ***P < 0.001. c–f Primary cultured calvarial pre-osteoblasts were transduced with Ad-Hif1a or Ad-Hif2a on day 3, and then cultured in osteogenic DM until day 6. Total RNAs were extracted and then used for RNA-seq analysis (n = 3). Hierarchical clustering heatmap for DEGs was visualized (c). Venn diagram for DEGs of RNA-seq analysis (d). GO enrichment analysis (e). Top 10-ranked enriched GO terms were listed in the bubble chart. Enriched GO terms by Hif1a (top) or Hif2a overexpression (bottom). Venn diagram of selected DEGs associated with cell differentiation was visualized and the representative genes were listed. Absolute value of fold change (FC) > 2, normalized data (log2) > 4 (d, e) or normalized data (log2) > 2 (f), and p-value (paired t-test) < 0.05 were used as the cut-offs (f).
HIF-1α inhibits osteoblast differentiation via the TWIST2-RUNX2-OCN axis
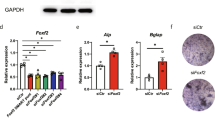
Ectopic expression of HIF-1α via the adenovirus transduction system showed inhibited differentiation of primary calvarial pre-osteoblasts. The expression of osteoblast marker genes, Ocn and Runx2, was reduced by HIF-1α overexpression (Fig. 2a), and mineralization and calcified nodule formation were inhibited as determined by ALP and ARS staining (Fig. 2b). The inhibitory effect of HIF-1α on osteoblast differentiation was confirmed by BMP-2-induced regeneration of calvarial bone defect models (Fig. 2c). Implantation with Ad-Hif1a in the calvarial defect regions considerably delayed BMP-2-induced bone regeneration (Fig. 2c). Knockdown of Hif1a via RNAi increased osteoblast marker gene expression (Fig. 2d), mineralization, and calcified nodule formation (Fig. 2e). Osteocalcin (OCN) encoded by Ocn is an essential factor for bone matrix mineralization, and RUNX2 is a well-known transcriptional regulator of Ocn. RUNX2 activity as a transcription factor was determined using two types of RUNX2-responsive luciferase reporters (6XOSE-Luc and OG2-Luc), and the results show that RUNX2 activity was significantly decreased by HIF-1α overexpression (Fig. 2f). Recently, we reported that HIF-2α inhibits osteoblast differentiation via the TWIST2-RUNX2-OCN axis23. In our current RNA-seq data (Fig. 1f), Twist2 was commonly upregulated by HIF-1α and HIF-2α overexpression. Similar to the effects of HIF-2α23, Twist2, but not Twist1, was upregulated by HIF-1α overexpression (Fig. 2g). Moreover, we determined that the regulatory cis-elements of Twist2 promoter contained putative HIF-1α binding sequences, 5′-(A/G)CGTG-3′, and ChIP results showed that Twist2 is a direct target of HIF-1α (Fig. 2h, Supplementary Fig. 3). Additionally, siRNA-mediated silencing of Hif1a in primary cultured calvarial osteoblasts confirmed that Hif1a regulates Twist2 expression (Fig. 2i). Twist2 knockdown with specific siRNA partially restored the decrease of Ocn and Runx2 by overexpression of Hif1a (Fig. 2j). These data show that HIF-1α and HIF-2α have similar effects on osteoblast differentiation by regulating the TWIST2-RUNX2-OCN axis.

a Osteogenic differentiation of calvarial pre-osteoblasts was induced by DM, and 200 or 400 MOI of Ad-Hif1a was infected at differentiation-inducing culture day 3. Subsequently, cells were harvested on the 6th day of differentiation. Relative mRNA levels of Hif1a, Ocn, and Runx2 were quantitated by qRT-PCR (n ≥ 3). b Osteogenic phenotypes were determined by ALP and ARS staining. The representative captured images of 24-well plates were displayed (n = 3). c 5-mm diameter critical-sized defects were created in 6-week-old male mice and covered with collagen sponges without (Veh) or with 300 ng BMP-2. For each group, collagen sponges containing Ad-C or Ad-Hif1a were applied. After 2 weeks, the size and bone volume of calvarial defects were measured. The representative μCT images were shown (n = 5). d Cells were transfected with siRNA against Hif1a or control-siRNA (si-C) on differentiation day 3 and cultured for 3 days. Transcript levels of Hif1a, Ocn, and Runx2 were determined by qRT-PCR (n = 3). e The representative images of ALP and ARS staining (n = 3). f The pre-osteoblasts were co-transfected with RUNX2-responsive luciferase reporter constructs (6XOSE-Luc or OG2-Luc) and infected together with indicated MOI of Ad-Hif1a. g The relative RNA levels of Twist1 and Twist2 isotypes in Hif1a-overexpressing cells (n = 4). h ChIP analysis showing the HIF-1α binding to the Twist2 promoter region. i mRNA level of Twist2 in pre-osteoblasts transfected with Hif1a siRNA (n = 4). j Cells were transfected with siRNA against Twist2 or si-C following infection of Ad-Hif1a. Transcript levels of Hif1a, Twist2, Ocn, and Runx2 were analyzed (n = 5). Values are presented as the mean ± SEM. *P < 0.05, **P < 0.01, and ***P < 0.001.
Osteoblast-specific conditional KO of Hif1a increases bone mass
To determine the direct role of HIF-1α in regulating bone homeostasis, we generated osteoblast-specific Hif1a-deficient mice by crossing Hif1afl/fl mice with Col1a1-Cre transgenic mice. Immunohistochemical staining data verified specific knockout of Hif1a in osteoblasts, but not in osteoclasts, of Hif1afl/fl;Col1a1-Cre (Hif1a conditional KO) mice (Fig. 3a). The three-dimensional (3D) microarchitecture of femoral trabecular bones in 4-month-old Hif1a conditional KO mice and their control Hif1afl/fl mice was analyzed using μCT. The μCT images exhibited an increase in cancellous trabeculae (Fig. 3b), supported by quantitative bone parameters, such as BMD, BV/TV, Tb.Th, Tb.Sp, and Tb.N (Fig. 3c). The BMD, BV/TV, Tb.Th, and Tb.N showed higher values, and Tb.Sp exhibited lower values in Hif1afl/fl;Col1a1-Cre mice than in Hif1afl/fl control mice (Fig. 3c). To complement the 3D-μCT data, we conducted histomorphometric analysis of morphometric parameters of osteoblasts in the metaphyseal regions using H&E staining. It also revealed that the bone parameters, BV/TV, N.Ob/B.Pm, and Ob.S/BS, showed higher values in Hif1afl/fl;Col1a1-Cre mice (Fig. 3d). Taken together, HIF-1α is a negative regulator in osteoblast differentiation.

