
Abstract
Measuring neuronal activity is important for understanding neuronal function. Ca2+ imaging by genetically encoded calcium indicators (GECIs) is a powerful way to measure neuronal activity. Although it revealed important aspects of neuronal function, measuring the neuronal membrane voltage is important to understand neuronal function as it triggers neuronal activation. Recent progress of genetically encoded voltage indicators (GEVIs) enabled us fast and precise measurements of neuronal membrane voltage. To clarify the relation of the membrane voltage and intracellular Ca2+, we analyzed neuronal activities of olfactory neuron AWA in Caenorhabditis elegans by GCaMP6f (GECI) and paQuasAr3 (GEVI) responding to odorants. We found that the membrane voltage encodes the stimuli change by the timing and the duration by the weak semi-stable depolarization. However, the change of the intracellular Ca2+ encodes the strength of the stimuli. Furthermore, ODR-3, a G-protein alpha subunit, was shown to be important for stabilizing the membrane voltage. These results suggest that the combination of calcium and voltage imaging provides a deeper understanding of the information in neural circuits.

Similar content being viewed by others


Introduction
Measuring neuronal activities in non-invasive unanesthetized animals is important for understanding neuron function. Recent progress in genetically encoded calcium indicators (GECI) has allowed simultaneous measurement of the activities of multiple neurons in living animals1,2,3,4,5. Among those, GCaMPs are GECIs applied to animal models1,3,6. Improved GECIs have faster time constants with strong fluorescence and high dynamic ranges to detect faster Ca2+ changes7,8. However, as changes of membrane voltage are followed by intracellular Ca2+ changes, measuring the membrane voltage would be theoretically advantageous for analyzing neuronal activities. Therefore, to better understand neuronal activation, membrane voltage measurements are essential.
The membrane voltage has been typically investigated using electrophysiological techniques9, while, for live imaging, genetically encoded voltage indicators (GEVIs) were developed using voltage-sensitive proteins10,11. Although these GEVIs are bright, their time constants were not high enough for sensitive detection of fast voltage changes10,12. In this decade, new GEVIs derived from 7TM proteins, such as Arch for optogenetics, were developed12,13,14, although these GEVIs show dim fluorescence. Together with recent developments in image processing and detector systems, these GEVIs allow fast voltage changes to be detected in non-invasive conditions. Particularly, paQuasAr313 and SomArchon4 are very useful for the analyses because of their large dynamic ranges and fast time constants. In addition, the fluorescence of these GEVIs derived from Arch is in the near far-red so that it can be easily distinguished from that of the GCaMP. Therefore, these kinds of GEVIs are suitable for the simultaneous, non-invasive measurement of Ca2+ and membrane voltage in living animals15.
In various model organisms, including C. elegans, neuronal activities in non-invasive and unanesthetized conditions have been extensively studied using GECI at single cellular and multicellular levels16,17. However, since the Ca2+ change and membrane voltage relationships are not yet fully understood, simultaneous measurement of Ca2+ and membrane voltage provides important information to interpret the Ca2+ imaging data and better understand the informational processing in neuronal circuits. In C. elegans, neuron activity has been analyzed using electrophysiology18,19, GECIs16,17, and GEVIs15. Moreover, as their neuronal circuit structure has been thoroughly described at the electron microscopic level20, they are used as a model organism to understand neuronal functions in behavioral and genetic analyses21,22. Although no voltage-dependent Na+ channels exist in the C. elegans genome, voltage-dependent Ca2+ channels function as regulators for membrane voltages and intracellular Ca2+. Indeed, in the olfactory neurons AWA, voltage-dependent Ca2+ channel, EGL-19 is essential for the all-or-none action potentials19, although the regulation of intracellular Ca2+ in neuronal activation can also be regulated through intracellular Ca2+ stores. Therefore, the simultaneous measurements of intracellular Ca2+ and membrane voltage lead to understanding how the Ca2+ signals are regulated through membrane voltage changes.
In this report, in a non-invasive and unanesthetized condition, we analyzed stimulation-dependent voltage changes and Ca2+ changes using paQuasAr313 and GCaMP6f6, respectively. By simultaneously measuring the mutants defective in odor perception, we found that a G-protein, ODR-3, mitigated spontaneous fluctuations in membrane voltage and the subsequent alterations in Ca2+ levels, thereby facilitating adequate responses to odorant stimuli. The simultaneous measurements enabled us to understand the neuronal mechanisms important for the regulation of neural activities.
Results
Simultaneous imaging of GEVI and GECI
We used paQuasAr3 as a GEVI in C. elegans to detect changes in membrane voltage because paQuasAr3 shows a significant fluorescent change according to the membrane voltage change12,13. For GECI, we chose GCaMP6f for the simultaneous imaging. The major time constants of paQuasAr3 are 0.8 ms (τup) and 0.9 ms (τdown)13, whereas those of GCaMP6f are 80 ms (τrise) and 400 ms (τdecay)6. For the simultaneous analyses of the membrane voltage and intracellular Ca2+, animals expressing paQuasAr3 and GCaMP6f in AWA olfactory neurons were produced. These animals could sense attractive odorants like diacetyl and pyrazine in AWA22. Since the changes in membrane voltage induce intracellular Ca2+ changes, Liu et al.19 measured changes induced by current injection in membrane voltage in AWA by electrophysiology and found that the current injection induces the plateau depolarization and the action potentials. The plateau depolarization is also stable unless the action potentials are induced. Furthermore, the higher current injection could not induce action potentials19. Since they could not directly measure the changes in membrane voltage of AWA induced by the odorant stimuli, they deduced those from the changes in GCaMP fluorescence by mathematical deconvolution. Although the mathematical deconvolution of the experimental data on GCaMP can serve to estimate the voltage changes induced by the odorant19, the current injection is an artificial approach to characterize neuronal activities. Therefore, simultaneous real-time imaging data of Ca2+ and membrane voltage dependent on odorants could help understand neuronal activation mechanisms in natural conditions. For expression in AWA, we used the odr-10 promoter. In turn, we used the gpa-4 promoter to analyze the mutant and control animals, as the odr-10 promoter is inactive in some mutants (see Materials and Methods).
