
Abstract
Long intergenic non-coding RNAs (lincRNAs) are emerging as regulators of protein-coding genes (PCGs) in many plant and animal developmental processes and stress responses. In this study, we characterize the genome-wide lincRNAs in potatoes responsive to a vascular bacterial disease presumably caused by Candidatus Liberibacter solanacearum (CLso). Approximately 4397 lincRNAs were detected in healthy and infected potato plants at various stages of zebra chip (ZC) disease progression. Of them, ~65% (2844) were novel lincRNAs, and less than 1% (9) were orthologs of Arabidopsis and rice based on reciprocal BLAST analysis, suggesting species-specific expansion. Among the proximal lincRNAs within 50 kbp from a PCG, ~49% were transcribed from the same strand, while ~39% and ~15% followed convergent (head-to-head) and divergent (tail-to-tail) orientations, respectively. Approximately 30% (1308) were differentially expressed following CLso infection, with substantial changes occurring 21 days after infection (DAI). Weighted Gene Co-expression Network Analysis (WGCNA) of lincRNAs and PCGs identified 46 highly correlated lincRNA–PCG pairs exhibiting co-up or co-downregulation. Furthermore, overexpression of selected lincRNAs in transgenic potato hairy roots resulted in perturbation of neighboring PCG expression and conferred tolerance to CLso infection. Our results provide novel insights into potato lincRNAs’ identity, expression dynamics, and functional relevance to CLso infection.
Similar content being viewed by others

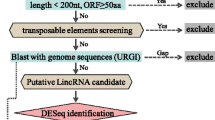
Introduction
Long non-coding RNAs (lncRNAs) are non-coding transcripts usually ≥200 bp long, lacking coding potential, and are primarily species-specific1,2. LncRNAs play a crucial role in regulating gene expression via diverse mechanisms in many complex developmental processes and stress responses3,4,5,6. Based on the genomic position and orientation with proximal protein-coding genes (PCGs), lncRNAs are classified into long intergenic non-coding RNAs (lincRNAs), long non-coding natural antisense transcripts (lincNATs), sense-overlapping lincRNAs and intronic lincRNAs4,7,8. Among them, lincRNAs represent over 50% of the lncRNA population9. LincRNAs share similar features with mRNAs, such as capping at 5′ end, polyadenylation at 3′ end, and splicing9,10. These characteristics allow lincRNAs to be identified using transcriptome-wide approaches (i.e., RNA-seq) designed to monitor mRNAs1,9,10,11. For instance, the FANTOM project identified 13,105 lincRNAs among 19,175 functional lncRNAs in humans. However, the human lincRNA repertoire could be close to 100,000 based on lincRNAs found in developmental stages, tissue types, and disease-specific RNA-Seq datasets in humans and rodents12. The GENCODE consortium (ENCODE project) also released 14,880 evidence-based lncRNA transcripts in humans13.
The different roles of lncRNAs in plants have only recently been investigated. With the availability of high-throughput technologies, lncRNAs were detected in various tissues and growth conditions in Arabidopsis8,14,15. However, functional characterization has been revealed only for a few lncRNAs. For instance, the function of MAS and SVALKA lncRNAs were found to be implicated in cold stimulus by governing the expression of their associated PCGs encoding MADS AFFECTING FLOWEIRNG4 (MAF4) and C-repeat/dehydration-responsive element binding factor (CBF), respectively8,16. In addition to the direct role of lincRNAs in regulating PCG expression, some can also act as microRNA (miRNA) decoys (target mimics) in post-transcriptional gene regulation. For instance, lincRNAs can compete with mRNAs and bind to specific miRNAs, thus interfering with the cleavage or translation of endogenous mRNA targets. However, the repertoire of plant lincRNAs, their involvement in gene regulation, and regulating plant–microbe responses still need to be better understood11,17.
Besides Arabidopsis, numerous lncRNAs were identified in maize (1724), rice (98), Populus trichocarpa (2542), foxtail millet (19), and switchgrass (1597) in response to drought stress5,6,18,19,20. In potatoes, ~2897 lincRNAs were identified during tuber sprouting21. Only a few studies of lncRNAs are available in response to biotic stress in plants. A strand-specific RNA-seq approach was employed to identify natural antisense transcripts (lncNATs) responsive to Fusarium oxysporum in Arabidopsis. Intriguingly, lncNATs were found in ~20% of the Arabidopsis transcripts, suggesting a crucial role of lncRNAs in governing the expression level of the PCGs22. Likewise, ~3181 lncRNAs were identified from the Sclerotinia sclerotiorum-infected Brassica napus and suggested their role in governing response to biotic stress23. LncRNAs from the tomato plants infected with yellow leaf curl virus (YLCV) were identified, and the plausible role of the lncRNAs as competing endogenous target mimics (eTMs) against the miRNAs has been determined24. In potatoes, ~17 lncRNAs were identified in response to infection with Pectobacterium carotovorum (P. carotovorum) (Pcb1692)17. In another study, about ~1565 lncRNAs were identified in response to YLCV infection in tomato24. The results suggest that lncRNAs may play an essential role in diverse biological processes, including host immune response against pathogen invasion via governing expression of stress-responsive PCGs via orchestrated regulatory networks.
Potato is a commercially important vegetable crop providing the bulk of calorie intake25,26. However, the productivity of potatoes is threatened by emerging diseases such as zebra chip (ZC) disease, which is associated with the phloem-limited bacterium Candidatus Liberibacter solanacearum (CLso) and is transmitted by the potato psyllid, Bactericera cockerelli (B. cockerelli). In this study, we characterized potato’s long intergenic RNA (lincRNA) landscapes and studied their role in mediating PCG expression and defense responses to CLso.
Results
Genome-wide identification of potato lincRNAs
To identify and understand the function of lincRNAs during the progression of ZC disease, we performed RNA-seq of healthy (CT; control) and infested potato plants with psyllid vector carrying CLso [CLso(+)] and psyllids without CLso [CLso(−)] temporally at 7, 14, and 21 days after infection (DAI). In total, ~298 million paired-end reads were generated from the temporal RNA-seq datasets (nine conditions having three biological replicates in each condition), covered with ~11.4 million paired-end reads in each biological replicate. The raw sequence data were subjected to quality filtering and mapped to the reference potato genome (Solanum tuberosum (S. tuberosum) v4.04). Of these, ~99% of the reads were high quality, and 54–61% mapped uniquely to the reference genome27 (Supplementary Table 1). Transcripts were assembled using the Read mapping and transcript assembly workflow28, and lincRNAs were identified using Evolinc29. After identifying putative lincRNAs, those expressed below 0.1 transcripts per million were considered artifacts and removed from further analysis.