a Osteoblast-specific, but not osteoclasts, depletion of Hif1a in Hif1afl/fl;Col1a1-Cre mice and their wild-type littermates, Hif1afl/fl was verified by immunohistochemistry with anti-HIF-1α antibody (n = 3). The arrows indicate osteoclasts, and the dotted line indicates osteoblasts. GP, growth plate; BM, bone marrow; TB, trabecula bone; CB, cortical bone. Scale bar, 25 μm. b μCT images of femurs captured by the transverse (top), sagittal (middle), and coronal (bottom) planes. Red boxes indicate regions of interest. c Representative images of μCT reconstructions of trabecular bones and quantitative analyses of BMD, BV/TV, Tb.Th, Tb.Sp, and Tb.N of femora (n = 8). d H&E staining results and bone histomorphometric analysis, including BV/TV, N.Ob/B.Pm, and Ob.S/BS (n = 8). Scale bar, 100 μm. Values are presented as the mean ± SEM. Unpaired t-test was performed. *P < 0.05, **P < 0.01, and ***P < 0.001.
HIF-1α indirectly promotes RANKL-mediated osteoclastogenesis via HIF-2α
The regulation of osteoblast-mediated osteoclastogenesis via the regulatory role of RANKL and OPG, a decoy receptor for RANKL, is essential to maintain bone homeostasis8. To determine whether the inhibitory function of HIF-1α on osteoblast differentiation affects the crosstalk between osteoblasts and osteoclasts. Expression of Rankl and Opg by Hif1a-overexpressing osteoblasts was examined for RANKL-mediated osteoclast maturation. HIF-1α overexpression significantly increased Rankl expression (but not Opg) in osteoblasts (Fig. 4a). Rankl to Opg ratio at the transcript level (Fig. 4b), immunofluorescence staining of RANKL proteins (Fig. 4c), and secreted level of RANKL (Fig. 4d) confirmed upregulation of RANKL by HIF-1α in osteoblasts. However, unexpectedly, specific siRNA-mediated silencing of Hif1a did not affect Rankl expression (Fig. 4e). Furthermore, TRAP staining of trabecular bones of Hif1afl/fl;Col1a1-Cre mice showed no differences compared to Hif1afl/fl control mice (Fig. 4f). These results led to the hypothesis that HIF-1α-mediated Rankl upregulation may be indirect. Based on the results of Fig. 1f, we compared the expression of Hif1a and Hif2a by adenovirus mediated overexpression of each isoform of HIFs. Consistent with the RNA-seq analysis, Hif2a expression was significantly increased by HIF-1α overexpression, whereas Hif1a was slightly increased by HIF-2α overexpression, while ectopic expression of each isoform of HIF-α showed an increase in Rankl expression (Fig. 5a, Supplementary Fig. 3). Promoter analysis of Hif2a revealed the existence of putative HIF-α binding sequence, and direct binding of HIF-1α to this regulatory element of Hif2a promoter was determined by ChIP assay, suggesting that Hif2a is a direct target gene of HIF-1α (Fig. 5b, Supplementary Fig. 3). Interestingly, silencing of HIF-2α blocked Ad-Hif1a-mediated Rankl expression (Fig. 5c), whereas knockdown of HIF-1α did not affect Ad-Hif2a-mediated Rankl expression (Fig. 5d), indicating that HIF-1α-induced Rankl expression was mediated by HIF-2α. We set up the co-culture system to examine the effects of indirect regulation of Rankl expression by the HIF-1α-to-HIF-2α axis on osteoblast-mediated osteoclastogenesis. Calvarial pre-osteoblasts and BMMs, precursor cells of osteoclasts, were co-cultured in the same culture dish, and osteoblast differentiation was induced by L-AA, β-Gp, and VitD3 and then osteoclast differentiation was measured by TRAP staining (Fig. 5e). Osteoclastogenesis of co-cultured BMMs with pre-osteoblasts transduced with Ad-Hif2a was more enhanced than co-culture with HIF-1α overexpressing cells. This was further evidenced by an increase in the number of TRAP-positive multinucleated cells (Fig. 5e). In terms of regulating osteoblast differentiation, specific silencing of Hif2a restored Ad-Hif1a-mediated reduction of Ocn and Runx2 expression (Fig. 5f) and knockdown of HIF-1α also recovered Ad-Hif2a-mediated inhibition of osteoblast marker gene expression (Fig. 5g). These data suggest a differential but complementary role of HIF-1α and HIF-2α in regulating RANKL-mediated osteoclastogenesis, although the effects of both isoforms of HIF-α were similar on osteoblast differentiation.

a, b Primary pre-osteoblasts were infected with Ad-C or Ad-Hif1a on differentiation day 3 and cultured for 3 days. Relative mRNA levels of Opg and Rankl (a) and the Rankl/Opg ratio (b) were quantitated (n ≥ 3). c RANKL expression at the protein level was examined by immunofluorescence staining with anti-RANKL antibody and Alexa-594 (red), and nuclei were stained with DAPI (blue) (n = 3). Scale bar, 100 μm. d Secreted RANKL was measured by ELISA using a culture medium (n = 5). e qRT-PCR of Hif1a and Rankl following siRNA-mediated silencing of Hif1a during osteogenic differentiation (n = 3). f Bone histomorphometric analysis of osteoclast parameters such as N.Oc/B.Pm and Oc.S/BS after TRAP staining (n = 8). Scale bar, 100 μm. Values are presented as the mean ± SEM. N.S. not significant, *P < 0.05, and ***P < 0.001.

a Primary cells were infected with the indicated MOI of Ad-Hif1a or Ad-Hif2a on the 3rd day of differentiation and cultured until day 6. Expression pattern of Hif1a, Hif2a, and Rankl was determined using RT-PCR, and representative data were shown (n = 3). b ChIP was performed with anti-HIF-1α antibody and primers spanning the HRE motif of the promoter region of the Hif2a gene (n = 3). c, d qRT-PCR analyses of Hif1a, Hif2a, and Rankl in pre-osteoblasts transfected with Hif2a siRNA in Hif1a-overexpressing cells (c) and transfected with Hif1a siRNA in Hif2a-overexpressing cells (d) (n ≥ 3). e Calvarial pre-osteoblasts infected with Ad-C, Hif1a, or Hif2a adenovirus were cultured with BMMs in a medium containing L-AA (50 μg/ml), β-Gp (5 mM), and VitD3 (10 nM) for 5 days. TRAP staining and quantitative analysis of multinucleated cells are shown (n = 6). Scale bar, 100 μm. f, g qRT-PCR analyses of Ocn and Runx2 in pre-osteoblasts transfected with Hif2a siRNA in HIF-1α-overexpressing cells (f) and transfected with Hif1a siRNA in HIF-2α-overexpressing cells (g) (n = 4). Values are presented as the mean ± SEM. *P < 0.05, **P < 0.01, and ***P < 0.001.