Since paQuasAr3 is excited at 640 nm and is further intensified by blue light13, we employed two distinct laser light sources for our simultaneous imaging: one at 470 nm and another at 640 nm. Notably, the 470 nm laser also serves to excite GCaMP6f. In our system, the fluorescence of paQuasAr3 and GCaMP6f were separated by a dichroic mirror and captured every 50 ms (20 Hz) side-by-side in one cMos camera (Fig. 1A, B). The captured images were processed in a pipeline, including region of interest (ROI) detection, ROI tracking, and quantifying fluorescent intensities. In addition, because the degree of photobleaching of paQuasAr3 was substantially more prominent than that of GCaMP6f and each sample showed a slightly different photo-bleaching rate, we applied a nonparametric spectral estimation method called singular spectrum analysis (SSA)23 to correct photo-bleaching (Fig. 1C, Supplementary Fig. 1A). Unlike wavelet analysis or Fourie analysis, SSA is a data-driven approach that does not rely on predefined basis functions or frequency bands. Instead, it adapts to the intrinsic structure of the time series data, likely enabling more flexible photobleaching correction for paQuasAr3. A basic methodology of SSA23 is summarized in the supplementary methods.

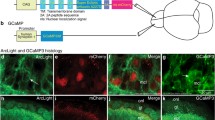
A Young adult animals in an olfactory chip under the microscope captured by cMOS camera through a microscopic system. GCaMP6f and paQuasAr3 were excited by 470 nm and 640 nm lasers, respectively; each fluorescence signal was split by the dichroic mirror (580 nm) and was captured via emission filters (512 and 750 nm, respectively) side-by-side using the cMos camera. B Representative GCaMP6f and paQuasAr3 images expressed in AWA neurons by odr-10 promoter. Yellow squares show the region of interest (ROI) for data analyses. Scale bar indicates 25 μm. C Image processing pipeline. D Dose dependency of GCaMP6f and paQuasAr3 fluorescence imaging to different diacetyl concentrations (10−5–10−9) in wild-type animals. The blue bars indicate the period of the diacetyl stimulation. Average traces in wild-type animals (n > 8) are shown with the standard error of the mean (SEM) as shades. No. of samples were shown in the panels. E Simultaneous imaging of GCaMP6f and paQuasAr3 expressed in AWA neurons of wild-type, N2, and odr-10 animals. Blue bars indicate the period of the diacetyl stimulation. Average traces of wild-type and odr-10 are shown with the standard error of the mean (SEM) as shades. No. of samples were shown in the panels. F Adaptation to diacetyl attenuates the change of Ca2+ and membrane voltages. Box graphs of the maximum responses in naïve and conditioned animals. These graphs are based on the data of (D) and Supplemental Fig. 2A. p-value by T-test is shown in the graphs.
First, by using this system, we carried out simultaneous imaging of GCaMP6f and paQuasAr3 expressed in AWA neurons by odr-10 promoter and found that diacetyl stimulation induced the Ca2+ increase and the membrane depolarization (Fig. 1D). To confirm that these responses are truly caused by diacetyl, sensed through ODR-10 receptor, we analyzed wild-type and odr-10 animals expressing paQuasAr3 and GCaMP6f in AWA neurons driven by the gpa-4 promoter. The fluorescence of paQuasAr3 and GCaMP6f were increased by the diacetyl stimuli in wild-type animals, but not in odr-10 animals, which have defects in the diacetyl receptor gene expressed in AWA (Fig. 1E), suggesting that the ODR-10 receptor is required for the depolarization and Ca2+ increase in AWA triggered by diacetyl. To confirm that this system can discriminate between the fluorescence of paQuasAr3 and GCaMP6f, we captured images from both paths by excitation at 470 nm or 640 nm. In this experiment, we could detect GCaMP6f changes only with the 470 nm excitation and paQuasAr3 changes only with 640 nm (Supplementary Fig. 1B), suggesting that this system can differentiate between the two fluorescence types. Furthermore, to examine whether the expression of GCaMP6f and paQuasAr3 affects the behavioral responses to the odorant, we analyzed the chemotaxis of the strains toward the odorants in the presence or absence of the transgene and found that the transgene did not affect the behavioral responses (Supplementary Fig. 1C). These findings led us to conclude that simultaneous imaging using GCaMP6f and paQuasAr3 detects the changes in intracellular Ca2+ and membrane voltage in AWA, respectively.
The changes of intracellular Ca2+ and membrane voltage by the odor stimulation
To elucidate the membrane voltage and intracellular Ca2+ changes dependent on the odor stimulus, we analyzed the dose responses to diacetyl in this system (Fig. 1D). By using the odr-10 promoter for expression of the reporters, we could measure the intensities of both reporters every 50 ms and found that the stimulation of 10−6 diacetyl induced Ca2+ increase and membrane depolarization and that those signals could not be detected in 10−9 diacetyl stimulation (Fig. 1D). These results suggest that the simultaneous imaging enabled us to analyze the intracellular Ca2+ and the membrane depolarization depending on the stimulation in AWA neurons.
