
Abstract
Histone lysine methyltransferases (KMTs) are biomedically important epigenetic enzymes that catalyze the transfer of methyl group from S-adenosylmethionine to lysine’s nucleophilic ε-amino group in histone tails and core histones. Understanding the chemical basis of KMT catalysis is important for discerning its complex biology in disease, structure-function relationship, and for designing specific inhibitors with therapeutic potential. Here we examine histone peptides, which possess simplest lysine analogs with different nucleophilic character, as substrates for human KMTs. Combined MALDI-TOF MS experiments, NMR analyses and molecular dynamics and free-energy simulations based on quantum mechanics/molecular mechanics (QM/MM) potential provide experimental and theoretical evidence that KMTs do have an ability to catalyze methylation of primary amine-containing N-nucleophiles, but do not methylate related amide/guanidine-containing N-nucleophiles as well as simple O- and C-nucleophiles. The results demonstrate a broader, but still limited, substrate scope for KMT catalysis, and contribute to rational design of selective epigenetic inhibitors.
Similar content being viewed by others


Introduction
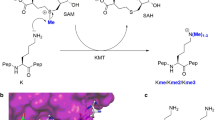
Histone proteins undergo numerous posttranslational modifications, which lead to a dynamic and complex epigenetic landscape that regulates the activity of genes in humans and other eukaryotes1,2,3. Nε-methylation of lysine residues has been found on unstructured N-terminal histone tails as well as on core histones, and could lead to transcriptional activation or repression, depending on the locations of methylated lysine residues and methylation states4,5,6,7. As established for most histone posttranslational modifications, lysine methylation is a dynamic process that is regulated by three classes of epigenetic proteins, i.e., histone lysine methyltransferases (KMTs), histone lysine demethylases (KDMs), and Nε-methyl lysine binding epigenetic proteins1. Histone Nε-lysine methylation is catalyzed by S-adenosylmethionine (SAM)-dependent histone lysine methyltransferases, which also catalyze the methylation of many non-histone proteins of biomedical importance (Fig. 1a)8. KMT-catalyzed methylation of lysine residues can produce monomethyllysine (Kme), dimethyllysine (Kme2), and trimethyllysine (Kme3) residues with an unaltered positive charge and an increased hydrophobicity (Fig. 1a)5,9,10. With the exception of DOT1L, all characterized KMTs contain a highly conserved SET (Su(var)3-9, enhancer-of-zeste, trithorax) domain, which is essential for the enzymatic activity8,11.

Histone lysine methyltransferase-catalyzed methylation of lysine. a Methylation of lysine residue by histone lysine methyltransferases using SAM as a methyl donor. b View on the crystal structure of SETD8 with H4K20 peptide (magenta) and SAH (yellow). c View on the crystal structure of GLP with H3K9me peptide (magenta) and SAH (yellow). d Unnatural lysine analogs possessing functionalities with a different nucleophilic character
Mechanistic and structural studies revealed basic molecular requirements for KMT-catalyzed Nε-lysine methylation of histones and other proteins8,11,12,13,14,15,16. Binding of SAM cosubstrate to a methyltransferase proceeds before histone substrate association, which induces a significant conformational change of the post-SET (located at the C terminus of SET) domain. Upon the formation of the tertiary complex, the lysine side chain occupies a narrow, hydrophobic channel, most often comprised of side chains of Phe and Tyr residues (Fig. 1b, c). The nucleophilic ε-amino group of lysine is positioned toward the electrophilic methyl group of SAM (Fig. 1b, c). Although lysine exists in a protonated form at physiological conditions, an active-site Tyr or a water channel have been proposed to act as a general base for deprotonation of the ammonium ion of lysine, thus leading to a neutral form of lysine with a strong nucleophilic character17,18. Examinations of the active-site variants demonstrated that Phe and Tyr residues control the final methylation state of lysine15. Kinetic isotope measurements provided evidence that the transfer of the methyl group from SAM to Nε-lysine takes place via the SN2 reaction19,20.
To understand the mechanism of KMT-catalyzed SAM-dependent methylation of lysine residues at an unprecedent level of detail, recent work explored the KMT active-site residues that possibly play an important role in enzymatic catalysis15. Moreover, examinations of sterically demanding SAM analogs as cosubstrates for recombinantly expressed KMTs, and in cellular assays showed that KMTs not only catalyze the methylation reaction, but also related alkylation reactions21,22,23. At present, however, it is still unknown whether KMTs do have an ability to catalyze methylation of lysine analogs that possess nucleophilic groups other than the Nε amino group. Here we report integrated experimental and computational studies on human KMT-catalyzed methylation of the simplest lysine analogs that possess N-, O-, and C-nucleophiles (Fig. 1d). The results demonstrate that, whereas O- and C-nucleophiles are not methylated, some of the N-nucleophiles can be substrates for KMTs.