Osteoblast-specific depletion of Hif1a delayed osteoporotic phenotypes
To examine the effects of HIF-1α on osteoporotic bone loss, OVX (Fig. 6a–c) and age-related (Fig. 6d–f) experimental animal models were applied. μCT images exhibited delay of OVX-induced bone loss in Hif1afl/fl;Col1a1-Cre mice compared to Hif1afl/fl control mice, supported by the parameters of quantitative analyses, such as BMD, BV/TV, Tb.Th, Tb.Sp, and Tb.N (Fig. 6a). Bone histomorphometric analyses with H&E (Fig. 6b) and TRAP staining results (Fig. 6c) revealed that osteoblast-specific depletion of Hif1a prevented OVX-induced reduction of osteoblasts (Fig. 6b) but not OVX-induced bone resorption (Fig. 6c). In particular, osteoblast-specific depletion of Hif1a reduced TWIST2 expression in osteoblasts of the OVX model, while the effect of Hif1a deficiency on HIF-2α and RANKL expression was not notably significant (Supplementary Fig. 2). Consistent with the data from OVX experimental mice, aging (12-month-old)-induced osteoporosis was also delayed in Hif1afl/fl;Col1a1-Cre mice (Fig. 6d–f). Taken all together, we concluded that HIF-1α is a potent regulator of bone homeostasis, similar to HIF-2α, but HIF-1α acts as an upstream of HIF-2α in regulating RANKL-mediated osteoclastogenesis.

a–c Quantitative μCT analysis of femoral trabecular bones in OVX- or sham-operated 3-month-old Hif1afl/fl and Hif1afl/fl;Col1a1-Cre mice (n = 8). d–f μCT analysis in 12-month-old Hif1afl/fl and Hif1afl/fl;Col1a1-Cre mice (n = 8). Representative images of μCT reconstructions (a, d), H&E staining, Scale bar, 100 μm (b, e), and TRAP staining, Scale bar, 100 μm (c, f). Bone-forming parameters, such as BMD, BV/TV, Tb.Th, Tb.Sp, Tb.N, N.Ob/B.Pm, and Ob.S/BS, were assessed using µCT measurements (a, d), bone histomorphometric analysis (b, e), and bone resorption parameters, N.Oc/B.Pm and Oc.S/BS were obtained from TRAP staining of the metaphyseal regions of femurs (c, f). Values are presented as means ± SEM (N.S. not significant, *P < 0.05; **P < 0.01, and ***P < 0.001). The effects of OVX and genotypes (Hif1afl/fl, Hif1afl/fl;Col1a1-Cre) and their interaction were tested using two-way ANOVA (a, BMD: interaction = 0.0404, OVX < 0.0001, genotype = 0.0162).
Discussion
Bone is a highly dynamic tissue that continuously undergoes remodeling to reflect an equilibrium of functional and metabolic demands even after bone development. Bone diseases, such as osteoporosis, osteopetrosis, and fragile bone fracture, are mainly caused by disturbances in bone homeostasis maintained by a balance between bone-forming osteoblasts and bone-resorbing osteoclasts. Our previous study demonstrated the critical role of HIF-2α as a catabolic regulator in bone remodeling and HIF-2α as a novel intrinsic mediator of age-related bone loss24, which led to the investigation of HIF-1α functions in regulating bone homeostasis. The activities of the HIF-α/β dimer on the transcriptional regulation of target genes depend on the stability of the HIF-1α subtype. So far, three isotypes of HIF-α have been identified; of these, HIF-1α and HIF-2α exhibit nearly the same amino acid sequence homology, while HIF-3α has relatively short sequences. Since both HIF-1α and HIF-2α have very similar structures and can recognize and bind the same HRE, 5′-(A/G)CGTG-3′, sequences in a promoter of specific target genes, many papers have reported similar functions of these two isotypes of HIF-α. However, several studies have supported distinct roles of HIF-1α and HIF-2α in different cellular processes. Such reports show that HIF-1α is associated with acute hypoxic response, whereas HIF-2α is related to chronic hypoxic response25. Moreover, HIF-1α contributes to the maintenance of chondrocyte phenotypes and metabolic adaptation to the hypoxic environment, whereas HIF-2α accelerates osteoarthritic processes26. This study determined similar and independent regulation of HIF-1α and HIF-2α during osteoblast differentiation but differential regulation of HIF-1α depending on HIF-2α in osteoclast activation via crosstalk with osteoblasts.
It is well-known that HIF-α stability is very sensitive to oxygen. HIF-α is hydroxylated by PHD27 and acetylated by ARD-128, resulting in the proteasomal degradation of HIF-α under normoxia. However, some investigations have reported that normoxic HIF-1α stabilization is caused by local inflammatory factors, such as TNF-α, INF-γ, IL-1β, and IGF-1, in human coronary endothelial cells13, and nitric oxide also promotes normoxic HIF-1α stabilization by inhibition of PHD29. Consistent with this finding, inflammatory cytokines, such as IL-1β, IL-17, IL-21, and TNF-α, induced increased expression of HIF-2α in mouse articular chondrocytes under normoxia30. The effect of inflammation on bone is mediated by inflammatory cytokines, such as IL-1β, IL-6, IL-17, and TNF-α, which promote osteoclastogenesis and skew bone homeostasis toward pathological bone resorption31,32,33,34. Interestingly, our previous study24 showed that HIF-1α and HIF-2α were stabilized under normoxia in cells cultured without any cytokines in osteogenic differentiation medium and in trabecular bone tissues during osteoporotic bone loss. It cannot be entirely ruled out that the stabilization of HIF-1α under normoxic conditions, as posited in our current research, may be influenced by locally occurring hypoxic conditions in in vivo pathological settings. The vascularized bone microenvironment, influenced by systemic circulation, is not hypoxic compared to various other organs35,36. Kim et al. directly measured in vivo oxygen levels in the bone architecture of rats, revealing that vascularized bone is a high-O2 area compared to vessel-free cartilage with low-O2 levels and bone marrow with medium-O2 levels37. They suggested that the higher expression of HIF-1α in high-O2 areas is associated with ROS signaling. In our study, we conducted all experiments under normoxia and observed stabilization of HIF-1α and HIF-2α during osteoblast differentiation of pre-osteoblasts under these conditions. We hypothesized that certain factors in the normal physiological or pathological environment could induce stabilization of the HIF-α subunit under normoxia. We investigated the effects of osteogenic differentiation-induced normoxic stabilization of HIF-1α in osteoblasts on regulating osteoblast differentiation and osteoblast-mediated osteoclast activation. Although we did not specifically investigate the signaling pathway of HIF stabilization during osteoblast differentiation under normoxia in the current study, we recognize the importance of clarifying this aspect in further studies.