We identified a total of ~4397 lincRNAs from the nine samples of potatoes representing healthy (CT), CLso(+), and CLso(−) conditions at 7, 14, and 21 DAI (Supplementary Table 1). Previous studies showed that lincRNAs are mostly species-specific9,30,31. For instance, only ~12% of human lincRNAs show sequence conservation with non-human species32. Consistent with this, when we compared potato lincRNAs with those of Arabidopsis and rice (downloaded from the CANTATAdb v2.0 database), we observed poor inter-species conservation (<1%) of the lncRNAs. In contrast, ~35% of potato lincRNAs were conserved with the lncRNAs of other Solanaceous plants (Fig. 1a; Supplementary Data 1a). Moreover, 4379 lincRNAs identified in this study were substantially higher than the previously reported lincRNAs under temporal infection with P. carotovorum subsp. brasiliense in the tolerant and susceptible potato cultivars17. On the contrary, about 3175 lncRNAs were detected from the apical buds of potato tubers21. Therefore, this study expanded the potato lincRNAs repertoire to include novel ones perturbed during biotic stress response.

a A Venn diagram compares potato lncRNAs available in the CANTATAdb v2.0 database with those detected in the current study. b The distribution of lincRNAs in the 12 potato chromosomes is shown in a pie chart. The fraction (%) of the highest and least number of lincRNAs is depicted. c Lengthwise distribution of the lincRNAs and comparison with protein-coding genes (PCGs) is shown via kernel density plot.
Genomic attributes of potato lincRNAs
The identified lincRNAs were distributed across all chromosomes (Chr) of the potato, with the most found on Chr 1 (12.66%) and the least on Chr 5 (5.93%) (Fig. 1b; Supplementary Data 1b). The transcript length of the lincRNAs ranged from 201 to 5095 bp with an average size of 491 bp, and ~95% of them were shorter than 1 kb. The length distribution of lincRNAs and PCGs revealed that the length of lincRNAs (average length 491 bp) was significantly shorter than that of PCGs (average length 1415 bp) (p value < 0.05, Welch’s two-sided t-test) (Fig. 1c; Supplementary Data 1c). Likewise, we analyzed the structural organization of the lincRNAs. In general, lincRNAs were predominantly mono- or di-exonic, whereas >50% of PCGs contained three or more exons (Fig. 2a; Supplementary Data 2). Consistent with other plant species, including Arabidopsis maize and soybean (Glycine max)3,7,8,11, the proportion of potato lincRNAs containing a single exon (~58%) was higher than that of lincRNAs containing multiple exons.

a The distribution of exons in lincRNAs and comparison with PCGs is shown in bar plot. b Types of lincRNAs based on orientation concerning their proximal PCGs are graphically depicted.
Previous studies suggested that lincRNAs can influence the expression of their proximal PCGs10,17. We analyzed PCGs located in the proximal (≤50 kb) regions in the up- or downstream regions of the lincRNAs. The average distance of the lincRNAs from their nearest PCGs was ~25 kb away. About 89% of the lincRNAs were located within 50 kb regions of their nearest PCGs, and therefore, we assigned these genes as proximal PCGs hereafter. Further, we analyzed the orientation of the lincRNAs relative to their proximal PCGs. Almost half (49–51%) of the lincRNAs were found to be oriented either in the same or opposite direction respective to proximal PCGs. Among the ~51% of lincRNAs oriented in the opposite direction, convergent type (~36%) represented a major fraction as compared to divergently (~15%) oriented lincRNAs (Fig. 2b). A previous study also showed a higher representation of convergently orientated lincRNAs with respect to their associated proximal PCGs33.
Functional relevance of potato lincRNAs as plausible miRNA decoys
Some lincRNAs could act as miRNA decoys by mimicking the target mRNA sequences, and consequently, the target mRNAs can be protected from degradation34,35,36. For instance, in Arabidopsis, the lincRNA Induced by Phosphate Starvation1 (IPS1) serves as a decoy to ath-miR399. It interferes with the binding of ath-miR399 to its primary target PHO2 transcript and rescue from post-transcriptional silencing in governing phosphate homeostasis in Arabidopsis37. However, lincRNAs that may act as miRNA decoys in potatoes have not been explored. The 4397 potato lincRNAs identified in this study were compared for sequence homology against the known potato mature miRNA sequences in the CANTATAdb v2.0 database. Six lincRNAs (lincRNA.ID.00006654, lincRNA.ID.00002569, lincRNA.ID.00018937, lincRNA.ID.00004487, lincRNA.ID.00010417, and lincRNA.ID.00012834) were predicted to harbor complementary sequences to known miRNAs and could serve as potential miRNA decoys (target mimics). Among them, lincRNA.ID.00006654 and five PCGs harbored binding sites of stu-miR482a-5p. Likewise, lincRNA.ID.00012834 along with three PCGs and lincRNA.ID.00002569, lincRNA.ID.00018937, lincRNA.ID.00004487, and lincRNA.ID.00010417 each with two PCGs harbored binding sites of same miRNAs (Supplementary Fig. 1a; Supplementary Data 3, 8). The PCGs encoded ATP binding protein, expansin, DNA repair protein, multidrug resistance, carboxylic ester hydrolase, a ubiquitin ligase, methyladenine DNA glycosylase, pentatricopeptide repeat protein, and potassium transporter.
Of the six, two lncRNAs (lincRNA.ID.00002569 and lincRNA.ID.00004487) were downregulated in CLso(+) infected tissues at 21 DAI based on the transcriptome analysis (Supplementary Fig. 1b; Supplementary Data 3). However, only one lincRNA.ID.00002569 showed significant downregulation by RT-qPCR (Supplementary Fig. 1c). This lincRNA had complementarity to a potato stu-miR8005a miRNA and similarity to the target PCGs: PGSC0003DMT400008676 (encoding Rad50 ATPase) and PGSC0003DMT400011415 (encoding Multidrug resistance pump protein). The RT-qPCR results also showed that the downregulation of lincRNA.ID.00002569 in CLso(+) infection paralleled the downregulation of PGSC0003DMT400011415 but not PGSC0003DMT400008676 (Supplementary Fig. 1; Supplementary Data 3), suggesting lincRNA.ID.00002569 could act as a potential miRNA decoy of PGSC0003DMT400011415.