Results
Selection of lysine analogs
Lysine’s structural elements comprise l-stereochemistry, four methylene groups, and the nucleophilic and basic Nε amino group at the end of the side chain. Our recent studies demonstrated that the l-stereochemistry, the chain length, and the main chain all determine a degree of KMT-catalyzed methylation, highlighting that lysine possesses the optimal l-configuration and the length of side chain for efficient KMT-catalyzed methylation24,25,26,27. The central question that needs to be addressed concerns the importance of the nucleophilic character of lysine’s Nε amino group for KMT catalysis. Therefore, determination of the effects on the methyl transfer due to simplest chemical perturbations on the lysine’s side chain, while maintaining the l-configuration and the same chain length, would advance our fundamental understanding of KMT catalysis and form the basis for designing chemical probes for KMTs. Our panel of lysine analogs includes: (i) Primary N-nucleophiles Kaza and Koxy; (b) Resonance-stabilized N-nucleophiles hGln and nArg; (c) O-nucleophiles KOH and KCOOH; and (d) C-nucleophiles Kalkyne and Kalkene (Fig. 1d).
The synthesis of the required building block Fmoc-Kaza(Boc)2-OH 1 was based on an established protocol (Fig. 2)28. Perbenzylation of l-glutamic acid 9 to intermediate 10, followed by selective DIBAL-H-mediated reduction of the side chain ester to alcohol afforded 11 in 87% yield. Subsequently, Swern oxidation furnished the corresponding amino aldehyde 12, which was then treated with tert-butylcarbazate to give the intermediate hydrazone 13 in excellent 93% yield. Next, sodium cyanoborohydride was used to reduce hydrazone to hydrazide 14 in 87% yield. The synthesis was continued with the Boc-protection of the ε-nitrogen yielding compound 15, deprotection of benzyl groups, and a final Fmoc-protection to generate the appropriate unnatural building block 1 (77% yield in two steps). The synthetic route for the preparation of Fmoc-Koxy(Boc)-OH 2 followed the same first two steps as for 1, according to previously described protocol (Fig. 2)29. The preparation of the hydroxylamine-containing amino acid proceeded through the activation of alcohol with N-hydroxyphthalimide employing Mitsunobu reaction to give compound 16. Deprotection of the phthalimide to produce a benzylated oxylysine, followed by installation of Boc group afforded 17 in 93% yield. Removal of the benzyl groups, followed by direct Fmoc-protection at the α-amino group, yielded the final Fmoc-Koxy(Boc)-OH 2 in 66% yield. Benzylation of carboxylic group of Fmoc-allylglycine-OH 18, followed by olefin cross metathesis with allyl-tert-butyl ether, produced alkene 19 in 41% yield. A subsequent hydrogenation step furnished the novel unnatural building block 3 in 48% yield. Similarly, the synthesis of the compound 4 was accomplished in three steps from 18 (Fig. 2). Benzylation of carboxylic acid 18, followed by the cross metathesis of the allyl side chain with tert-butyl acrylate yielded product 20. Subsequently, reduction of the resulting alkene via catalytic hydrogenation with the concomitant generation of the free acid liberated the building block 4 in excellent 87% yield. The building blocks 5, 6, 7, and 8 were commercially available.

Synthesis of Fmoc-protected lysine analogs. a (i) BnBr, K2CO3, NaOH, H2O, reflux, 2.5 h, 50%; (ii) DIBAL-H,THF, 0 °C, 1 h, Ar, 87%; (iii) Swern oxidation, −78 °C, 1 h, 88%; (iv) tert-butylcarbazate, HF, 16 h, rt, 93%; (v) NaBH3CN, p-TsOH, rt, 30 mins, 87%; (vi) Boc2O, Et3N, DCM, rt, 16 h, 80%; (vii) Pd/C, MeOH, H2 (1 atm), rt, 16 h; (viii) Fmoc-OSu, K2CO3, dioxane/water, 0 °C, 6 h, 77%; (ix) DEAD, Ph3P, N-hydroxyphthalimide, THF, 5 h, 85% (two steps); (x) NH2OH, DCM, 0 °C; (xi) Boc2O, Et3N, THF, rt, 16 h, 93% (two steps); (xii) Pd/C, MeOH, H2 (atm), rt, 16 h; (xiii) Fmoc-OSu, NaHCO3, dioxane/water, 0 °C, 2.5 h, 66% (two steps). b (xiv) BnBr, NaHCO3, DMF, rt, N2 (1 atm), 16 h, 57%; (xv) 10 mol% HGII, allyl-tert-butyl ether, DCM, reflux, 16 h, 41%; (xvi) Pd/C, H2 (1 atm), EtOH, 12 h, 48%. c (xvii) BnBr, NaHCO3, DMF, rt, N2 (1 atm), 16 h, 57%; (xviii) 5 mol% HGII, tert-butyl acrylate, DCM, reflux, 2 h, 82%; (xix) Pd/C, H2 (1 atm), EtOH, 1 h, 87%
To examine whether the unnatural amino acids are methylated by human KMTs, histone peptides that contain the above eight lysine analogs (Fig. 1d) were synthesized by solid-phase peptide synthesis (SPPS) (H4 residues 13–27, GGAKRHRX20VLRDNIQ; H3 residues 1–15, ARTKQTARX9STGGKA). We also synthesized 15-mer H4K20me for NMR studies and 14-mer H3K9 for competition experiments in MALDI-TOF MS studies. All histone peptides were obtained in high purity by preparative HPLC, and all purified histone peptides were further examined by analytical HPLC and ESI–MS (Supplementary Figs. 1–12 and Supplementary Tables 1 and 2).