In this study, we determined similar but slightly different regulation by HIF-1α compared to HIF-2α in regulating bone remodeling. The RNA-seq analysis to determine gene expression profile by HIF-1α and HIF-2α overexpression during osteoblast differentiation (Fig. 1) showed that about 62.7% DEGs of a total of 40,213 genes were similarly regulated, and about 10.9% and 26.4% DEGs were differentially regulated by HIF-1α and HIF-2α, respectively (Fig. 1d). Performing additional experiments comparing the RNA-seq results from osteoblasts with knockdown of each HIF isoform would provide more robust evidence for conclusive insights. In the current study, HIF-1α and HIF-2α expression increased continuously until about the 15-day, starting from the 3-day in vitro culture with osteogenic differentiation medium before osteoblast marker gene, OCN expression (Supplementary Fig. 1, Supplementary Fig. 4). We observed that overexpression of HIF-1α delayed BMP-2-induced bone regeneration by inhibiting osteogenic differentiation (Fig. 2c), and HIF-1α overexpression inhibited the expression and activity of RUNX2, which resulted in the inhibition of Ocn expression, a transcriptional target of RUNX2. Regarding the studies showing that hypoxia signaling inhibits osteogenic differentiation of mesenchymal stem cells by suppressing RUNX2 via transcriptional repression of Twist238 and HIF-2α directly binds to the promoter of Twist2 gene23, we found that HIF-1α was also a transcriptional activator of Twist2 gene. Thus, HIF-1α inhibited osteoblast differentiation by regulating the TWIST2-RUNX2-OCN axis, in a similar way of HIF-2α.
In our previous report23, HIF-2α expression was increased during in vitro osteoclast differentiation of BMMs, whereas no significant differences in HIF-1α expression were observed. We found that overexpression of HIF-2α resulted in increased osteoclast formation and the number of multinucleated giant cells23. However, overexpression of HIF-1α did not lead to any changes in osteoclast formation and resorption. The effects of HIF-1α on osteoclast formation and activation have been controversial. Some studies have observed an increase in osteoclast differentiation following HIF-1α stimulation39,40,41, while others have noted a decrease42,43. Shirakura et al. 44 and Wang et al. 45 suggested the possibility that the increase in osteoclast differentiation under hypoxia is the result of crosstalk between osteoblasts and osteoclasts. HIF-1α exhibited a slightly different signaling pathway on the crosstalk between osteoblasts and osteoclasts. The ratio of RANKL and OPG expressed by osteoblasts is the key regulator for the interplay between osteoblasts and osteoclasts in bone homeostasis46. RANKL expressed by osteoblasts binds to its receptor RANK on osteoclast precursors and induces osteoclast activation47. OPG produced by osteoblasts acts as a soluble decoy receptor for RANKL and modulates the balance between osteoblasts and osteoclasts8,48. Hif1a overexpression increased the ratio of Rankl to Opg expression by osteoblasts (Fig. 4a–d), but, unexpectedly, osteoblast-specific depletion of HIF-1α did not affect osteoclast activation (Fig. 4f). We also found that the knockdown of Hif1a with siRNA did not block Rankl expression, although HIF-1α remarkably upregulated RANKL expression. Our previous study showed that HIF-2α directly promotes Rankl transcription in osteoblasts23. Furthermore, HIF-2α was identified as one of the genes upregulated by Hif1a overexpression from the data of RNA-seq analysis (Fig. 1f). Thus, we thought that HIF-1α could be a direct transcriptional activator of Hif2a and it was verified by the results of the ChIP assay and reciprocal overexpression and knockdown experiments as shown in Fig. 5. The knockdown of Hif2a significantly blocked the Hif1a overexpression-mediated increase of Rankl expression, whereas the knockdown of Hif1a showed no effect on increased Rankl expression by Hif2a overexpression. The co-culture experiment showed osteoclast activation by HIF-1α-HIF-2α axis-mediated RANKL expression. Osteoclast activation of BMMs co-cultured with Hif1a overexpressed osteoblasts was lesser than those with Hif2a overexpressed cells, which indicates that HIF-1α is an upstream effector of HIF-2α, a critical factor for RANKL-mediated osteoclastogenesis23. The doubt that HIF-1α-mediated inhibition of osteoblast differentiation might be also mediated by HIF-2α was eliminated by the data of Fig. 5f, g. Through the reciprocal overexpression and knockdown experiments, the knockdown of Hif2a significantly restored Hif1a overexpression-mediated suppression of Runx2 and Ocn expression, and the pair of Hif1a siRNA and Ad-Hif2a also showed the same restored effect. Thus, HIF-1α and HIF-2α exhibited the same effect on the inhibition of osteoblast differentiation by sharing the TWIST2-RUNX2-OCN axis, but it was independent regulation by HIF-1α and HIF-2α. However, HIF-1α showed slightly different signaling pathways in the osteoblast-mediated osteoclastogenesis, but it was dependent on HIF-2α.
The results obtained in this study yielded somewhat challenging interpretations, among which is the difference in bone mass as depicted in Figs. 3 and 6. 4-month-old male Hif1afl/fl;Col1a1-Cre mice exhibited an increase in bone mass (Fig. 3), whereas 3-month-old female sham groups of Hif1afl/fl and Hif1afl/fl;Col1a1-Cre mice in OVX experiments showed no discrepancy in bone mass (Fig. 6a, b). One possible explanation for this inconsistency is gender-dependent differences in responses associated with specific sex steroids and growth hormone levels49,50,51. However, further studies are required to clarify this aspect. Additionally, in our study, bone analyses were conducted using osteoblast-specific Hif-1α knockout mice, revealing different findings compared to those of previous studies. In our study, depletion of osteoblast-specific Hif-1α in mice resulted in a significant increase in bone mass through modulation of osteoblast functions. This inconsistency could be explained in two ways. Firstly, we utilized a different Cre transgenic model, Col1a1-Cre, for generating osteoblast-specific conditional knockout mice, in contrast to the Ocn-Cre transgenic mice used by previous studies. Col1a1 is known to be expressed earlier than Ocn during osteogenesis. To further examine the expression patterns of osteogenic markers and HIF-1α, we assessed Hif-1α, Col1a1, and Ocn levels during osteogenesis in human mesenchymal stem cells. Consistent with the results obtained using pre-osteoblast cells (Supplementary Fig. 1a), the mRNA expression pattern of Hif-1α closely resembled that of Col1a1 but decreased at the Ocn-expressing stage. In addition, conflicting results were reported by Wu et al. 52 They suggest that HIF-1α is necessary within the context of combined PHD2 and PHD3 inactivation to inhibit bone resorption through an osteoblastic mechanism of OPG. Inconsistent results on the direct regulation of Opg by HIF-1α were presented in our current study (Fig. 4a). In their report, Osterix (Osx)-Cre mice were used for generation of osteoblast-specific conditional KO mice. The mouse Osx-Cre transgenic line expresses the Cre recombinase in committed osteoblast progenitors in both endochondral and membranous-derived bones2. Immature osteoblasts can be targeted by using the Osx-Cre transgenic mice, while mature osteoblasts can be targeted by using the Col1a1-Cre transgenic mice (especially the 2.3 kb Col1a1-Cre mice used in our experiment) based on their rich expression of type I collagen, the main constituent of bones53. This finding supports the differing phenotypes observed between our study and earlier ones. Another possible explanation for the discrepant results could be the variations in the ages of mice used for experiments. It has been suggested the HIF may exert differential effects depending on age and bone cycle (modeling vs. remodeling) in our previous study23. While previous studies analyzed juvenile or young adult mice, we utilized mature mice to assess the regulatory role of HIF-1α in bone remodeling and osteoporotic bone loss. These hypotheses should be further investigated using various Cre mouse lines and multiple time points (ages) in future studies.