Differentially expressed potato lincRNAs in response to CLso and psyllid
LincRNAs are important in regulating diverse biological processes via complex regulatory networks with PCGs. To examine the role of lincRNAs under pathogen infection, we identified sets of differentially expressed lincRNAs under conditions of psyllid challenges without CLso [CLso(−)/CT], psyllid carrying CLso [CLso(+)/CT] and pathogen-specific [CLso(+)/CT]/[CLso(−)/CT] temporally at 7, 14, and 21 DAI. A total of 775 unique lincRNAs were found to be differentially expressed in any of the 9 different comparisons (Supplementary Data 4). A substantially higher number of lincRNAs were found to be differentially expressed at 21 DAI preferentially under [CLso(+)/CT] followed by [CLso(+)/CT]/[CLso(−)/CT] condition (Fig. 3a; Supplementary Data 4). Furthermore, we performed principal component analysis (PCA) of the nine sets of differentially expressed lincRNAs. The first component (PC1) variation was up to 88.4%. We ruled out the extent of variation due to outliers and lack of reproducibility among the biological replicates since we used the differentially expressed values in the log2 scale. Moreover, variation due to time of sampling is unlikely as we observed a distinct and diverse differential expression profile of lincRNAs specifically at 21 DAI (Fig. 3b). The results suggest a dynamic and plausible role of lincRNAs in response to CLso and psyllid infection at the terminal stage of ZC disease in potato.

a Differential expression profile of lincRNAs under CLso(−)/CT, CLso(+)/CT, and CLso(+)/CLso(−) conditions at 7 days after infection (DAI), 14 DAI, and 21 DAI is shown via heatmap. The scale represents differential expression in log2 fold change. b The similarity/difference among the nine sets of differentially expressed lincRNAs described in (a) is shown via a PCA plot. Each data point represents the differential expression status of individual lincRNA.
Further, we specifically determined up- and downregulated lincRNAs among the three sets of comparisons [CLso(−)/CT, CLso(+)/CT, and CLso(+)/CLso(−)] within each stage of infection. For instance, at 7 DAI, 8 and 11 lincRNAs were specifically up- and downregulated under CLso(−)/CT condition, suggesting a psyllid-specific response. Likewise, four and six lincRNAs exhibited up- and downregulation under CLso(+)/CT condition at 7 DAI, suggesting either a psyllid or CLso-specific response. However, only one lincRNA showed upregulation under the CLso(+)/CLso(−) condition at 7 DAI (Fig. 4a; Supplementary Data 4), suggesting a CLso-specific response. Similarly, quite a few differentially expressed lincRNAs exhibiting specific expression in the three sets of comparisons were detected at 14 DAI, too (Fig. 4b; Supplementary Data 4). At 21 DAI, the number of specifically up- and downregulated lincRNAs was substantially high under CLso(+)/CT [up: 133 and down: 161] followed by CLso(+)/CLso(−) condition [up: 20 and down: 34] (Fig. 4c; Supplementary Data 4). These results suggest a dynamic regulation of lincRNAs at the subsequent stages of ZC disease in response to CLso infection preferentially at 21 DAI. Furthermore, we examined the PCGs located in the proximal regions of the specifically up- and downregulated lincRNAs under CLso(+)/CT condition at 21 DAI as they constitute a major fraction (294) of the differentially expressed lncRNAs. Intriguingly, PCGs encoding TFs (HBP and HAT), signaling transduction components (auxin/ethylene signaling genes and serine/threonine protein kinase), and those involved in stress responses, including, cytochrome P450 monooxygenase CYP736B, disease resistance protein BS2, glutathione S-transferase (GST), lipoxygenase, resistance protein PSH-RGH6 and SAUR family protein were detected at the proximal regions of the differentially expressed lncRNAs. The dynamic expression of the lincRNAs may influence the regulation of these PCGs and could impact disease tolerance.

Several common and unique lncRNAs exhibiting differential expression under CLso(−)/CT, CLso(+)/CT, and CLso(+)/CLso(−) conditions at 7 DAI (a), 14 DAI (b), and 21 DAI (c) are depicted via Venn diagrams. Labels on top (black), bottom left (red), and bottom right (blue) indicate the number of differentially expressed, upregulated, and downregulated lncRNAs, respectively.
Transcriptional regulatory networks of potato lincRNAs and protein-coding genes
The interactions of lincRNAs and PCGs are complex and poorly understood. However, a few studies revealed that lincRNAs can also influence the expression of their proximal PCGs4,10. We examined to identify sets of co-up and co-downregulated lincRNAs–PCGs pairs located proximal to each other under CLso(−)/CT, CLso(+)/CT, and CLso(−)/CLso(+) conditions at 7, 14, and 21 DAI. We did not observe co-differentially expressed lincRNAs and proximal PCG pairs at 7 and 14 DAI. However, at least 25 co-up and 21 co-downregulated lincRNA and proximal PCG pairs were detected under CLso(+)/CT condition at 21 DAI (Supplementary Data 9). Pearson’s correlation for the set of co-upregulated (r = 0.77, p < 0.01) lincRNA–PCG pairs was higher than the set of co-downregulated pairs (r = 0.31). Among the 46 co-regulated lincRNA–PCG pairs, a majority of them were oriented in opposite directions (29), which were further categorized into convergent (16) and divergent (13) types. Conversely, the remaining 17 lincRNA–PCG pairs were oriented in the same direction. Many of the co-differentially expressed lncRNAs, along with their proximal PCGs, were involved in biotic stress responses38,39,40,41,42,43 such as wound-inducible carboxypeptidase, SAUR, GST, F-box, and chitinases (Supplementary Data 9).
Further, to examine the co-expression networks among transcripts, we performed weighted gene co-expression network analysis (WGCNA)44 to determine sets of co-upregulated transcripts under CLso(−)/CT, CLso(+)/CT, and CLso(+)/CLso(−) conditions at 7, 14, and 21 DAI. At least four distinct modules (turquoise, blue, brown, and gray) representing distinct sets of differentially co-expressed transcripts were detected (Fig. 5a, b). The turquoise module was the largest among the different modules and comprised 331 transcripts (lincRNAs and PCGs) (Supplementary Data 5). The turquoise module was significantly associated with the CLso(+) treatment at 21 DAI (r = 0.93 and p = 1e − 12) (Fig. 5b). Further, we examined the functional relevance of the co-expressed transcripts belonging to the turquoise module by determining the enrichment of gene family and gene ontology (GO) terms using the corresponding PCGs as input. Functional terms related to acyl lipid metabolism superfamily, APETALA2 (AP2)/ethylene-responsive element binding proteins (EREBP), nucleoside transporter, GST, protein kinase and small auxin upregulated RNA (SAUR) processes were significantly enriched (Supplementary Data 10). Previous studies have shown that genes encoding AP2/EREBP, GST, and SAUR play crucial roles in plant defense against pathogen invasion41,42,43. In addition, GO terms involved in cell wall modification (GO:0009827), defense response to bacteria (GO:1900424), ion transport (GO:0006811), DNA binding (GO:0003677), glutathione peroxidase activity (GO:0004602), cell wall (GO:0005618), and plasma membrane (GO:0005886) were found to be significantly enriched (Supplementary Data 10). The results suggest the possible contribution of lincRNAs in governing different biological processes involved in biotic stress responses.