Examining KMT-catalyzed methylation by MALDI-TOF MS
Histone peptides that bear lysine and its unnatural analogs were examined as substrates for KMTs employing MALDI-TOF MS assays. We previously established the standard conditions at which the natural histone sequences are efficiently methylated by various human KMTs24. At standard conditions, a mixture of KMT enzyme (2 µM), H3/H4 histone peptide (100 µM), SAM (200 μM for monomethylation and 500 μM for trimethylation), buffered in 50 mM Tris at pH 8.0 was incubated for 1 h at 37 °C, and then examined by MALDI-TOF MS. SETD8 (SET domain containing lysine methyltransferase 8) catalyzed almost quantitative monomethylation of H4K20 (Fig. 3a, top panel, and Supplementary Fig. 13), whereas G9a and GLP (G9a-Like Protein) catalyzed the predominant formation of H3K9me3 (Fig. 3b, c, top panels, and Supplementary Figs. 14 and 15). Initially, we examined whether SETD8 could catalyze the methylation of the H4Kaza20 and H4Koxy20 peptides. In the presence of SETD8 and SAM, these two peptides underwent quantitative monomethylatation (H4Kaza20me and H4Koxy20me), similar to the H4K20 peptide (Fig. 3a and Supplementary Fig. 13). To verify that the formation of monomethylated species is due to the enzyme activity in the presence of SAM, control experiments were carried out in the absence of SETD8 and SAM with both unnatural substrates. As expected, the results manifested that no detectable methylation was observed (Supplementary Figs. 16 and 17). Enzyme kinetics analyses revealed that H4K20, H4Kaza20 and H4Koxy20 exhibit similar kinetic parameters for SETD8-catalyzed methylation reactions (Supplementary Fig. 18). Measured substrate efficiencies (kcat/Km) were: 38.5 mM−1 min−1 for H4K20, 38.9 mM−1 min−1 for H4Kaza20, and 30.3 mM−1 min−1 for H4Koxy20 (Table 1). Both kcat values (4.14–4.89 min−1) and Km values (114–137 μM) were comparable for H4K20, H4Kaza20, and H4Koxy20, indicating similar substrate binding affinity and efficiency of the methyl transfer reaction (Table 1). We subsequently screened the remaining six peptides as possible substrates for SETD8 (Supplementary Fig. 19). H4nArg20 (positively charged) and H4hGln20 (neutral) did not undergo SETD8-catalyzed methylation within limits of detection (<5%), demonstrating that not all N-nucleophiles act as substrates for KMTs. Next, enzymatic reactions were undertaken to determine whether the replacement of the positively charged Nε-amino group of lysine by simplest O-nucleophiles (i.e., –OH and –COOH) leads to SETD8-catalyzed methylation. Nevertheless, no O-methylation was detected within detection limits by MALDI–MS. Similarly, the electron-rich triple and double bonds did not react with SAM in the presence of SETD8. Moreover, a prolonged incubation (1 and 5 h) in the presence of high concentration of SETD8 (10 µM) and SAM (1 mM) also did not lead to observable formation of methylated products (Supplementary Figs. 20 and 21).