The ultimate question of our study was attributed to the similar regulation in osteoblast differentiation but different regulation in osteoblast-mediated osteoclast activation. Both HIF-1α and HIF-2α expression was increased during osteoblast differentiation, and their effects on osteoblast differentiation were the same; therefore, both HIF-1α and HIF-2α expression during osteoblast differentiation seemed to be redundant. However, we found their different effects on osteoclast activation. In the results of in vivo experiments using OVX- or aging-induced osteoporosis model, osteoblast-specific knockout of HIF-1α (Hif1afl/fl;Col1a1-Cre mice) showed no differences in osteoclast differentiation and activation compared to control, Hif1afl/fl mice, although Hif1a cKO did show a significant increase in BMD and bone parameters (Fig. 6). In a previous report23, HIF-2α expression was increased during osteoclastogenesis of BMM, but not HIF-1α, and HIF-2α specifically promoted osteoclastogenesis by directly promoting Traf6 and RANKL-mediated osteoclastogenesis. This finding indicates that HIF-2α has more catabolic function in regulating bone remodeling than HIF-1α.
In conclusion, in the normal physiology of bone remodeling, HIF-1α could fine-tune osteoblast-mediated osteoclastogenesis by regulating HIF-2α expression, enabling the maintenance of bone homeostasis. Especially in the bone pathophysiologic microenvironment, some inflammatory cytokines may enhance the normoxic stabilization of HIF-1α and/or HIF-2α and skew the equilibrium of bone homeostasis toward bone resorption because HIF-2α promotes osteoclastogenesis and RANKL-mediated osteoclast activation and HIF-1α enhances HIF-2α function. We suggest that the normoxic stabilization of HIF-1α and HIF-2α during osteoblast differentiation is essential to osteoporotic bone loss. Especially in bone pathophysiology, several factors, including inflammatory cytokines, have the potential to stimulate the stabilization of HIF-1α and HIF-2α, which contributes to disturbances in bone homeostasis. In this study, although we didn’t investigate what factors induce normoxic stabilization of HIF-1α and HIF-2α and it should be further studied, we can emphasize the need to block both HIF-1α and HIF-2α to promote bone formation and regeneration. However, it is more necessary to inhibit HIF-2α to delay or inhibit osteoporotic bone loss. This study provides therapeutic advances in other human diseases correlated with bone loss.
Methods
Ethics statement and experimental mice
Hif1afl/fl mice were obtained from Jackson Laboratory (#007561, Sacramento, CA, USA), and Col1a1-Cre mice were kindly donated by Dr. Je-Yong Choi (Kyungpook National University, Daegu, Korea). To establish osteoblast-specific Hif1a knockout mice, Hif1afl/fl mice were backcrossed against Col1a1-Cre mice. Critical-sized (5-mm diameter) defects were created in 6-week-old C57BL/6 J male mice for calvarial defect models as previously described in ref. 23. 300 ng bone morphogenetic protein (BMP)-2 containing collagen sponges were applied to cover the defects. After 2 weeks, calvarial defect size was measured and further analyzed. Male mice were used for all experiments except for the ovariectomized (OVX) osteoporosis models. For the OVX models, a 5-mm dorsal incision and sham operation (as a control) were performed using 8-week-old female mice. After four weeks, the OVX mice were sacrificed for further analysis. All procedures for animal care and experiments were approved by the Institutional Animal Care and Use Committee (IACUC) of Chonnam National University (Gwangju, Korea).
Primary culture of pre-osteoblasts and osteogenic differentiation
All experiments conducted in vitro culture in our study were under normoxic conditions. In general cell culture models, normoxic conditions are maintained in standard humidified cell culture incubators with 5% CO2, where the oxygen concentration is approximately 18% O2 (v/v), equivalent to an oxygen tension or partial pressure (pO2) of 138 mmHg54,55. Primary culture of calvarial pre-osteoblasts and induction of osteoblast differentiation were performed as previously described in refs. 23,24. In brief, calvarial bones were isolated from 3-day-old pups of mice and enzymatically digested with dulbecco’s modified eagle’s medium (DMEM) containing 0.1% type II collagenase (Sigma–Aldrich, St. Louis, MO, USA) and 0.25% trypsin/EDTA at 37 °C for 20 min. The isolated pre-osteoblasts were cultured in complete media, DMEM (GIBCO, Grand Island, NY, USA) containing 10% fetal bovine serum and 1% penicillin/streptomycin. After 3 days, 1 × 105 cells were plated in a 35-mm culture dish and cultured in osteogenic differentiation media (DM) containing 50 μg/ml L-ascorbic acid (L-AA, A0278, Sigma–Aldrich) and 5 mM β-glycerophosphate (β-Gp, sc-220452A Chemcruz, Dallas, TX, USA). Adenoviral infection was performed on day 3 at the indicated MOI (Multiplicity of infection). For siRNA-mediated knockdown, primary cells were transfected with Hif1a siRNA, Hif2a siRNA, or Twist2-siRNA (Dharmacon, La Fayette, CO, USA) on differentiation day 3 using Lipofectamine RNAiMAX (Invitrogen, Carlsbad, CA, USA) following the manufacturer’s recommendations. Non-targeting siRNA (scrambled; Bioneer, Daejeon, Korea) was used as a negative control. After gene overexpression and knockdown, cells were harvested on 6 days of osteoblast differentiation for further experiment.
Co-culture of osteoblasts and bone marrow-derived macrophages (BMMs)
To perform the co-culture of osteoblasts and BMMs, primary calvarial pre-osteoblasts and BMMs were prepared as follows: Bone marrow was isolated from the long bones of 6- to 8-week-old mice and flushed with serum free α-MEM. Bone marrow cells were cultured in complete α-MEM for 24 h, after which non-adherent cells were collected and cultured in complete α-MEM in the presence of 30 ng/ml of M-CSF (Peprotech, Rocky Hill, NJ, USA) for 3 days. Adherent BMMs (2 × 104 cells per well in a 48-well plate) were then maintained in complete α-MEM containing 30 ng/ml of M-CSF for 24 h. Subsequently, they were co-cultured with primary calvarial pre-osteoblasts (4 × 103 cells/well) infected with Ad-C, Ad-Hif1a, or Ad-Hif2a in the presence of 100 ng/ml BMP-2, 50 μg/ml L-AA, 5 mM β-Gp, and 10 nM 1,25-dihydroxyvitamin D3 (VitD3) for 5 days23. Cells were fixed and applied for tartrate-resistant acid phosphate (TRAP) staining, and images were obtained using LAS (Leica ApplicationSuite) V4.1 program (Leica, Switzerland).