a A hierarchical clustering of the transcripts exhibiting a co-expression network based on topological overlap dissimilarity is shown via dendrogram. b Different sets (modules) of co-expressed transcripts in CT, CLso(−), and CLso(+) conditions at 7 DAI, 14 DAI, and 21 DAI are shown via heatmap. Labels in each module indicate Pearson’s correlation (R) and significance level (p value). The scale on the right side indicates Pearson’s correlation (R). c A co-expression network of the topmost 24 hub transcripts belonging to the turquoise module is shown. Light-green triangles indicate lncRNAs specific to CLso. Likewise, pink and blue ovals indicate lncRNAs and PCGs, respectively.
Further, we examined regulatory networks among the transcripts of either lincRNAs or PCGs within the turquoise module to identify hub transcripts. A total of 24 transcripts exhibited the most interactions among themselves, and we considered them hub transcripts. Among them, 11 transcripts belonged to lincRNAs, including seven lincRNAs specific to CLso (Fig. 5c). The remaining 13 transcripts were PCGs, including genes encoding expansin, GST, and SAUR. The role of expansin and SAUR genes was implicated in defense responses against different types of biotic and abiotic stresses43,45. Overexpression of expansin genes led to increased tolerance in response to varying abiotic stresses, such as heat, drought, and salinity45. In plants, the accumulation of reactive oxygen species and programmed cell death processes has been implicated in response to pathogen invasion. Glutathione peroxidase activity encoded by GSTs inhibits the spread of cell death in infected plants46. Likewise, the function of SAUR genes in cell wall biosynthesis has been implicated in biotic stress responses43,47. The results suggest an intricate network between coding and non-coding transcripts preferentially at the later stage (21 DAI) of ZC disease progression.
Overexpression of lincRNAs confers potato tolerance to CLso
Next, we sought to characterize if lincRNAs can mediate resistance to CLso infection in potatoes. Four lincRNAs were selected with criteria of (1) co-upregulated along with their proximal PCGs, (4) belonging to the hub transcripts, and (3) progressively induced (7 and 21 DAI) during the progression of ZC disease. The respective lincRNAs were cloned in an overexpression vector under the control of a double-enhanced CaMV35S promoter (DE35S-P). Moreover, the vector harbored a GFP reporter (Fig. 6a). The constructs were used for Rhizobium rhizogenes (R. rhizogenes)-mediated potato hairy root transformation48. After 28 days post-transformation, transgenic hairy roots were analyzed to confirm the overexpression of the four lincRNAs via RT-qPCR and GFP marker fluorescence (Fig. 6b, c; Supplementary Data 6). The transgenic hairy roots showed significantly higher expression of the lincRNAs than control plants (Fig. 6c; Supplementary Data 6).

a Schematic diagram showing plasmid vector employed for overexpression (OE) of the selected lincRNAs. b Photographs showing the phenotype of the transformed potato plants at 28 d post-transformation and GFP fluorescence. Scale bars indicate 1 cm (left) and 10 µm (right). c The relative expression level of the lincRNAs between the CT and OE plants estimated via RT-qPCR is shown in the bar plot. d Likewise, the relative expression level of the proximal PCGs located either in up- and downstream regions of the lincRNAs between CT and OE plants estimated via RT-qPCR is shown in bar plots. e Relative Candidatus Liberibacter spp. (CLso) titer between CT and the OE plants estimated via qPCR is shown via bar plot. Asterisks represent statistically significant (p value ≤ 0.05) differences compared to control (CT) as estimated by Student’s t-test. The error bars represent the standard error among the three biological replicates.
To examine the influence of overexpression of the lincRNAs on their proximal PCGs, we selected three PCGs, each located in the upstream and downstream regions of the lincRNA. The expression level of these proximal PCGs was compared between the control and overexpression lines via RT-qPCR. We observed a predominantly negative correlation between higher expression of the lincRNAs and their proximal PCGs expression. For instance, overexpressing lincRNA.ID.00017498 exhibited downregulation of all the six proximal PCGs compared to their controls. Interestingly, RNAi-mediated knockdown of lincRNA.ID.00017498 showed converse effects on the expression dynamics of the proximal PCGs, implicating a role of the lincRNAs in regulating PCG gene expression (Supplementary Fig. 2; Supplementary Data 7). Likewise, overexpression of lincRNA.ID.00013924 reduced the expression of the most proximal PCGs except one located in the upstream region. Conversely, the influence of overexpressing lincRNA.ID.00017902 and lincRNA.ID.00013620 represented both higher and lower expression of their proximal PCGs (Fig. 6d; Supplementary Data 6). The results suggest that lincRNAs could govern the expression level of their proximal PCGs both negatively and positively. Previous studies have also shown that lincRNAs can influence the expression of their proximal PCGs4,10. The functional annotations of the proximal PCGs, with altered expression in the lincRNA-overexpressed hairy roots, included genes encoding proteinases, ubiquitin-protein ligase components, B2 proteins, and ribosomal proteins. Many of these genes could impact plant biotic stress responses49,50,51. Next, we determined whether overexpression of candidate lincRNAs impacted potato tolerance to CLso. Overexpression of three of the four lincRNA exhibited significantly lower CLso titers in transgenic hairy roots compared to control roots transformed with an empty vector. The amplitude of CLso titer reduction was about sixfold in lincRNA.ID.0007902 and lincRNA.ID.0007498 overexpressing hairy roots, while about halffold in the lincRNA.ID.0003924 hairy roots (Fig. 6e; Supplementary Data 6). The results suggest that these three lincRNAs could also impact CLso accumulation.