KMTs catalyze methylation of lysine, azalysine, and oxylysine. a MALDI-TOF MS showing methylation of H4K20 (top panel), H4Kaza20 (middle panel), and H4Koxy20 (bottom panel) peptide in the presence of SETD8 and SAM. b MALDI-TOF MS showing methylation of H3K9 (top panel), H3Kaza9 (middle panel), and H3Koxy9 (bottom panel) peptide in the presence of G9a and SAM. c MALDI-TOF MS showing methylation of H3K9 (top panel), H3Kaza9 (middle panel), and H3Koxy9 (bottom panel) peptide in the presence of GLP and SAM
Attention was then focused on the examination of H3Kaza9 and H3Koxy9 as possible substrates for G9a and GLP. Strikingly, H3Kaza9 underwent predominant formation of trimethylated product H3Kaza9me3 in the presence of G9a; the same methylation level was observed with the natural H3K9 forming H3K9me3 under standard conditions (Fig. 3b, c, middle panels, and Supplementary Figs. 14 and 15). Lowering the concentration of G9a (200 and 500 nM) also resulted to the formation of higher methylation states of lysine and its analogs (Supplementary Figs. 22 and 23). Incubation for 1 h at high concentrations of G9a or GLP (10 µM) and SAM (2 mM) led to nearly quantitative formation of trimethylated species H3Kaza9me3 (Supplementary Figs. 24 and 25). Remarkably, H3Koxy9 underwent GLP/G9a-catalyzed methylation to produce H3Koxy9me2 as a main product under standard conditions (Fig. 3b, c, bottom panels, and Supplementary Figs. 18 and 19). Longer incubation times (1 and 5 h) using additional G9a or GLP (10 µM) and SAM (2 mM) still afforded only H3Koxy9me2, not forming significant amounts of H3Koxy9me3 (Supplementary Fig. 26). Control experiments in the absence of G9a/GLP and SAM with H3Kaza9 and H3Koxy9 peptides showed that no methylation was detected (Supplementary Figs. 27–30). Given the ability of both G9a and GLP to catalyze trimethylation of azalysine and dimethylation of oxylysine, we next sought to examine H3hGln9, H3nArg9, H3KOH9, H3KCOOH9, H3Kalkyne9, and H3Kalkene9 as possible substrates for G9a and GLP. G9a and GLP displayed no activity for the tested peptides under standard conditions (Supplementary Figs. 31 and 32), as well as upon prolonged incubation with additional G9a/GLP and SAM (Supplementary Figs. 33–36).
Competition studies were then carried out to determine whether the eight lysine analogs that display different nucleophilic character do inhibit G9a-catalyzed trimethylation of the 14-mer H3K9 peptide. The examination was performed in the presence of equimolar amounts of each of 15-mer histone peptides that possess unnatural lysine analogs and the 14-mer H3K9 natural sequence. The degree of methylation of 14-mer H3K9 during the competition experiments was compared to a control sample in the absence of competing peptide. Experiments revealed that all these analogs were able to bind the active site of G9a, thus leading to a partial inhibition G9a, as manifested by a reduced intensity of 14-mer H3K9me3 and increased intensities of H3K9me2 and H3K9me, and in some cases H3K9 signals (Supplementary Figs. 37–40). Following these competition experiments, we carried out additional inhibition studies aimed at providing IC50 values. All unmethylated histone peptides were screened for inhibition at 100 µM concentration in the presence of G9a or GLP (100 nM), SAM (20 µM) and 14-mer H3K9 peptide (5 µM) in glycine assay buffer pH 8.8 at 37 °C for 35 min. The initial rates of 14-mer peptide trimethylation in the samples were compared to a control sample in the absence of unmethylated histone peptides. MALDI-TOF MS data showed no significant (IC50 > 100 µM) inhibition of 14-mer H3K9 peptide trimethylation by the unnatural histone peptides within the examined range (Supplementary Figs. 41–43). Among the panel of histone peptides, H3hGln9 was observed to be the most potent inhibitor of G9a and GLP. A dose–response curve revealed that the H3hGln9 peptide inhibits G9a-catalyzed methylation of H3K9, with IC50 values of 88.8 and 76.0 µM for G9a and GLP, respectively.