RNA sequencing (RNA-seq) analysis
For RNA-seq analysis, RNA samples were prepared from primary cultured calvarial pre-osteoblasts infected with control adenovirus (Ad-C), Ad-Hif1a, or Ad-Hif2a. Adenoviral infection was performed on day 3, followed by culturing the cells in osteogenic DM until day 6. RNA-seq service was provided by Ebiogen (Seoul, Korea). Gene expression profiling and graphic visualization were performed using the ExDEGA tool and ExDEGA Graphic Plus software. For the analysis of differentially expressed genes (DEGs), significant gene selection was filtered using an absolute value of fold change (FC) > 2, normalized data (log2) > 4 (Fig. 1d, e) or normalized data (log2) > 2 (Fig. 1f), and p-value (paired t-test) < 0.05 as the cut-off.
Reverse transcription-polymerase chain reaction (RT-PCR) and quantitative real-time (qRT)-PCR
Total RNA was isolated using TRI reagent (TR118, MRC, Cincinnati, OH, USA), and cDNA was obtained with a reverse transcription kit (TOPscript RT DryMIX, Enzynomics, Daejeon, Korea). The AmpOneTM Tap DNA Polymerase Mix (GeneAll, Seoul, Korea) and Applied Biosystems and SYBR premix Ex Taq (RR420, TaKaRa, Japan) were used for conventional PCR and qRT-PCR, respectively. All primer pairs are for the mouse genes and the sequence information of the primers and PCR conditions are listed in Supplementary Table 1. The individual transcript levels of each target gene were normalized with Gapdh, and relative levels were represented as a fold change relative to the indicated controls.
Western blotting
Lysis buffer (RIPA) for western blotting was prepared using 50 mM Tris-HCl (pH 8.0), 150 mM NaCl, 5 mM NaF, 1% NP-40, 0.2% SDS, 0.5% deoxycholate, a protease inhibitor cocktail, and a phosphatase inhibitor cocktail (Roche, Basel, Switzerland). Protein samples were applied for SDS-PAGE and transferred to nitrocellulose membranes. After blocking the membrane with 5% skim milk, the samples were incubated with indicated primary antibodies at 4 °C overnight. The following antibodies were used: anti-HIF-1α (NB100-134; Novus biologicals, Centennial, CO, USA), anti-HIF-2α (NB100-122; Novus biologicals), anti-Lamin A/C (2032 S; Cell signaling, Davnvers, MA, USA), anti-OCN (AB10911, Merck, Darmstadt, Germany), and anti-β-Actin (A3584, Sigma–Aldrich). Protein levels were detected using horseradish peroxidase-conjugated secondary antibodies and an ECL detection system (RPN2235, Cytiva, Buckinghamshire, UK).
Enzyme-linked immunosorbent assay (ELISA)
Secreted RANKL protein levels in the culture medium were measured using a RANKL-ELISA kit (ab-100749, Abcam, Cambridge, UK), following the manufacturer’s instructions.
Immunocytochemistry
Cells cultured on 12-mm coverslips were fixed with 3.5% paraformaldehyde, permeabilized with 0.1% Triton X-100, and blocked with 1% BSA. Then, cells were incubated with primary antibodies, anti-HIF-1α, anti-HIF-2α or anti-RANKL antibodies, and an Alexa-594-conjugated secondary antibody. DAPI (4′,6-diamidino-2-phenylindole) was used for staining nuclei. Fluorescence images were captured by Zeiss microscope and analyzed by IMT isolution FL/Auto software. Positive staining cells were counted using the NIH ImageJ program (version 1.47, National Institutes of Health, Bethesda, MD, USA).
Chromatin immunoprecipitation (ChIP) assay
ChIP assays were performed as described previously24. In brief, crosslinks of genomic DNA and proteins were achieved by adding 1% formaldehyde to the cell culture medium at room temperature for 10 min. Cells were lysed with RIPA buffer and then sonicated to cause fragmentation of DNA. The samples were incubated with 2 μg of anti-HIF-1α antibody or anti-IgG at 4 °C overnight. The precipitated complex of antibody and chromatin fragments were applied for PCR with specific primers for Twist2 or Hif2a promoter. The primer sequences for the ChIP assay are listed in Supplementary Table 1.
Luciferase reporter assay
Primary calvarial pre-osteoblasts infected with Ad-Hif1a were transfected with pGL3-6xOSE or pGL3-OG2, RUNX2-responsive luciferase reporter constructs, plus a CMV-β-galactosidase construct as an internal control56. Cell lysates were used for measuring luciferase reporter activity using a luciferase reporter assay system (Promega, Madison, WI, USA). The individual values were normalized with those of β-galactosidase activity.
Alkaline phosphatase (ALP) and alizarin red-S (ARS) staining
For ALP staining, pre-osteoblasts were cultured for 7 days in DM media. After fixation with 4% formaldehyde, cells were rinsed with deionized water and stained with 5-bromo-4-chloro-3-indolyl phosphate (BCIP®)/nitro blue tetrazolium (NBT) Liquid Substrate solution (Sigma–Aldrich) for 30 min in a dark room. For ARS staining, cells were cultured for 14 days in DM media, fixed with 10% formalin for 15 min, and stained with 2% Alizarin red-S solution (ARS, Sigma–Aldrich) for 45 min at room temperature57.
X-ray microcomputed tomography (μCT) and bone histomorphometry analysis
Mouse femurs were fixed in 10% neutral buffered formalin and analyzed with high-resolution μCT (Skyscan 1172 system, Bruker, Aartselaar, Belgium) as described previously23. Image reconstruction software (NRecon; Bruker) was used with identical thresholds for all samples (0–6000 in Hounsfield units) for reconstructing serial cross-section images. For the trabecular bones in the proximal femurs, a region of interest comprising 300 total steps starting at 30 steps away from the growth plate was manually designated for the trabecular bones in the proximal femurs. Femoral morphometric parameters, such as bone mineral density (BMD), bone volume per tissue volume (BV/TV), trabecular thickness (Tb.Th), trabecular separation (Tb.Sp), and trabecular number (Tb.N), were determined with CTAn data analysis software. Quantitative histomorphometric parameters, the number of osteoblasts per bone perimeter (N.Ob/B.Pm), and the osteoblasts surface per bone surface (Ob.S/BS) were analyzed using the data of H&E staining with OsteoMeasure software (Osteometrics, Inc., Decatur, GA, USA). For osteoclast parameters, the number of osteoclasts per bone perimeter (N.Oc/B.Pm) and osteoclasts surface per bone surface (Oc.S/BS) were analyzed with TRAP staining data.