Discussion
Long intergenic RNAs are emerging as crucial players in the transcriptome programming of PCGs during various plant and animal developmental processes and stress responses. However, the study of lncRNAs is largely underrepresented, although they can be identified from the transcript sequences obtained from RNA-Seq data, a few studies employed strand-specific RNA sequencing to identify natural antisense transcripts (lincNATs)7,8,22. Intriguingly, lincNATs were found to be up to 20% in the Arabidopsis PCGs, implicating a crucial role of lncRNAs22. The repertoire of lncRNAs is further expanded due to variable regions of origin such as lincRNAs and intronic lincRNAs4,7,8, and also due to polymorphic alternatively spliced variants. Although less attention has been paid to lncRNAs earlier, leveraging the availability of a large number of RNA-Seq data can be employed to identify and unravel the roles of lncRNAs in different biological processes. A few plant lncRNAs have been characterized, and their roles are crucial in plant growth and development. For instance, vernalization-induced expression of the COLDAIR lncRNA triggered flower initiation by repressing the FLC locus by recruiting polycomb repressive complex 252. Another classic example is lncRNA PS1, which competes with PHO2 mRNA due to harboring a competing binding site of a miRNA (ath-miR399). Eventually, post-transcriptional degradation of the PHO1 mRNA is rescued under low phosphate stimulus in Arabidopsis37. LncRNAs can also govern the spread of heterochromatization to the neighboring genomic/chromatin region52. Prioritization of candidate lncRNAs is crucial to studying the role of lncRNAs. Therefore, genome-wide identification followed by assessment of dynamic regulation are prerequisites for understanding lncRNAs. Moreover, the role of lncRNAs in response to pathogens is largely unknown.
Here, we identified potato lincRNAs and studied their role in response to CLso infection. Our analysis identified ~4397 lincRNAs in healthy and infected potatoes at different stages of ZC disease progression. The fidelity of the identified lincRNAs was corroborated by shorter transcript lengths and structural features of the exons and introns33,53. The number of detected lincRNAs in this study was substantially higher than previously reported potato lncRNAs17,21. This could be due to the activation of several lincRNAs that respond primarily to the pathogen (CLso) and the insect vector (psyllid). Therefore, our study expanded the potato lincRNAs repertoire by uncovering novel plant–pathogen/insect-responsive lncRNAs.
Furthermore, by sequence comparisons, we identified six potato lincRNAs whose sequences had complementarity to known potato miRNAs (Supplementary Data 8). Previous reports showed that a few lncRNAs may act as potential decoys of miRNAs, thus influencing the miRNA primary target PCG(s) expression or protein accumulation. Interestingly, a lincRNA (lincRNA.ID.00002569), with a sequence similarity to a potato miRNA (stu-miR8005a), was downregulated in CLso infection and paralleled the expression of a PCG target encoding a Multidrug resistance pump protein (PGSC0003DMT400011415) (Supplementary Fig. 1). Although further biochemical and functional studies are needed to understand the cellular interactions of the lincRNAs, miRNAs, and the target PCGs, our results implicate the role of potato lincRNAs as potential miRNA decoys.
It is well established that the host immune responses against pathogen infection can be dynamic and oscillate during the disease progression54,55. Recent studies have implicated lincRNAs in regulating plant defense hormone pathways56,57. Here, we analyzed differentially expressed lincRNAs in nine different sets of comparisons described earlier. The majority, approximately 86.5% (671), of the differentially expressed lincRNAs were detected at 21 DAI stage under CLso+ condition [CLso(+)/CT]. Conversely, the fraction of differentially expressed lincRNAs under psyllid alone condition at 21 DAI [CLso(−)/CT] was substantially low (97; 12.5%). This suggests that most of the unique lincRNAs that are differential regulated are primarily a response to pathogen infection with greater preference under CLso stimulus at 21 DAI.
The role of lincRNAs in influencing the expression of PCGs is complex and poorly understood. Growing evidence showed that lincRNAs can govern nearby PCGs by proliferating similar chromatin structures locally10,58. Here, we identified several lincRNA-proximal PCGs that appear co-regulated with the lincRNAs (Supplementary Data 9). These PCGs had putative functions in biotic stress response, such as chitinase, GST, F-box, and wound-inducible carboxypeptidase. Likewise, using weighted gene co-expression analysis, we identified another set of PCGs co-expressed with lincRNAs (Fig. 5). The most significant and highest correlated module was detected under the CLso(+) condition at 21 DAI. These PCGs include EREBP, GST, and SAURs (Supplementary Data 10)41,42,59. These results suggest that the potato lincRNAs may influence the expression of PCGs involved in biotic stress and plant signaling.
The role of selected lincRNAs in potato defenses was evaluated by overexpressing four candidates in transgenic potato hairy roots (Fig. 6a–c). The overexpression of the lincRNAs modulated the expression of their respective neighboring PCGs (Fig. 6d). Interestingly, our analysis indicates that these disease-associated lincRNAs could predominantly act as transcriptional repressors of PCGs. Strikingly, we observed a significant reduction of CLso titer when three of the four lincRNA candidates were overexpressed in plant tissues (Fig. 6e), implicating these lincRNAs in resistance to CLso. These lincRNA–PCG modules are prime candidates for further mechanistic studies to determine their mode of action in disease resistance.
Methods
Plant materials, inoculation with pathogens, and RNA sequencing
Potato (S. tuberosum L. var. Atlantic) plants were grown inside growth chambers at 20 °C, 50% relative humidity with a periodic setting of 14 h light and 10 h dark. About 4–6 weeks old plants were challenged with psyllid alone [B. cockerelli (Šulc)] [CLso(−)] and CLso vectored by the psyllid [CLso(+)]. The control (CT) plants were unchallenged plants. Young leaf tissues from CT, [CLso(−)], and [CLso(+)] potato plants were collected at 7, 14, and 21 DAI in three independent biological replicates. The leaf tissues were immediately frozen in liquid nitrogen and stored at −80 °C until further use. Total RNA was extracted from the leaf samples using the Direct-zol RNA Miniprep kit (Zymo Research, Irvine, CA). The quality of the RNA was estimated by analyzing the absorbance ratio at 260/230, and the quantity was determined using a NanoDrop-1000 spectrophotometer (Thermo Fisher Scientific, Waltham, MA). RNA-seq library preparation and sequencing was performed at the Texas A&M AgriLife Genomics and Bioinformatics Services (College Station, TX). Libraries were prepared using TruSeq Stranded Library Synthesis with Ribo depletion (Illumina, San Diego, CA). Libraries were multiplexed and sequenced using a HiSeq 4000 instrument (Illumina, San Diego, CA) to obtain 150 bp long paired-end reads.