Examining KMT-catalyzed methylation by NMR spectroscopy
After investigating the methylation of different nucleophilic analogs of lysine in the presence of SETD8, G9a, and GLP, we then carried out detailed NMR studies to provide additional information about the level and site of methylation. Before NMR analysis of SETD8- and G9a-catalyzed methylation of lysine analogs, all synthetic peptides were fully characterized by 1D and 2D NMR analyses (Supplementary Figs. 44–49). 1H NMR spectrum of reaction mixture that contains the H4K20 peptide (400 μM), SAM (2 mM), and SETD8 (8 μM) in Tris-D11 buffer (50 mM, pD 8.0) showed a downfield shift for the CH2ε (2.94 ppm) of K20, a characteristic new singlet (2.62 ppm) for NMe, and a triplet for SAH (2.61 ppm) (Fig. 4a and Supplementary Fig. 50). This result is consistent with previously reported data on SETD7-catalyzed monomethylation of H3K4 (ref. 30). The Heteronuclear Single Quantum Coherence (HSQC) NMR experiment showed that chemical shifts change upon the installation of the methyl group on Nε. 1H NMR resonance at 2.94 ppm (13C: 48.3 ppm) is lysine CH2ε, and the 1H resonance at 2.62 ppm (13C: 32.0 ppm) is lysine NMe (Fig. 4d); these results were further supplemented with Total Correlated Spectroscopy (TOCSY) and Heteronuclear Multiple Bond Correlation HMBC analyses (Supplementary Fig. 51). To provide unambiguous evidence for the methylation of H4K20 by SETD8, we synthesized and characterized the H4K20me peptide by 1D and 2D NMR spectroscopy and compared the spectroscopic data with the enzymatically produced H4K20me (Supplementary Figs. 52 and 53). The characteristic proton resonances of chemically synthesized H4K20me have identical chemical shifts to those seen for the enzymatically produced species (Supplementary Fig. 54).

NMR analyses of SETD8-catalyzed methylation. a 1H NMR data and the assignment of indicative resonances of the SETD8-catalyzed monomethylation of H4K20 in the presence of SAM after 1 h at 37 °C. b 1H NMR data of the SETD8-catalyzed monomethylation of H4Kaza20. c 1H NMR data of the SETD8-catalyzed monomethylation of H4Koxy20. The zoomed areas are shown in dotted box. d 1H-13C HSQC data of the enzymatic mixture with H4K20 with the assignment of product cross-peaks. e 1H-13C HSQC data of the enzymatic mixture with H4Kaza20 with the assignment of product cross-peaks. f 1H-13C HSQC data of the enzymatic mixture with H4Koxy20 with the assignment of product cross-peaks
We then performed the analysis of methylation of H4Kaza20 and H4Koxy20 by SETD8. The appearance of new singlet resonances at 2.48 ppm for H4Kaza20 and 2.55 ppm for H4Koxy20 indicated that SETD8 catalyzed methylation of both lysine analogs (Fig. 4b, c, respectively). 1H-13C HSQC spectrum revealed that the 1H resonance at 2.76 ppm (13C: 47.8 ppm) is H4Kaza20me CH2δ, and the 1H resonance at 2.48 ppm (13C: 35.1 ppm) is NMe of H4Kaza20me (Fig. 4e). The site of methylation was confirmed by HMBC analysis; NMe resonance at 2.48 ppm did not show correlation with CH2δ at 47.8 ppm (Supplementary Fig. 55). A correct coupling network for H4Kaza20me was supported by TOCSY analysis (Supplementary Fig. 55). 1H-13C HSQC spectrum showed that the 1H resonance at 3.65 ppm (13C: 71.9 ppm) is H4Koxy20me CH2δ, and the 1H resonance at 2.55 ppm (13C: 37.6 ppm) is NMe of H4Koxy20me (Fig. 4f). The NMe resonance at 2.55 ppm did not show correlation with CH2δ at 71.9 ppm in HMBC spectrum, while the TOCSY spectrum further supported the coupling network for H4Koxy20me (Supplementary Fig. 56). The integral ratio between the arginine CH2δ at 3.11 ppm and the NMe resonance at 2.48 ppm (H4Kaza20me)/2.55 ppm (H4Koxy20me) was 6:3, whereas the ratio between arginine CH2δ and SAH-CH2γ at 2.61 ppm was 6:2. These results suggest that the methylation of H4Kaza20/H4Koxy20 by SETD8 is quantitative and also that the conversion of SAM to SAH is tightly coupled with the enzymatic process (Supplementary Figs. 57 and 58).