Immunohistochemistry
Isolated distal femurs were fixed with 10% neutral buffered formalin and decalcified in 0.5 M EDTA (pH 7.4). After serial dehydration procedures, bone tissues were embedded in paraffin. Sectioned slices at 5-μm thickness on slide glasses were incubated in 3% H2O2 to block endogenous peroxidase activity and treated with 0.1% trypsin for 40 min at 37 °C for antigen retrieval. After blocking with 1% BSA for 30 min, the sectioned tissues were incubated with anti-HIF-1α. EnVision-HRP (K5007, Dako, Denmark) and AEC substrate kit (SK-4200, Vector laboratories, Newark, CA, USA) were used for visualization, and hematoxylin (Dako) was used for counterstain.
Statistical analysis
All experiments were repeated at least three times. Results were reported as the mean ± SEM. μCT and histomorphometric parameters were presented in bar charts with scatter plots. All statistical analyses were performed using GraphPad Prism version 7 (GraphPad Software, Inc., San Diego, CA, USA). All quantified data were first tested for conformation to a normal distribution using the Shapiro–Wilk test, followed by analysis with two-tailed Student’s t-test for quantitative variables between the means of two groups or one-way ANOVA or two-way ANOVA followed by Tukey’s post hoc tests (multi-comparison) for variables between the means of three or more independent groups, as appropriate. The n-value was the number of independent experiments or mice. Results were considered statistically significant when p-values were less than 0.05.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
RNA-seq data was obtained from the Gene Expression Omnibus database under accession number GSE271730. Source data for the graphs and charts are provided as Supplementary Data 1. All data that support the findings of this study are available from the corresponding author upon reasonable request.
References
Gomathi, K., Akshaya, N., Srinaath, N., Moorthi, A. & Selvamurugan, N. Regulation of Runx2 by post-translational modifications in osteoblast differentiation. Life Sci. 245, 117389 (2020).
Nakashima, K. et al. The novel zinc finger-containing transcription factor osterix is required for osteoblast differentiation and bone formation. Cell 108, 17–29 (2002).
Li, Z. et al. Role of TCF/LEF transcription factors in bone development and osteogenesis. Int. J. Med. Sci. 15, 1415–1422 (2018).
Teitelbaum, S. L. Bone resorption by osteoclasts. Science 289, 1504–1508 (2000).
Boyle, W. J., Simonet, W. S. & Lacey, D. L. Osteoclast differentiation and activation. Nature 423, 337–342 (2003).
Soysa, N. S. & Alles, N. NF-κB functions in osteoclasts. Biochem. Biophys. Res. Commun. 378, 1–5 (2009).
Park, J. H., Lee, N. K. & Lee, S. Y. Current understanding of RANK signaling in osteoclast differentiation and maturation. Mol. Cells 40, 706–713 (2017).
Cawley, K. M. et al. Local production of osteoprotegerin by osteoblasts suppresses bone resorption. Cell Rep. 32, 108052 (2020).
Albadari, N., Deng, S. & Li, W. The transcriptional factors HIF-1 and HIF-2 and their novel inhibitors in cancer therapy. Expert Opin. Drug Discov. 14, 667–682 (2020).
Strowitzki, M. J., Cummins, E. P. & Taylor, C. T. Protein hydroxylation by Hypoxia-inducible factor (HIF) hydroxylases: unique or ubiquitous? Cells 8, 384 (2019).
Schofield, C. J. & Ratcliffe, P. J. Oxygen sensing by HIF hydroxylases. Nat. Rev. Mol. Cell Biol. 5, 343–354 (2004).
Thornton, R. D. et al. Interleukin 1 induces hypoxia-inducible factor 1 in human gingival and synovial fibroblasts. Biochem. J. 350, 307–312 (2000).
Abdi Sarabi, M. et al. Normoxic HIF-1α stabilization caused by local inflammatory factors and its consequences in human coronary artery endothelial cells. Cells 11, 3878 (2022).
Doi, K. et al. LSD1 metabolically integrates osteoclast differentiation and inflammatory bone resorption through HIF-1α and E2F1. Arthritis Rheumatol. 74, 948–960 (2022).
Woo, S. K. et al. Sequential activation of hypoxia-inducible factor 1 and specificity protein 1 is required for hypoxia-induced transcriptional stimulation of Abcc8. J. Cereb. Blood Flow Metab. 32, 525–536 (2012).
Biddlestone, J., Bandarra, D. & Rocha, S. The role of hypoxia in inflammatory disease (review). Int. J. Mol. Med. 35, 859–869 (2015).
Wang, Y. et al. The hypoxia-inducible factor α pathway couples angiogenesis to osteogenesis during skeletal development. J. Clin. Invest. 117, 1616–1626 (2007).
Shomento, S. H. et al. Hypoxia‐inducible factors 1α and 2α exert both distinct and overlapping functions in long bone development. J. Cell Biochem. 109, 196–204 (2010).
Frey, J. L., Stonko, D. P., Faugere, M.-C. & Riddle, R. C. Hypoxia-inducible factor-1α restricts the anabolic actions of parathyroid hormone. Bone Res. 2, 14005 (2014).
Riddle, R. C., Leslie, J. M., Gross, T. S. & Clemens, T. L. Hypoxia-inducible factor-1α protein negatively regulates load-induced bone formation. J. Biol. Chem. 286, 44449–44456 (2011).
Chen, D. et al. Synergistic inhibition of Wnt pathway by HIF-1α and osteoblast-specific transcription factor osterix (Osx) in osteoblasts. PLoS ONE 7, e52948 (2012).
Li, C., Zhao, R., Yang, H. & Ren, L. Construction of bone hypoxic microenvironment based on bone-on-a-chip platforms. Int. J. Mol. Sci. 24, 6999 (2023).
Lee, S. Y. et al. Controlling hypoxia-inducible factor-2α is critical for maintaining bone homeostasis in mice. Bone Res. 7, 14 (2019).
Lee, S. Y. et al. Hypoxia-inducible factor-2α mediates senescence-associated intrinsic mechanisms of age-related bone loss. Exp. Mol. Med. 53, 591–604 (2021).
Holmquist-Mengelbier, L. et al. Recruitment of HIF-1α and HIF-2α to common target genes is differentially regulated in neuroblastoma: HIF-2α promotes an aggressive phenotype. Cancer Cell 10, 413–423 (2006).
Zhang, F.-J., Luo, W. & Lei, G.-H. Role of HIF-1α and HIF-2α in osteoarthritis. Joint Bone Spine 82, 144–147 (2015).
Lando, D., Gorman, J. J., Whitelaw, M. L. & Peet, D. J. Oxygen-dependent regulation of hypoxia-inducible factors by prolyl and asparaginyl hydroxylation. Eur. J. Biochem. 270, 781–790 (2003).