To examine whether simultaneous imaging can detect plastic responses after adaptation, we analyzed AWA responses after adaptation. Like other animals, in C. elegans, exposure to high concentration of odorant induces an adaptation in which the behavioral response is weakened24. We simultaneously measured the membrane voltage and intracellular Ca2+ levels after conditioning for adaptation. Similar to previous reports, after conditioning, the Ca2+ responses in AWA decreased compared to those in naive animals (Fig. 1F, Supplemental Fig. 2A)7. The membrane voltage changes to the odor stimulus were also decreased after the stimulation. These results suggest that, in adapted animals, the responses of membrane voltage to odorants are less sensitive those in naive animals, and thereby the responses of Ca2+ are decreased.
To confirm that the responses were preserved to other odorants, we analyzed the responses to pyrazine, which is also sensed by AWA neurons22. As shown in Supplementary Fig. 2B, the changes in Ca2+ and membrane voltages induced by pyrazine stimulation were similar to those observed with diacetyl.
Fast simultaneous imaging of AWA neurons
Since the action potentials induced by current injections are faster than the signals detected in this condition19, we tested the faster simultaneous recordings, but we could not detect sufficient signals by using the odr-10 promoter because of dim fluorescence. To overcome this, we searched for a strong promoter specific to AWA by CeNGEN, a scRNA database for C. elegans neurons25. Among them, we found that ZC334.13 promoter gives a strong expression specific to AWA (Supplemental Fig. 3A). We made strains expressing GCaMP6f and paQuasAr3 under the ZC334.13 promoter and confirmed that their chemotactic behavior is similar to those of wild-type animals (Supplemental Fig. 1C).
By using this strain, we succeeded in simultaneous detection of the Ca2+ responses and voltage changes to diacetyl every 4 ms (250 Hz) (Fig. 2, Supplemental Fig. 3B, although the noises of the paQuasAr3 are relatively high compared with the slower measurement. In this condition, we measured Ca2+ responses and membrane voltage changes to various concentrations of diacetyl, although the delays of both responses were observed. Since these delays were also observed in the slower imaging, we supposed that these were due to multiple factors including the delay of odor delivery at the switching and the neuronal properties of AWA neurons. Ca2+ responses were induced by the stimulation of diacetyl ≥ 10−8 within a few seconds, and, on average, the stimulation with 10−5–10−6 gave the highest responses. On the measurement of membrane voltages, we could detect the single rapid-sharp depolarization within a few seconds from the onset of the stimulation (Supplemental Fig. 3B) as well as the semi-stable depolarization, which was maintained for the duration of the stimulation (Fig. 2). In the average traces, the stimulation of diacetyl in the range between 10−4 and 10−7 gave both the rapid-sharp depolarization and the semi-stable depolarization. In contrast, the weaker stimulation only gave semi-stable depolarization.

Dose dependency of GCaMP6f and paQuasAr3 at simultaneous fast imaging (250 Hz). The left panels showed the average traces of GCaMP6f and paQuasAr3 in the same panel. Red line with shed (SEM) indicates paQuasAr3 (left axis) and Green line with shed (SEM) indicates GCaMP6f (right axis). No. of samples were shown in the panels. The center panels showed traces of GCaMP6f. The right panels showed traces of paQuasAr3. In these panels, each trace was shifted and overlaid for display.
The changes in individual measurements were further analyzed, as shown in Fig. 2 and Supplemental Fig. 3B, which show all the measurement traces. In this analysis, a single rapid-sharp depolarization, sustained for about 0.5 sec, can be detected within a few seconds from the onset of the stimulation. These rapid-sharp depolarizations were always induced by the stimulation with the mid concentration (10−5–10−7), although diacetyl stimulation at higher concentrations (≥10−4) or at lower concentrations (≤10−8) did not always result in the rapid-sharp depolarization. In contrast, the semi-stable depolarization can be essentially observed in any concentration of diacetyl. To further understand the AWA neuronal properties, we analyzed the correlation of the peaks of paQuasAr3 intensities and those of GCaMP6f intensities (Supplemental Fig. 3C), and those showed a strong correlation (r = 0.80), although the changes of paQuasAr3 appeared to have maximum changes. Although the changes in fluorescence intensities of GEVIs and GECIs are not precisely proportional to membrane voltages and intracellular Ca2+ concentration, respectively, these results suggest that the higher membrane depolarization induces the higher Ca2+ increase and that Ca2+ increases do not occur without membrane depolarization. Because this high-time-resolution measurement essentially showed the same results as the slower measurement and gave noisy results, we used the slower measurements for the analyses of mutants.
Sequential stimulation with different odorant concentrations
The differences between intracellular Ca2+ and the membrane voltage change raise the possibility that the membrane voltage encodes distinct information from intracellular Ca2+. To test this possibility, we analyzed a sequential stimulation of different diacetyl concentrations. For this analysis, we modified the olfactory chip16 to stimulate animals using three different solutions (Supplementary Fig. 4). This custom chip allowed continuous sequential stimulation with three different solutions by regulating liquid valves. Using this chip, we analyzed the response to the same imaging solution with 10−6 diacetyl and confirmed that the change in the flow path itself did not cause changes in intracellular Ca2+ and membrane voltage (Fig. 3).

The left panel shows the result of the change in flow paths but not concentration. The center panel shows the result of the change in the concentration from 10−8–10−6 and the right panel shows the result of the change in the concentration from 10−6–10−8. Blue bars indicate the stimulation period, where the thickness represents the concentration. Each panel shows average traces with the SEM as shades. No. of samples were shown in the panels.