Next, G9a-catalyzed methylation of H3K9 peptides was investigated by NMR. We first performed the reaction of H3K9 with G9a and SAM. The NMR results of the reaction mixture that contains G9a (8 μM), the H3K9 peptide (400 μM), and SAM (2 mM) in Tris-D11 buffer (50 mM, pD 8.0) after 1 h at 37 °C, largely mirrored the results from experiments using functionally related GLP (Supplementary Fig. 6a, d, and Supplementary Fig. 59)25. This observation was further supported by 2D TOCSY and HMBC analyses (Supplementary Fig. 60). Under the same conditions, we then investigated the site and level of G9a-catalyzed methylation of H3Kaza9 and H3Koxy9. 1H NMR spectrum of the enzymatic reaction of H3Kaza9 showed a new singlet resonance at 3.25 ppm (9H) and a triplet resonance at 2.61 ppm (2H), which corresponds to SAH-CH2γ. In agreement with MALDI-TOF MS data, this result implies that H3Kaza9 underwent trimethylation by G9a (Fig. 5b). The 1H-13C HSQC spectrum showed chemical shift change observed at 2.98 ppm (13C: 42.1 ppm, CH2δ), and the 1H resonance at 3.25 ppm (13C: 54.1 ppm, NMe3) derived from H3Kaza9me3 (Fig. 5e). Additional HMBC and TOCSY analyses confirmed that G9a-catalyzed trimethylation of Kaza occurs on the terminal amine (Supplementary Fig. 61). The NMR spectrum of G9a-catalyzed methylation of H3Koxy9 showed a singlet resonance at 2.52 ppm (6H) and a triplet resonance at 2.61 ppm (2H) (SAH-CH2γ) (Fig. 5c and Supplementary Fig. 62); in line with MALDI-TOF results, this observation indicates that H3Koxy9 underwent dimethylation by G9a. Multiplicity-edited HSQC manifests that the 1H resonance at 2.52 ppm (13C: 46.6 ppm) is NMe2 of H3Koxy9me2 and the 1H resonance at 3.65 ppm (13C: 70.4 ppm) is CH2δ of H3Koxy9me2 (Fig. 5f). HMBC analysis did not show correlation between the NMe2 resonance at 2.52 ppm and CH2δ at 70.4 ppm, and TOCSY showed the proper coupling network (Supplementary Fig. 63).

NMR analyses of G9a-catalyzed methylation. a 1H NMR data and the assignment of indicative resonances of the G9a-catalyzed trimethylation of H3K9 in the presence of SAM after 1 h at 37 °C. b 1H NMR data of the G9a-catalyzed trimethylation of H3Kaza9. c 1H NMR data of the G9a-catalyzed dimethylation of H3Koxy9. The zoomed areas are shown in dotted box. d 1H-13C HSQC data of the enzymatic mixture with H3K9 with the assignment of product cross-peaks. e 1H-13C HSQC data of the enzymatic mixture with H3Kaza9 with the assignment of product cross-peaks. f 1H-13C HSQC data of the enzymatic mixture with H3Koxy9 with the assignment of product cross-peaks
After confirming that H4K20, H4Kaza20, and H4Koxy20 peptides, as well as H3K9, H3Kaza9, and H3Koxy9 peptides, underwent methylation reaction in the presence of SETD8 and G9a, respectively, we carried out additional 1H NMR analyses with histone peptides that bear other six unnatural lysine analogs. The absence of new characteristic resonances (singlets) in the spectra of enzymatic reactions with these analogs and a lack of an indicative triplet for SAH-CH2γ at 2.61 ppm implies that lysine analogs that bear N-amide, N-guanidine, O-nucleophiles, and C-nucleophiles were not methylated in the presence of SETD8 or G9a (Supplementary Figs. 64–69). Collectively, our NMR observations indicate that H4K20, H4Kaza20, and H4Koxy20 act as substrates for SETD8, and that H3K9, H3Kaza9, and H3Koxy9 are substrates for G9a. Other N-, O-, and C-nucleophiles in our panel of simplest lysine analogs, however, were not methylated by SETD8 and G9a. It is worth stressing that these NMR findings are in complete agreement with results from our MALDI-TOF-based assays.
Quantum mechanics/molecular mechanics studies
Computer simulations can provide additional information concerning the energetic and structural origins of the KMT’s activities on different lysine analogs. Previous computational studies have shown that the free-energy barriers for the methyl transfers are the key determinants for the product specificity and on whether the enzymes are active or not for catalyzing certain methylation processes17. The QM/MM MD and free-energy simulations were performed for SETD8 (GLP) complexed with H4K20 (H3K9) and the analogs containing the N-nucleophiles. The free-energy profiles for the first, second, and third methylation reactions in SETD8 involving lysine, methyllysine, and dimethyllysine in histone substrates, respectively, are plotted in Fig. 6a. The general trend of the free-energy barriers for the SETD8-catalyzed methyl transfers obtained here is quite similar to that obtained in our previous study31; i.e., the free-energy barrier increases by about 8 kcal mol−1 in Fig. 6a and by 6.5 kcal mol−1 in the earlier study, respectively, in going from monomethylation to dimethylation. Both results suggest that the enzyme is a monomethyltransferase. Some structural information obtained from the simulations is also provided here. The average active-site structure of the reactant complex for the first methylation is given in Fig. 6b (Supplementary Fig. 70), which shows that the active-site structure has the lone pair of electrons on Nε of the target lysine (based on the sp3 hybridization) well aligned with the transferable methyl group of SAM with a relatively short r(CM···Nε) distance (~ 3.4 Å). Supplementary Fig. 71c shows that, for the reactant complex of the third methylation, the average distance between Nε and the methyl group (∼4.4 Å) became significantly larger compared to that for the first methylation, and the S–CH3 group of SAM cannot be well aligned with the lone pair of electrons on Nε for the third methyl transfer. Thus, the efficiency of the corresponding methyl transfer is likely to be significantly compromised. This conclusion is consistent with the results in Fig. 6a, which shows that the free-energy profile before reaching the transition state (TS) is shifted to the left of that of the first methylation and that the free-energy barrier for the third methylation is increased by ~5 kcal mol−1. The similar discussions can be made for the second methylation reaction involving the monomethyllysine substrate (Supplementary Fig. 71a). Figure 6c shows that there is a strengthening for the interactions with the substrate near the transition state, which may contribute to lowering the free-energy barrier for the methyl transfer.