Identification and differential expression analysis of lincRNAs
The raw paired-end reads were processed to remove low-quality reads, adapter sequences, and reads containing excessive uncalled bases60. The filtered high-quality reads were mapped to the S. tuberosum reference genome (DM v4.04)27 using HISAT261,62. The SAM files were converted into sorted BAM files using SAMtools (v1.3.1)63. LincRNAs were identified from the mapped BAM files via modules implemented in Read Mapping-Transcript Assembly-Evolinc29. The high-quality reads mapped uniquely to the reference genome were used to estimate read counts via the featureCounts tool64. Subsequently, the read count matrixes of two samples (conditions) were used as input to identify differentially expressed lincRNAs using DESeq2 (v1.28.1)65. In total, nine different sets of differentially expressed lincRNAs were determined under CLso(−)/CT, CLso(+)/CT, and CLso(+)/CLso(−) conditions each at 7 DAI, 14 DAI, and 21 DAI. Transcripts with ≥1 and ≤ −1 log2 fold change and <0.05 p value adjusted with the Benjamini–Hochberg method were considered as differentially expressed. Similarity/difference among the nine sets of differentially expressed lincRNAs was analyzed using PCA with factoextra (v1.0.7) and FactoMineR (v2.4) packages implemented in the R program. We used the log2 transformed differential expression values of the nine comparisons under the CLso(+)/CT, CLso(+)/CT, and CLso(+)/CLso(−) conditions at 7, 14, and 21 DAI for the PCA analysis. To examine common and unique sets of up- and downregulated lincRNAs within each disease stage, we employed the Venn Diagram (v1.6.20) package implemented in the R program. The Nextflow pipeline was run on the Texas A&M High-Performance Research Computing (Grace) cluster with 50 computing nodes (48 cores and 384 GB RAM for each node).
Analysis of lincRNAs harboring binding sites of microRNAs (miRNAs)
The lincRNAs that act as potential miRNA decoys (miRNA target mimics) were predicted using the psMimic standalone tool36. The parameters used for miRNA decoy prediction include no more than four mismatches and two mismatches allowed between the second and eighth positions of miRNA. The putative target of identified miRNAs was identified using the psRNATarget36 using potato transcriptome sequences (Phytozome 13, 448_v4.03). We selected the top transcript targets based on the expectation I value (<3). Subsequently, we examined the expression level of those lincRNAs and PCGs in CT, CLso(−), and CLso(+) conditions at 21 DAI via RT-qPCR, and the primer sequences have been provided in Supplementary Table 2.
Differential co-expression network analysis
To identify co-up and co-downregulated lincRNA and PCG pairs under CLso(−)/CT, CLso(+)/CT, and CLso(+)/CLso(−) conditions at 7 DAI, 14 DAI, and 21 DAI, WGCNA was performed44. The log2 fold change differential expression values of the 9 comparisons were used as an input with the criteria of soft thresholding power of 5 and minimum size of 30 using the dynamic tree cut method. Distinct sets/modules of co-up and co-downregulated lincRNAs and PCGs pairs were visualized using plotDendroAndColors. Moreover, the topmost 24 hub transcripts (either lincRNAs or PCGs) based on the highest number of interactions found within the turquoise module were identified and displayed in Cytoscape (v3.8.2)66. Further, we examined the functional relevance of the co-up and co-downregulated PCGs via enrichment analysis of GO terms and gene family via GenFam tool (Supplementary Data 10)67,68.
Overexpression and knockdown of lincRNAs in potato hairy roots
For overexpression, the selected four lincRNAs were commercially synthesized and cloned at StuI and KpnI restriction sites in a binary vector containing the GFP reporter gene (pBIN-mGFP) under the control of a double-enhanced CaMV35S promoter (DE35S-P) and 35S terminator (35S-T). The constructs were subsequently used for R. rhizogenes-mediated potato hairy root transformation48. For RNAi-mediated knockdown of lincRNA, a hairpin RNA containing a PDK (pyruvate orthophosphate dikinase) intron flanked by the sense and antisense lincRNA sequence was commercially synthesized and cloned at StuI and KpnI restriction sites in the pBIN-mGFP binary vector. Briefly, CLso-infected potato shoots were transformed with either the lincRNA overexpression or RNAi constructs and the empty vector (GFP alone) using R. rhizogenes (ATCC 43056). The composite plants’ newly formed transgenic hairy roots were verified by microscopy based on GFP fluorescence at ~28 days post-transformation. RT-qPCR confirmed the overexpression or knockdown of the lincRNAs in the transformed hairy roots. Total RNA was extracted from the transgenic hairy roots (three biological replicates) using the Direct-zol RNA Miniprep Plus kit (Zymo Research, Irvine, CA). First-strand cDNAs were synthesized from 1.0 µg of total RNA using Superscript IV First-Strand Synthesis System (Thermo Fisher Scientific, Waltham, MA) followed by qPCR analysis. Further, the expression of the six nearest PCGs (three each located in up- and downstream regions of the lincRNAs) was analyzed by RT-qPCR to determine their expression changes.
RT-qPCR and qPCR analysis
To examine the expression level of lincRNAs and PCGs, we employed RT-qPCR. Primers were designed using NCBI and Primer3web (v4.1.0)69 (Supplementary Tables 3, 4). Relative CLso titer was estimated at ~28 days post-transformation using qPCR. All qPCR and RT-qPCR reactions were performed using iTaq SYBR Green Supermix (Bio-Rad Laboratories, Hercules, CA) with three biological and two technical replicates. The potato housekeeping gene RPL2 was used as an internal reference. Relative expression and bacterial titers were analyzed using the ∆∆Ct method.
Statistics and reproducibility
The details of the statistical tests to identify the significance level of differences have been described in the “Results”, “Methods”, and figure legends. The RNA-seq data from nine different samples under CT, CLso(−), and CLso(+) conditions at 7 DAI, 14 DAI, and 21 DAI in three independent biological replicates were processed to identify lincRNAs. The differential expression of the lincRNAs between two samples/conditions was detected using default algorithms implemented in DESeq2 (v1.28.1). Furthermore, distinct modules of co-expressed transcripts were identified with default algorithms implemented in WGCNA. The significance of the difference in the expression level of the lincRNAs and their proximal PCGs between the wild type and mutant plants (overexpression/RNAi) was detected using Student’s t-test.
Reporting summary
Information on the research design and ethics is available in the Nature Research Reporting Summary linked to this article.
Code availability
We have not used in-house or custom codes/algorithms to draw the conclusions of the study. The details of the publicly available software and parameters employed in this study have been described in the “Methods” section.
References
Nelson, A. D. L. et al. A genomic analysis of factors driving lincRNA diversification: lessons from plants. G3 Genes Genom. Genet. 6, 2881–2891 (2016).
Wang, K. C. & Chang, H. Y. Molecular mechanisms of long noncoding RNAs. Mol. Cell 43, 904–914 (2011).