Next, we analyzed the responses to a sequential diacetyl of increase from 10−8, 10−7–10−6, or decrease from 10−6, 10−7–10−8. With increasing diacetyl, the intracellular Ca2+ levels increased as well. On the other hand, the membrane voltage was rapidly depolarized as the stimuli increased, but the increase showed no dependence on the concentration of the stimuli (Fig. 3). This phenomenon is similar to the results of the electrophysiolosical study; the increase of the current injection also induced the all-or-none action potentials19. This result suggested that the intracellular Ca2+ changes in accordance with the magnitude of stimuli and that the membrane voltage encodes the increase in the stimulation. Alternatively, the responsiveness of membrane voltage adapts more rapidly to the stimuli than that of Ca2+, and thereby, the coding appears different between them.
In contrast, with the sequential decrease in diacetyl concentration (10−6, 10−7–10−8), the prominent Ca2+ signal increase and the depolarization of the membrane voltage was observed only with the first stimulation, although repolarization could be observed with stimulation drop/removal (Fig. 3). These results also suggest that the rapid-sharp depolarization mainly encodes the beginning of the stimulation, and the semi-stable depolarization encodes the duration of the stimulation, while the change in Ca2+ signals encodes the diacetyl concentration.
Voltage-dependent Ca2+ channels and TRP channels regulate the changes in membrane voltage depending on the stimulation
To understand the mechanisms underlying the changes in membrane voltage, we analyzed perception defective mutants in AWA sensory neurons. Liu et al.19 showed that EGL19, an L-type voltage-gated calcium channel26, is required for the all-or-none responses observed with electrophysiology in AWA. However, its role in the response to the odorant remains unclear. Therefore, we first analyzed the responses of an egl-19 mutant to diacetyl using simultaneous imaging with GCaMP6f and paQuasAr3 expressed under the control of odr-10 promoter. As shown in Fig. 4A, increased Ca2+ levels triggered by diacetyl in egl-19 mutant animals were significantly smaller than those detected in wild-type animals. However, we could not detect a significant difference in the semi-stable depolarization in egl-19 as compared to the wild-type, although the rapid-sharp depolarization was slightly lower in egl-19 (Fig. 4A). These results suggest that the EGL-19 Ca2+ channel is required for the adequate intracellular Ca2+ change triggered by the odorant stimulation and that other channels, rather than EGL-19, at least partially induce rapid-sharp depolarization at the onset of the stimulation. In addition, the weak semi-stable depolarization was considered independent of the Ca2+ influx through EGL-19.

A Averaged traces are shown in wild-type animals (using dark green for GCaMP6f and purple for paQuasAr3) and egl-19 (light green for GCaMP6f and red for paQuasAr3), with the standard error of the mean (SEM) as shades. No. of samples were shown in the panels. The right panels show the boxplots of maximum values during the stimulation. Asterisks indicate a significant difference. B Averaged traces are shown for wild-type animals (dark green for GCaMP6f and purple for paQuasAr3) and unc-2 animals (light green for GCaMP6f and red for paQuasAr3) with the SEM as shades. No. of samples were shown in the panels. The right panels show the boxplots of maximum values during the stimulation. C Averaged traces are shown for wild-type animals (dark green for GCaMP6f and purple for paQuasAr3) and osm-9 animals (light green for GCaMP6f and red for paQuasAr3) with the SEM as shades. No. of samples were shown in the panels. The right panels show the boxplots of maximum values during the stimulation. Asterisks indicate significant differences.
Next, we analyzed a mutant of another voltage-dependent Ca2+ channel, UNC-2, which is not required for the depolarization induced by current injection19. As shown in Fig. 4B, in this mutant, the increase in intracellular Ca2+ levels and the rapid-sharp depolarization were not significantly different from those of the wild-type animals. This result suggests that UNC-2 is dispensable for the Ca2+ increase, consistent with the previous report.
Further, we analyzed a mutant of a Transient Receptor Potential (TRP) channel, OSM-9, required for diacetyl perception27. In this mutant, we used a gpa-4 promoter for the expression in AWA and found that osm-9 mutant animals did not show any responses in membrane voltage nor intracellular Ca2+ upon the stimulation, suggesting that a TRP channel, OSM-9, is essential for the rapid-sharp depolarization and the semi-stable depolarization triggered by diacetyl sensation, possibly relaying the signal from the ODR-10 receptor (Fig. 4C).
Since ODR-3, a trimeric G-protein alpha subunit is involved in diacetyl sensation28,29, we analyzed the responses of an odr-3 mutant by using the gpa-4 promoter. As shown in Fig. 5A, in odr-3 mutant animals, the increase in Ca2+ levels and membrane voltage changes appeared very weak compared to the wild-type. However, as shown in Supplementary Fig. 5a, individual animals randomly showed a spontaneous increase in Ca2+ signal and depolarization, even without stimulation. These results raise the possibility that notable peaks in the mean activity cannot be detected due to random activation.

A Dose responses to the change in GCaMP6f and paQuasAr3 in wild-type (dark green for GCaMP6f and purple for paQuasAr3) and odr-3 (light green for GCaMP6f and red for paQuasAr3) animals with the standard error of the mean (SEM) as shades. Blue bars indicate the period of the diacetyl stimulation. B Fluorescent responses, with maximum increases of F/F0 > 0.08, similar to wild-type standard responses (average responses by diacetyl 10−6), are shown in blue (correlation coefficients > 0.6) and red (correlation coefficients > 0.85). Each line shows the result of an individual animal. The vertical solid line shows the beginning of stimulation by diacetyl. Bold horizontal bars indicate the duration of stimulation. C Averaged paQuasAr3 traces to the various concentrations of odr-3 animals when the most similar GCaMP6f to the standard responses (Supplemental Fig. 5B). t0 indicates the beginning of the most similar responses to the standard. Average traces of GCaMP6f and paQuasAr3 in odr-3 are shown with the standard error of the mean (SEM) as shades. D ODR-3 was important in stabilizing the neuronal activities of AWA neurons. The spontaneous activation without stimulation is much more frequently observed in odr-3 deficient mutant animals (Student T test p = 0.00020).