Computational analyses on SETD8-catalyzed methylation. a Free-energy (potential of mean force) profiles for the first, second, and third methylation reactions in SETD8 involving K, Kme, and Kme2, respectively, as a function of the reaction coordinate [R = r(CM···Sδ) – r(CM···Nε)]; the designation of CM, Sδ, and Nε is shown in Fig. 6b. First methylation: blue line with a free-energy barrier of 19.4 kcal mol−1; second methylation: gray line with a free-energy barrier of 27.5 kcal mol−1; third methylation: orange line with a free-energy barrier of 24.1 kcal mol−1. b Representative active-site structure of the reactant complex of SETD8 for the first methylation containing SAM and lysine. Non-relevant hydrogen atoms are not shown for clarity. SETD8 is shown in sticks, and SAM and lysine are in balls and sticks. Some average distances are given in Å. c Representative active-site structure near the transition state of the SETD8 complex for the first methylation of H4K20. d Free-energy profile for the first methylation reaction involving H4Kaza20. e Representative active-site structure of the reactant complex of SETD8 for the first methylation containing SAM and H4Kaza20. f Representative active-site structure near the transition state of the SETD8 complex for the first methylation of H4Kaza20. g Free-energy profile for the first methylation reaction involving H4Koxy20. h Representative active-site structure of the reactant complex of SETD8 for the first methylation containing SAM and H4Koxy20. i Representative active-site structure near the transition state of the SETD8 complex for the first methylation of H4Koxy20. j Free-energy profiles for the first methylation reactions involving H4hGln20. k Representative active-site structure of the reactant complex of SETD8 for the first methylation containing SAM and H4hGln20. l Representative active-site structure near the transition state of the SETD8 complex for the first methylation of H4hGln20. m Free-energy profiles for the first methylation reactions to Nη1 involving H4nArg20. n Representative active-site structure of the reactant complex of SETD8 for the first methylation containing SAM and H4nArg20. o Representative active-site structure near the transition state of the SETD8 complex for the first methylation of H4nArg20
The free-energy profile for the methylation reaction involving H4Kaza20 in SETD8 is given in Fig. 6d, which shows that the free-energy profile for the methylation reaction has a free-energy barrier of 18.0 kcal mol−1. The free-energy profile for the methylation reaction involving H4Koxy20 in SETD8 is given in Fig. 6g. Comparison of Fig. 6a, g shows that there is a small increase in the free-energy barrier (by about 2 kcal mol−1) in going from K20 to Koxy20 for the first methylation reaction. Although Koxy is an α-nucleophile, this result seems to be consistent with the fact that the oxygen atom in Koxy20 is more electronegative than the carbon atom and therefore has a higher tendency to draw electrons from the neighboring N atom. This would lead to a decrease of nucleophilicity of NH2 and make it more difficult to accept the methyl group from SAM. Nevertheless, the increase of the barrier is rather small, and this is consistent with the enzyme kinetics data given above (Table 1), which showed that monomethylation reaction can occur for K20, Kaza20, and Koxy20. Figure 6h shows that the active-site structure for the reactant complex with Koxy20 is quite similar to that with K20 (Fig. 6b). For instance, the distances between the methyl donor (CM) and acceptor (N) are 3.36 Å and 3.37 Å for the cases involving Koxy20 and K20, respectively, and in both cases the lone pair of electrons on Nε is well aligned with the methyl group of SAM. Tyr245 forms a relatively stronger hydrogen bond with the ε-amino group of Koxy20 (i.e., a hydrogen bond distance of 3.06 Å compared to 3.20 Å in Fig. 6b). This observation is consistent with the suggestion that there is a decrease of nucleophilicity for NH2 (i.e., H may carry a more positive partial charge and be able to form a stronger hydrogen bond). The structure near the transition state with Koxy20 (Fig. 6i) is also quite similar to that with K20 (Fig. 6c). It should be pointed out that the changes of reactivity measured experimentally or free-energy barriers determined computationally can be the results of changing different factors, including, but not limited to, the alternations of electronic structures among different substrates (leading to different intrinsic reactivity) and the structural fits of substrates to the active sites. The free-energy profiles for the first methylation reactions involving H4hGln20 (Fig. 6j) and H4nArg20 in SETD8 (Fig. 6m, Supplementary Fig. 72), respectively, show that the free-energy barriers are very high (e.g., 43 kcal mol−1 for H4hGln20), suggesting that the methylation reactions cannot occur for these two substrate analogs, consistent with the experimental observations.