Li, L. et al. Genome-wide discovery and characterization of maize long non-coding RNAs. Genome Biol. 19, 13059 (2018).
Xu, W. et al. Differential expression networks and inheritance patterns of long non-coding RNAs in castor bean seeds. Plant J. 95, 324–340 (2018).
Zhang, C. et al. Long non-coding RNAs of switchgrass (Panicum virgatum L.) in multiple dehydration stresses. BMC Plant Biol. 18, 79 (2018).
Qi, X., Xie, S., Liu, Y., Yi, F. & Yu, J. Genome-wide annotation of genes and noncoding RNAs of foxtail millet in response to simulated drought stress by deep sequencing. Plant Mol. Biol. 83, 459–473 (2013).
Wang, H. et al. Genome-wide identification of long noncoding natural antisense transcripts and their responses to light in Arabidopsis. Genome Res. 24, 444–453 (2014).
Zhao, X. et al. Global identification of Arabidopsis lncRNAs reveals the regulation of MAF4 by a natural antisense RNA. Nat. Commun. 9, 5056 (2018).
Ransohoff, J. D., Wei, Y. & Khavari, P. A. The functions and features of intergenic lncRNAs. Physiol. Behav. 176, 100–106 (2016).
Gil, N. & Ulitsky, I. Regulation of gene expression by cis-acting long non-coding RNAs. Nat. Rev. Genet. 21, 102–117 (2020).
Golicz, A. A., Singh, M. B. & Bhalla, P. L. The long intergenic noncoding RNA (LincRNA) Landscape of the soybean genome. Plant Physiol. 176, 2133–2147 (2018).
Hon, C. C., Ramilowski, J. A. & Harshbarge, J. An atlas of human long non-coding RNAs with accurate 5′ ends. Nature 543, 199–204 (2017).
Derrien, T. et al. The GENCODE v7 catalog of human long noncoding RNAs: analysis of their gene structure, evolution, and expression. Genome Res. 22, 1775–1789 (2012).
Blein, T. et al. Landscape of the noncoding transcriptome response of two Arabidopsis ecotypes to phosphate starvation. Plant Physiol. 183, 1058–1072 (2020).
Severing, E. et al. Arabidopsis thaliana ambient temperature responsive lncRNAs. BMC Plant Biol. 18, 145 (2018).
Kindgren, P., Ard, R., Ivanov, M. & Marquardt, S. Transcriptional read-through of the long non-coding RNA SVALKA governs plant cold acclimation. Nat. Commun. 9, 4561 (2018).
Kwenda, S., Birch, P. R. & Moleleki, L. N. Genome-wide identification of potato long intergenic noncoding RNAs responsive to Pectobacterium carotovorum subspecies brasiliense infection. BMC Genomics 17, 614 (2016). 18.
Shuai, P. et al. Genome-wide identification and functional prediction of novel and drought-responsive lincRNAs in Populus trichocarpa. J. Exp. Bot. 65, 4975–4983 (2014).
Zhang, W. et al. Identification of maize long non-coding RNAs responsive to drought stress. PLoS ONE 9, e98958 (2014).
Chung, P. J. et al. Transcriptome profiling of drought responsive noncoding RNAs and their target genes in rice. BMC Genomics 17, 563 (2016).
Hou, X. et al. Genome-wide analysis of long non-coding RNAs in potato and their potential role in tuber sprouting process. Int. J. Mol. Sci. 19, 101 (2018).
Zhu, Q. H., Stephen, S., Taylor, J., Helliwell, C. A. & Wang, M. B. Long noncoding RNAs responsive to Fusarium oxysporum infection in Arabidopsis thaliana. New Phytol. 201, 574–584 (2014).
Joshi, R. K., Megha, S., Basu, U., Rahman, M. H. & Kav, N. N. Genome wide identification and functional prediction of long non-coding RNAs responsive to sclerotinia sclerotiorum infection in Brassica napus. PLoS ONE 11, e0158784 (2016).
Wang, J. et al. Genome-wide analysis of tomato long non-coding RNAs and identification as endogenous target mimic for microRNA in response to TYLCV infection. Sci. Rep. 5, 16946 (2015).
Hameed, A., Zaidi, S. Se. A., Shakir, S. & Mansoor, S. Applications of new breeding technologies for potato improvement. Front. Plant Sci. 9, 925 (2018).
Levy, J. G., Mendoza, A., Miller, J. C., Tamborindeguy, C. & Pierson, E. A. Global gene expression in two potato cultivars in response to ‘Candidatus Liberibacter solanacearum’ infection. BMC Genomics 18, 960 (2017).
Xu, X. et al. Genome sequence and analysis of the tuber crop potato. Nature 475, 189–195 (2011).
Peri, S. et al. Read mapping and transcript assembly: a scalable and high-throughput workflow for the processing and analysis of ribonucleic acid sequencing data. Front. Genet. 10, 1361 (2019).
Nelson, A. D. L. et al. A tool for the identification and evolutionary comparison of long intergenic non-coding RNAs. Front. Genet. 8, 52 (2017).
Ulitsky, I. & Bartel, D. P. LincRNAs: genomics, evolution, and mechanisms. Cell 154, 26–46 (2013).
Statello, L., Guo, C. J., Chen, L. L. & Huarte, M. Gene regulation by long non-coding RNAs and its biological functions. Nat. Rev. Mol. Cell Biol. 22, 96–118 (2021).
Ransohoff, J. D., Wei, Y. & Khavari, P. A. The functions and unique features of long intergenic non-coding RNA. Nat. Rev. Mol. Cell Biol. 19, 143–157 (2018).
Wang, H. L. V. & Chekanova, J. A. Long noncoding RNAs in plants. Adv. Exp. Med. Biol. 1008, 133–154 (2017).
Fan, C., Hao, Z., Yan, J. & Li, G. Genome-wide identification and functional analysis of lincRNAs acting as miRNA targets or decoys in maize. BMC Genomics 16, 793 (2015).
Franco-Zorrilla, J. M. et al. Target mimicry provides a new mechanism for the regulation of microRNA activity. Nat. Genet. 39, 1033–1037 (2007).
Wu, H. J., Wang, Z. M., Wang, M. & Wang, X. J. Widespread long noncoding RNAs as endogenous target mimics for microRNAs in plants. Plant Physiol. 161, 1875–1884 (2013).
Kuo, H. F. & Chiou, T. J. The role of microRNAs in phosphorus deficiency signaling. Plant Physiol. 156, 1016–1024 (2011).