To address this possibility, we analyzed the time course of GCaMP6f fluorescence by comparing the average responses of GCaMP6f to 10−6 diacetyl stimulation in wild-type animals (standard response, Supplementary Fig. 5b). Figure 5B shows the time points where changes similar to the wild-type standard response were observed in each recording. As shown in Fig. 5B, in odr-3 mutant animals, the response of GCaMP6f was similar to the standard response of wild-type in shape, and the amount of change was observed independent of the stimulation. We also analyzed the changes in membrane voltage during the Ca2+ responses in odr-3 mutant animals and found that the paQuasAr3 fluorescent changes coincided with them. This suggests that the membrane depolarization triggers the Ca2+ level increase even in odr-3 (Fig. 5C). To confirm that the ODR-3 suppresses neuronal activation, we compared the intracellular Ca2+ in non-stimulus conditions and found that, in odr-3 animals, the rapid-sharp depolarization is more prominent than that in wild-type animals (Fig. 5D, Supplemental Fig. 5C, D). Therefore, ODR-3 may stabilize the membrane voltage and, thereby, it can inhibit spontaneous depolarization of AWA and regulate the timing and magnitude of responses to diacetyl, upstream of the ion channels, OSM-9 and EGL-19.
Discussion
In these few decades, the development of GECI has enabled the assessment of neuronal activities through measuring intracellular Ca2+ in living animals. Since the change in the intracellular Ca2+ level was regulated by the change of membrane voltage, to understand neuronal activities, it is important to correctly interpret the results in Ca2+ imaging experiments. In this study, we simultaneously measured intracellular Ca2+ and membrane voltage levels in unanesthetized C. elegans using GCaMP6f (GECI)6 and paQuasAr3 (an Arch-derived GEVI)13. To achieve simultaneous imaging, we used high-power lasers for excitation and an image-splitting system, as well as correction of fluorescence photo-bleaching using a nonparametric spectral estimation method, termed SSA23 (Fig. 1C). The results of our experiments suggested that the dynamics in Ca2+ signals encode distinct information from that in the membrane voltage.
We measured AWA neurons of C. elegans in a microfluidic device called an olfactory chip16 for imaging in unanesthetized conditions because AWA exhibits all-or-none action potentials19. The present study detected the semi-stable and rapid-sharp depolarization in AWA neurons by stimulation with an odorant. We found that the stimulation with 10−5–10−6 diacetyl stimulation could consistently induce the rapid-sharp depolarization, but, by the stimulation with higher or lower concentration from the optimum, the rapid-sharp depolarization is less likely to be induced. This phenomenon is similar to that of the action potentials being efficiently induced by the optimal range of current injection but less efficiently by a larger or smaller range. Therefore, the rapid-sharp depolarization observed in this study was very similar to the action potentials induced by the current injection observed in the previous study, although they seem to correlate with the Ca2+ increases in the limited range. Furthermore, we found that, in egl-19 mutant animals, the rapid-sharp depolarization was not prominently induced, although the semi-stable depolarization sustained during the stimulation could be observed, suggesting that the rapid-sharp depolarization requires voltage-dependent Ca2+ channel encoded by egl-19 gene. This phenotype of egl-19 observed in voltage imaging is similar to those observed in electrophysiological studies because the current injection could induce the semi-stable depolarization but not the action potentials. Taken together, these properties of the rapid-sharp depolarization observed in this study are very similar to those of the action potentials observed in the electrophysiology. On the other hand, electrophysiological studies show that the optimal current injection sometimes induces multiple action potential peaks, although we could observe only single depolarization but not multiple peaks. This difference can be due to the difference in the time resolution, although the time constant of the voltage indicator and the time resolution may be sufficient for the single action potential. Another possibility is that the odor stimulation cannot induce the multiple spikes induced by the current injection.
The previous reports showed that the current injection can trigger all-or-none action potentials in AWA neurons and that these action potentials are generated through voltage-dependent Ca2+ channel, EGL-1919. In this report, we detected the rapid-sharp depolarizations at the onset of the stimulation by paQuasAr3 (Figs. 1F, 2, Supplemental Fig. 3B), raising the possibility that these depolarizations are all-or-none action potentials. This possibility is supported by some observations. The magnitudes of the rapid-sharp depolarization are not fully dependent on the stimuli concentration when observed and the optimum strength of the stimulus exists for their induction. On the other hand, we found some differences between the action potentials triggered by the current injection and the rapid-sharp depolarization at the onset of the stimulation detected by paQuasAr3. First, the action potentials triggered by the current injection are fully dependent on a voltage-dependent Ca2+ channel, EGL-19, although the weak rapid-sharp depolarization can be observed in egl-19 mutant animals. Second, the current injection sometimes induces multiple action potentials; the increase induced by the odorant stimuli was detected only once by paQuasAr3. In our study, two types of measurement were used for the simultaneous measurement of Ca2+ and membrane voltage: 50 ms exposure (20 Hz) and 4 ms exposure (250 Hz). Both procedures can detect similar changes in the membrane voltage: the single rapid-sharp depolarization at the onset of the stimulation and the semi-stable depolarization sustained during the stimulation. The time resolution of our fast capture system (4 ms) was higher than the intervals of action potentials (10–20 ms) induced by the current injection, suggesting that rapid-sharp depolarization occurs once after AWA neurons sense odor stimuli in natural conditions. Therefore, further studies will be required to conclude that the rapid-sharp depolarizations detected by paQuasAr3 in response to odorant stimulation are the same as the all-or-none action potentials by the current injection detected by electrophysiology.