Jeong, J. W. et al. Regulation and destabilization of HIF-1α by ARD1-mediated acetylation. Cell 111, 709–720 (2002).
Metzen, E., Zhou, J., Jelkmann, W., Fandrey, J. & Brüne, B. Nitric oxide impairs normoxic degradation of HIF-1α by inhibition of prolyl hydroxylases. Mol. Biol. Cell 14, 3470–3481 (2003).
Yang, S. et al. Hypoxia-inducible factor-2α is a catabolic regulator of osteoarthritic cartilage destruction. Nat. Med. 16, 687–693 (2010).
Ruscitti, P. et al. The role of IL-1β in the bone loss during rheumatic diseases. Mediators Inflamm. 2015, 782382 (2015).
Luo, G., Li, F., Li, X., Wang, Z.-G. & Zhang, B. TNF-α and RANKL promote osteoclastogenesis by upregulating RANK via the NF-κB pathway. Mol. Med. Rep. 17, 6605–6611 (2018).
Kudo, O. et al. Interleukin-6 and interleukin-11 support human osteoclast formation by a RANKL-independent mechanism. Bone 32, 1–7 (2003).
Kotake, S. et al. IL-17 in synovial fluids from patients with rheumatoid arthritis is a potent stimulator of osteoclastogenesis. J. Clin. Invest. 103, 1345–1352 (1999).
Gan, E. S. & Ooi, E. E. Oxygen: viral friend or foe? Virol. J. 17, 115 (2020).
Hannah, S. S., McFadden, S., McNeilly, A. & McClean, C. “Take My Bone Away?” Hypoxia and bone: a narrative review. J. Cell Physiol. 236, 721–740 (2021).
Kim, H.-S. et al. O2 variant chip to simulate site-specific skeletogenesis from hypoxic bone marrow. Sci. Adv. 9, eadd4210 (2023).
Yang, D.-C. et al. Hypoxia inhibits osteogenesis in human mesenchymal stem cells through direct regulation of RUNX2 by TWIST. PLoS ONE 6, e23965 (2011).
Miyauchi, Y. et al. HIF1α is required for osteoclast activation by estrogen deficiency in postmenopausal osteoporosis. Proc. Natl. Acad. Sci. USA 110, 16568–16573 (2013).
Tian, Y. et al. HIF-1α regulates osteoclast activation and mediates osteogenesis during mandibular bone repair via CT-1. Oral Dis. 28, 428–441 (2022).
Wang, D. et al. Hypoxia-inducible factor 1α enhances RANKL-induced osteoclast differentiation by upregulating the MAPK pathway. Ann. Transl. Med. 10, 1227 (2022).
Hulley, P. A. et al. Hypoxia-inducible factor 1-alpha does not regulate osteoclastogenesis but enhances bone resorption activity via prolyl-4-hydroxylase 2. J. Pathol. 242, 322–333 (2017).
Knowles, H. J. Distinct roles for the hypoxia-inducible transcription factors HIF-1α and HIF-2α in human osteoclast formation and function. Sci. Rep. 10, 21072 (2020).
Shirakura, M. et al. Activation of the hypoxia-inducible factor-1 in overloaded temporomandibular joint, and induction of osteoclastogenesis. Biochem. Biophys. Res. Commun. 393, 800–805 (2010).
Wang, G. et al. Short-term hypoxia accelerates bone loss in ovariectomized rats by suppressing osteoblastogenesis but enhancing osteoclastogenesis. Med. Sci. Monit. 22, 2962–2971 (2016).
Boyce, B. F. & Xing, L. Biology of RANK, RANKL, and osteoprotegerin. Arthritis Res. Ther. 9, S1 (2007).
Roux, S. & Orcel, P. Bone loss. Factors that regulate osteoclast differentiation: an update. Arthritis Res. 2, 451–456 (2000).
Han, Y., You, X., Xing, W., Zhang, Z. & Zou, W. Paracrine and endocrine actions of bone-the functions of secretory proteins from osteoblasts, osteocytes, and osteoclasts. Bone Res. 6, 16 (2018).
Kim, B.-T. et al. The structural and hormonal basis of sex differences in peak appendicular bone strength in rats. J. Bone Miner. Res. 18, 150–155 (2003).
Martin, R. B. Size, structure and gender: lessons about fracture risk. J. Musculoskelet Neuronal Interact. 2, 209–211 (2002).
Yao, X. et al. Gender-dependence of bone structure and properties in adult osteogenesis imperfecta murine model. Ann. Biomed. Eng. 41, 1139–1149 (2013).
Wu, C. et al. Oxygen-sensing PHDs regulate bone homeostasis through the modulation of osteoprotegerin. Genes Dev. 29, 817–831 (2015).
Liu, F. et al. Expression and activity of osteoblast-targeted Cre recombinase transgenes in murine skeletal tissues. Int. J. Dev. Biol. 48, 645–653 (2004).
Wenger, R. H. et al. Frequently asked questions in hypoxia research. Hypoxia 3, 35–43 (2015).
Martinez, C.-A. et al. A cell culture model that mimics physiological tissue oxygenation using oxygen-permeable membranes. Bio. Protoc. 9, e3371 (2019).
Jeong, B.-C. et al. The orphan nuclear receptor estrogen receptor-related receptor γ negatively regulates BMP2-induced osteoblast differentiation and bone formation. J. Biol. Chem. 284, 14211–14218 (2009).
Kim, J.-W. et al. Chemical inhibitors of c-Met receptor tyrosine kinase stimulate osteoblast differentiation and bone regeneration. Eur. J. Pharmacol. 806, 10–17 (2017).
Acknowledgements
This work was supported by National Research Foundation of Korea (NRF) grants funded by the Korean government (MSIT) (2019R1A5A2027521, 2021R1A2C3005727, 2021R1F1A1062446, 2022R1C1C2013357), and a Korean Fund for Regenerative Medicine (KFRM) grant funded by the Korean government (the Ministry of Science and ICT, the Ministry of Health & Welfare) (22A0104L1).
Author information
Authors and Affiliations
Contributions
J.-H.R. and Y.H.H. conceived and designed the study. S.Y.L. and S.-J.K. performed the in vitro and in vivo experiments and analyzed data. S.-J.K. prepared cells for RNA-seq. S.Y.L. performed computational analysis for RNA-seq. K.H.P., G.L., and Y.O. helped with the data analysis. Y.H.H. wrote the manuscript. All authors revised and agreed on the final version of the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Communications Biology thanks Bethan Davies and the other, anonymous, reviewer for their contribution to the peer review of this work. Primary Handling Editors: Martina Rauner and Joao Valente. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Lee, S.Y., Kim, SJ., Park, K.H. et al. Differential but complementary roles of HIF-1α and HIF-2α in the regulation of bone homeostasis. Commun Biol 7, 892 (2024). https://doi.org/10.1038/s42003-024-06581-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s42003-024-06581-z
- Springer Nature Limited