Quilis, J. et al. A potato carboxypeptidase inhibitor gene provides pathogen resistance in transgenic rice. Plant Biotechnol. J. 5, 537–553 (2007).
Maldonado-Calderón, M. T., Sepúlveda-García, E. & Rocha-Sosa, M. Characterization of novel F-box proteins in plants induced by biotic and abiotic stress. Plant Sci. 186, 208–217 (2012).
Ali, M. et al. Chitinase gene positively regulates hypersensitive and defense responses of pepper to Colletotrichum acutatum infection. Int. J. Mol. Sci. 21, 6624 (2020).
Najafi, S., Sorkheh, K. & Nasernakhaei, F. Characterization of the APETALA2/Ethylene-responsive factor (AP2/ERF) transcription factor family in sunflower. Sci. Rep. 8, 16–18 (2018).
Han, Q. et al. A glutathione S-transferase gene from Lilium regale Wilson confers transgenic tobacco resistance to Fusarium oxysporum. Sci. Hortic. 198, 370–378 (2016).
Deng, G. et al. Identification and expression of saur genes in the cam plant Agave. Genes 10, 555 (2019).
Langfelder, P., Horvath, S. & WGCNA An R package for weighted correlation network analysis. BMC Bioinform. 9, 559 (2008).
Chen, Y. et al. A comprehensive expression analysis of the expansin gene family in potato (Solanum tuberosum) discloses stress-responsive expansin-like B genes for drought and heat tolerances. PLoS ONE 14, 0219837 (2019).
Gullner, G., Komives, T., Király, L. & Schröder, P. Glutathione S-transferase enzymes in plant-pathogen interactions. Front. Plant Sci. 871, 1836 (2018).
Wong, J. H., Spartz, A. K., Park, M. Y., Du, M. & Gray, W. M. Mutation of a conserved motif of PP2C.D phosphatases confers SAUR immunity and constitutive activity. Plant Physiol. 181, 353–366 (2019).
Irigoyen, S. et al. Plant hairy roots enable high throughput identification of antimicrobials against Candidatus Liberibacter spp. Nat. Commun. 11, 5802 (2020).
Ying, M. Role of HECT ubiquitin protein ligases in Arabidopsis thaliana. J. Plant Sci. Phytopathol. 2, 20–30 (2018).
Carter, A. P. et al. Functional insights from the structure of the 30S ribosomal subunit and its interactions with antibiotics. Nature 407, 340–348 (2000).
Figueiredo, L., Santos, R. B. & Figueiredo, A. Defense and offense strategies: the role of aspartic proteases in plant–pathogen interactions. Biology 10, 75 (2021).
Kim, D. H., Xi, Y. & Sung, S. Modular function of long noncoding RNA, COLDAIR, in the vernalization response. PLoS Genet. 13, e1006939 (2017).
Chodroff, R. A. et al. Long noncoding RNA genes: conservation of sequence and brain expression among diverse amniotes. Genome Biol. 11, R72 (2010).
Jones, J. D. G. & Dangl, J. L. The plant immne syetem. Nature 444, 323–329 (2006).
Ngou, B. P. M., Ding, P. & Jones, J. D. G. Thirty years of resistance: Zig-zag through the plant immune system. Plant Cell 34, 1447–1478 (2022).
Liu, N. et al. A lncRNA fine-tunes salicylic acid biosynthesis to balance plant immunity and growth. Cell Host Microbe 30, 1124–1138.e8 (2022).
Huang, J., Zhou, W., Zhang, X. & Li, Y. Roles of long non-coding RNAs in plant immunity. PLoS Pathog. 19, e1011340 (2023).
Greenstein, R. A. et al. Noncoding RNA-nucleated heterochromatin spreading is intrinsically labile and requires accessory elements for epigenetic stability. Elife 7, e32948 (2018).
Ghanashyam, C. & Jain, M. Role of auxin-responsive genes in biotic stress responses. Plant Signal. Behav. 4, 846–848 (2009).
Bedre, R., Avila, C. & Mandadi, K. HTSQualC is a flexible and one-step quality control software for high-throughput sequencing data analysis. Sci. Rep. 11, 18725 (2021).
Pertea, M., Kim, D. & Pertea, G. Transcript-level expression analysis of RNA-seq experiments with HISAT, StringTie, and Ballgown. Nat. Protoc. 11, 1650–1667 (2016).
Kim, D., Langmead, B. & Salzberg, S. L. HISAT: a fast spliced aligner with low memory requirements. Nat. Methods 12, 357–360 (2015).
Li, H. et al. The sequence alignment/map format and SAMtools. Bioinformatics 25, 2078–2079 (2009).
Liao, Y., Smyth, G. K. & Shi, W. FeatureCounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 30, 923–930 (2014).
Love, M. I., Huber, W. & Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15, 550 (2014).
Demchak, B. et al. Cytoscape: the network visualization tool for GenomeSpace workflows. F1000Res. 3, 151 (2014).
Ashburner, M., Ball, C. A. & Blake, J. A. Gene ontology: tool for the unification of biology. Nat. Genet. 25, 25–29 (2000).
Bedre, R. & Mandadi, K. GenFam: a web application and database for gene family-based classification and functional enrichment analysis. Plant Direct 3, 1–7 (2019).
Untergasser, A. et al. Primer3-new capabilities and interfaces. Nucleic Acids Res. 40, e115 (2012).
Acknowledgements
This study was supported in part by funds from USDA-NIFA (2021-70029-36056, HATCH TEX09621), Texas A&M AgriLife Research Insect-vectored Disease Seed Grants (124190-96210), Texas A&M AgriLife Institute for Advancing Health Through Agriculture to K.M. We acknowledge the support of the Texas A&M AgriLife Genomics and Bioinformatics Services for the RNA sequencing and the Texas A&M High-Performance Research Computing (HPRC) for bioinformatics resources. We thank Denise Rossi and Victoria Garza (Texas A&M AgriLife Research) for their technical assistance with plant and insect propagation.
Author information
Authors and Affiliations
Contributions
K.M. conceived and supervised the project. R.B., A.N., N.R.K., M.R., M.S.R., and S.I. performed and interpreted the experiments. All authors contributed to the preparation of the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Communications Biology thanks Erik Alexandersson and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. Primary Handling Editors: Leena Tripathi and David Favero. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Bedre, R., Kavuri, N.R., Ramasamy, M. et al. Long intergenic non-coding RNAs modulate proximal protein-coding gene expression and tolerance to Candidatus Liberibacter spp. in potatoes. Commun Biol 7, 1095 (2024). https://doi.org/10.1038/s42003-024-06763-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s42003-024-06763-9
- Springer Nature Limited