Our analyses of mutants defective in diacetyl perception clarified that a diacetyl receptor, ODR-10, and a TRP channel, OSM-9, are essential for this process. On the other hand, a voltage-dependent Ca2+ channel, EGL-19, is essential for the changes in intracellular Ca2+ and important for the rapid-sharp depolarization at the onset of the stimulation, but not for weak semi-stable depolarization sustained during the stimulation. These results may be consistent with previous reports (19) suggesting that EGL-19 is required for the action potentials and OSM-9 is not for the action potentials triggered by the current injection. The weak semi-stable depolarization triggered by odorant stimulation may open the voltage-dependent Ca2+ channel EGL-19. UNC-2, another voltage-dependent Ca2+ channel dispensable for action potentials, was not important for the response to odorants in AWA neurons in natural conditions; hence, further studies are necessary to unveil the function of UNC-2 in odorant perception.
In this study, we found that a G-protein alpha subunit, ODR-3, was required to suppress spontaneous activation, suggesting that ODR-3 stabilize the neuronal activity of AWA even in the absence of stimuli. In addition, ODR-3 was needed for the depolarization triggered by the odorant stimuli, suggesting that ODR-3 regulates the neuronal activities upstream of ion channels for the responses to the stimulation. These two functions may belong to two distinct signaling pathways; hence, further studies are required to elucidate the function of ODR-3.
This study provides insights into the encoding differences between Ca2+ and membrane voltage in C. elegans neurons, thus demonstrating the essential importance of measuring not only intracellular Ca2+ but also membrane voltage in neurons to understand the regulation of their activities by signaling molecules. Although our simultaneous imaging unveiled neuronal coding in sensory neurons, the simultaneous imaging of interneurons is essential to understanding the neuronal code of sensory neurons to regulate the downstream circuits. Furthermore, simultaneous imaging of sensory neurons and interneurons will enable us to understand the informational processing in the neuronal circuit, especially informational coding in neuronal circuits.
Materials and methods
C. elegans strains and culture
All C. elegans strains were cultured on nematode growth medium (NGM) agar plates seeded with Escherichia coli strain OP50 and grown at 20 °C before experiment21 unless otherwise indicated. In all experiments, we used young adult hermaphrodites. All mutants and wild-type animals used in this study are shown in Supplementary Table 1.
Transgenic animals
For the expression of GCaMP6f and paQuasAr3, the coding sequences were synthesized as optimized codons for C. elegans and inserted downstream of AWA promoters. We used an odr-10 promoter for expression except in some mutants (odr-10, osm-9, odr-3) and control animals for the analyses of those mutants, where we used a gpa-4 promoter since the odr-10 promoter is activity-dependent and is not active in those mutants. In those mutants, we used a gpa-4 promoter for the expression. The odr-10 promoter drives expression exclusively in AWA neurons, whereas the gpa4 promoter drives expression in AWA and ASI neurons. To ensure the measurement of AWA neurons, animals exhibiting expression in both AWA and ASI neurons were used, and AWA neurons were assigned according to their location.
For fast recordings, the AWA-specific strong promoter was selected from the scRNA data of AWA in CeNGEN database25. 2.3 kb of 5’ upstream region of ZC334.13 gene were amplified using primers (TACTTTATACAGAACCCCCTC, AACTCAGAAAGTGAGCTAGAG) and inserted upstream of the reporter genes.
Microscopy and image capture
L4 animals expressing GCaMP6f and paQuasAr3 were picked and cultured on NGM plates containing 2 mM all-trans retinal for 24 h at 25 °C. Animals were washed with imaging buffer (50 mM NaCl, 5 mM CaCl2, 1 mM MgCl2, 25 mM HEPES NaOH (pH 6.0), 0.02% gelatin) once and captured into the olfactory chip16.
GCaMP6f and paQuasAr3 fluorescence was observed with an Olympus BX53 microscope equipped with a 60× objective (UPLSAPO60XS2). GCaMP6f and paQuasAr3 were excited at 470 nm (1000 mW) and 640 nm (400 mW) with a laser (LDI, 89North) using a dichroic mirror (488/543/635 Semrock). At the front aperture of the objective lens, the laser power was 3 mW (low-speed imaging) to 12 mW (high-speed imaging) at 470 nm and 4 mW (low-speed imaging) to 33 mW (high-speed imaging) at 640 nm. Fluorescent images of GCaMP6f and paQuasAr3 were separated using a dichroic mirror (FF580-FDI01 Semrock) in Gemini (Hamamatsu) and were captured every 50 ms (20 fps) through emission filters (512/25 for GCaMP6f and 708/75 for paQuasAr3) (Semrock) by ORCA-Flash4.0 (Hamamatsu). An ND filter (10%) was used for GCaMP6f fluorescence reduction. Fluorescent images were processed using original software written in MATLAB (Mathworks). Briefly, the cell body of AWA neurons was manually delineated as ROI (Region of Interest) and tracked. The average intensities of the top 10% pixels in the ROI were used for the intensities of AWA neurons. The resulting time-series data were corrected for the photobleaching by SSA (Singular Spectrum Analysis)23.
For the fast recordings, the fluorescent images exposed for 4 ms were captured every 4 ms (250 fps) by the same system, except that the emission filter (720/75) was used for paQuasAr3. The captured images were processed as above, except that the timeline data was filtered with a median filter (n = 5) in Fig. 2 and without the filter in Supplemental Fig. 2B.
Animals were stimulated with various concentrations of odorants in imaging buffer using MPS (World Precision Instruments). For sequential stimulation, we altered the olfactory chip to have three lines instead of one in the original chip (Supplementary Fig. 4).
Image processing
In our system, GCaMP6f and paQuasAr3 were captured side-by-side in one image, so they had a standard positional coordinate system. First, an ROI surrounding the cell body was manually defined in the initial frame of the GCaMP6f images. Second, our software automatically defined the corresponding ROI in the initial frame of the paQuasAr3. For subsequent frames, ROIs in the GCaMP6f and paQuasAr3 images were automatically tracked by our software. Briefly, our software searches the minimum of the difference of the ROI (ArgMinΣ(Aij-Bij)2, where Aij ∈ ROI and Bij ∈ ROI of the next frame).
Then, the spatial average of the top 10% of the fluorescent intensity within the ROI was calculated at every frame for GCaMP6f and paQuasAr3, respectively. After that, SSA was applied to remove DC trends that reflect fluorescence photobleaching from the paQuasAr3 data. Finally, the software normalized each GCaMP6f and paQuasAr3 data by the value obtained in the 1 s interval before the first diacetyl stimulation.
Singular spectrum analysis (SSA)
We briefly describe the methodology of SSA22. Let us denote the original time series data as \({x}_{1},{x}_{2},\cdots ,{x}_{N}\), where \(N\) is the length of the time series. The time series data is embedded into a trajectory matrix, X, of lagged vectors. The trajectory matrix with a window size L can be constructed as follows:
Here, each column of X represents a lagged vector of the time series. Then the trajectory matrix X is decomposed into three matrices by applying Singular Value Decomposition:
where \(U\) is an \(N\times L\) matrix whose columns are the left singular vectors, \(\Sigma\) is an \(L\times L\) diagonal matrix containing the singular values \({\sigma }_{1}^{2},{\sigma }_{2}^{2},\cdots ,{\sigma }_{L}^{2}\), \(V\) is an \(L\times L\) matrix whose columns are the right singular vectors. Through this decomposition, a slowly varying DC component at time i can be reconstructed by
For photobleaching correction, we chose empirical parameters of SSA as \(L=300,r=1\) for the low-speed imaging and \(L={{\mathrm{3,750}}},r=1\) for the high-speed imaging.
Analyses of ODR-3 responses
The average of normalized fluorescent responses of GCaMP to 10−6 diacetyl stimulation (15–25 s) in wild-type animals was used as the standard response (Supplemental Fig. 5B). The correlation coefficient of each GCaMP fluorescence of 10 s bins to the standard responses was calculated. The time points where the correlation coefficients are > 0.6 and 0.85 and the maximum changes to the fluorescent ratio are > 0.08 are visualized in Fig. 5B. The time points at which the maximum correlation coefficients were observed in each animal were used to calculate the average paQuasAr3 fluorescent changes at the time of the Ca2+ increase.
Chemotaxis assay
Chemotaxis assay toward attractive odorants was performed as described previously22, with the exception that the assay plates contained 50 mM NaCl30. We used 1% diacetyl (0.11 M; Sigma-Aldrich) and 10 mg/ml (W/V) pyrazine. For adaptation, animals were incubated in S-Basal buffer (100 mM NaCl, 50 mM K2HPO4 [pH 6], 0.02% gelatin) containing 0.02% diacetyl at 20 °C for 1.5 h. Animal responses were measured within 30 min after conditioning.
Statistical analyses
F/F0 was calculated as the fluorescent intensities relative to the basal fluorescent intensity during 1 s of the application of the odorant. For comparison, the data were analyzed by t-test (Fig. 4) or one-way ANOVA with Tukey–Kramer post hoc test (Figs. 1F, 5D) by using Bellcurve for Excel. Sample sizes are noted in the figure legends.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
All raw data and MATLAB programs for image processing that used in this study have been deposited in the QIR repository and can be found at https://doi.org/10.48708/7183604.
References
Tian, L. et al. Imaging neural activity in worms, flies and mice with improved GCaMP calcium indicators. Nat. Methods 6, 875–881 (2009).
Grienberger, C. & Konnerth, A. Imaging Calcium in Neurons. Neuron vol. 73 862–885 https://doi.org/10.1016/j.neuron.2012.02.011 (2012).
Nakai, J., Ohkura, M. & Imoto, K. A high signal-to-noise Ca2+ prove composed of a single green fluorescent protein. Nat. Biotechnol. 19, 137–141 (2001).
Piatkevich, K. D. et al. Population imaging of neural activity in awake behaving mice. Nature 574, 413–417 (2019).
Zong, W. et al. Large-scale two-photon calcium imaging in freely moving mice. Cell 185, 1240–1256.e30 (2022).
Chen, T. W. et al. Ultrasensitive fluorescent proteins for imaging neuronal activity. Nature 499, 295–300 (2013).
Inoue, M. et al. Rational engineering of XCaMPs, a multicolor GECI suite for in vivo imaging of complex brain circuit dynamics. Cell 177, 1346–1360.e24 (2019).
Zhang, Y. et al. Fast and sensitive GCaMP calcium indicators for imaging neural populations. Nature (2023) https://doi.org/10.1038/s41586-023-05828-9 (2023).
Scanziani, M. & Häusser, M. Electrophysiology in the age of light. Nature 461, 930–939 (2009).
Akemann, W. et al. Innovative methodology imaging neural circuit dynamics with a voltage-sensitive fluorescent protein. J. Neurophysiol. 108, 2323–2337 (2012).
Tsutsui, H., Karasawa, S., Okamura, Y. & Miyawaki, A. Improving membrane voltage measurements using FRET with new fluorescent proteins. Nat. Methods 5, 683–685 (2008).
Knöpfel, T. & Song, C. Optical voltage imaging in neurons: moving from technology development to practical tool. Nat. Rev. Neurosci. 20, 719–727 (2019).
Adam, Y. et al. Voltage imaging and optogenetics reveal behaviour-dependent changes in hippocampal dynamics. Nature 569, 413–417 (2019).
Tian, H. et al. Video-based pooled screening yields improved far-red genetically encoded voltage indicators. Nat. Methods (2023) https://doi.org/10.1038/s41592-022-01743-5 (2023).
Hashemi, N. A. et al. Rhodopsin-based voltage imaging tools for use in muscles and neurons of Caenorhabditis elegans. Proc. Natl Acad. Sci. USA 116, 17051–17060 (2019).
Chronis, N., Zimmer, M. & Bargmann, C. I. Microfluidics for in vivo imaging of neuronal and behavioral activity in Caenorhabditis elegans. Nat. Methods 4, 727–731 (2007).
Wen, C. et al. 3deecelltracker, a deep learning-based pipeline for segmenting and tracking cells in 3D time lapse images. Elife 10, e59187 (2021).
Goodman, M. B., Hall, D. H., Avery, L. & Lockery, S. R. Active currents regulate sensitivity and dynamic range in C. elegans neurons. Neuron 20, 763–772 (1998).
Liu, Q., Kidd, P. B., Dobosiewicz, M. & Bargmann, C. I. C. elegans AWA olfactory neurons fire calcium-mediated all-or-none action potentials. Cell 175, 57–70.e17 (2018).
White, J. G., Southgate, E., Thomson, J. N. & Brenner, S. The structure of the nervous system of the nematode Caenorhabditis elegans. Philos. Trans. R. Soc. Lond. B Biol. Sci. 314, 1–340 (1986).
Brenner, S. The genetics of Caenorhabditis elegans. Genetics 77, 71–94 (1974).
Bargmann, C. I., Hartwieg, E. & Robert Horvitz, H. Odorant-selective genes and neurons mediate olfaction in C. elegans. Cell 74, 515–527 (1993).
Vautard, R. & Ghil, M. Singular spectrum analysis in nonlinear dynamics, with applications to paleoclimatic time series. Phys. D. 35, 395–424 (1989).
Colbert, H. A. & Bargmann, C. I. Odorant-specific adaptation pathways generate olfactory plasticity in C. elegans. Neuron. 14, 803–812 (1995).
Hammarlund, M., Hobert, O., Miller, D. M. III & Sestan, N. The CeNGEN project: the complete gene expression map of an entire nervous system. Neuron 99, 430–433 (2018).
Jospin, M., Jacquemond, V., Mariol, M. C., Ségalat, L. & Allard, B. The L-type voltage-dependent Ca2+ channel EGL-19 controls body wall muscle function in Caenorhabditis elegans. J. Cell Biol. 159, 337–347 (2002).
Colbert, H. A., Smith, T. L. & Bargmann, C. I. OSM-9, a novel protein with structural similarity to channels, is required for olfaction, mechanosensation, and olfactory adaptation in Caenorhabditis elegans. J. Neurosci. 17, 8259–8269 (1997).
Ferkey, D. M., Sengupta, P. & L’Etoile, N. D. Chemosensory signal transduction in Caenorhabditis elegans. Genetics 217, iyab004 (2021).
Tanimoto, Y. et al. Calcium dynamics regulating the timing of decision-making in C. elegans. Elife 6, e21629 (2017).
Arai, M., Kurokawa, I., Arakane, H., Kitazono, T. & Ishihara, T. Regulation of diacylglycerol content in olfactory neurons determines forgetting or retrieval of olfactory memory in Caenorhabditis elegans. J. Neurosci. 42, 8039–8053 (2022).
Acknowledgements
We thank Dr. Takayuki Teramoto for making the new PDMS olfactory chip (Supplemental Fig. 4). We thank the Caenorhabditis Genetics Center funded by the US National Institutes of Health National Center for Strains. This work was supported by JST PRESTO Grant Numbers JPMJPR1875 to T.T., and by JSPS KAKENHI Grant Numbers, J19H03326, and 25115009 and NTT-Kyushu University Collaborative Research to T.I. We also thank Dr. Manabi Fujiwara for the discussions and critical reading of the manuscript.
Author information
Authors and Affiliations
Contributions
T.T. and T.I. designed the study. N.S. performed most of the experiments. M.A. and T.I. carried out chemotaxis and supported the experiments. T.N. supported data analyses, including photobleaching correction. T.T. and T.I. wrote the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interest.
Peer review
Peer review information
Communications Biology thanks the anonymous reviewers for their contribution to the peer review of this work. Primary Handling Editors: Asuka Takeishi and Benjamin Bessieres.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Tokunaga, T., Sato, N., Arai, M. et al. Mechanism of sensory perception unveiled by simultaneous measurement of membrane voltage and intracellular calcium. Commun Biol 7, 1150 (2024). https://doi.org/10.1038/s42003-024-06778-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s42003-024-06778-2
- Springer Nature Limited