The free-energy profiles for the first, second, and third methylation reactions in GLP involving K, Kme, and Kme2 as the substrates, respectively, are given in Fig. 7a. The free-energy barriers for the three methylation reactions are quite similar and all rather low. For instance, the free-energy barriers of the first, second and third methylation are 17.0, 17.8 and 17.0 kcal mol−1, respectively. The similar and low free-energy barriers indicate that all the three methylation reactions occur. The results are consistent with the experimental observations from this work, but different from some previous computational investigations. In our earlier study32, we found that GLP could only produce mono- and dimethyllysine products. Nevertheless, the earlier simulations were based on older SCC-DFTB parameters with an empirical scaling and two different X-ray structures for different methylation reactions (which might lead to some inconsistency). Interestingly, the active-site structure for the reactant complex of the third methylation (Fig. 7b, Supplementary Fig. 73) seems to be rather similar to that obtained in the earlier simulations, and the both structures obtained here and earlier showed that the lone pair of electrons could not be well aligned with the transferable methyl group, with an average r(CM···Nε) distance of about 4.1–4.5 Å. The existence of the similar free-energy barriers in Fig. 7a for all the three methylation reactions in GLP suggests that some additional transition-state stabilization may exist for the third methylation reaction (to offset the poor reactant structure for the third methyl transfer) (Supplementary Fig. 74). Figure 7c shows the active-site structure near the transition state is stabilized through strengthening the CH···O interactions as well as by the presence of cation–π interactions involving F1209 and Y1124. A similar explanation has been used to understand the substrate/product specificities of Suv4–20h2 (ref. 33).

Computational analyses on GLP-catalyzed methylation. a Free-energy profiles for the first, second, and third methylation reactions in GLP involving K, Kme, and Kme2, respectively. The color scheme for the profiles is the same as in a. Free-energy barrier of the first methylation: 17.0 kcal mol−1; second methylation: 17.8 kcal mol−1; third methylation: 17.0 kcal mol−1. b Representative active-site structure of the reactant complex of GLP for the third methylation containing SAM and H3K9me2. Non-relevant hydrogen atoms are not shown here for clarity. c Representative active-site structure near the transition state of the GLP complex for the third methylation of H3K9me2. d Free-energy profiles for the first, second, and third methylation reactions in GLP involving H3Kaza9, H3Kaza9me, and H3Kaza9me2, respectively. Free-energy barrier of the first methylation: 15.9 kcal mol−1; second methylation: 17.2 kcal mol−1; third methylation: 16.5 kcal mol−1. e Representative active-site structure of the reactant complex of GLP for the second methylation containing SAM and H3Kaza9me. f Representative active-site structure of the reactant complex of GLP for the third methylation containing SAM and H3Kaza9me2. g Free-energy profiles for the first, second, and third methylation reactions in GLP involving H3Koxy9, H3Koxy9me, and H3Koxy9me2, respectively. Free-energy barrier of the first methylation: 17.5 kcal mol−1; second methylation: 18.8 kcal mol−1; third methylation: 21.3 kcal mol−1. h Representative active-site structure of the reactant complex of GLP for the second methylation containing SAM and H3Koxy9me. i Representative active-site structure of the reactant complex of GLP for the third methylation containing SAM and H3Koxy9me2. j Free-energy profile for the first methylation reaction involving H3hGln9. k Representative active-site structure of the reactant complex of GLP for the first methylation containing SAM and H3hGln9. l Representative structure near-transition state of the GLP complex for the first methylation of H3hGln9
The free-energy profiles for the first, second, and third methylation reactions in GLP involving H3Kaza9, H3Kaza9me, and H3Kaza9me2, respectively, are shown in Fig. 7d; the active-site structures for the reactant complexes of the second and third methylation reactions are given in Fig. 7e, f, respectively (Supplementary Fig. 75). The free-energy barriers for all the three methylation reactions are similar and rather low (15.9–17.2 kcal mol−1), indicating that GLP is a trimethyltransferase for H3Kaza9, in agreement with the experimental data. The free-energy profiles for the first, second, and third methylation reactions of H3Koxy9 in the presence of GLP are given in Fig. 7g. While the free-energy barriers for the first and second methylation reactions are close to each other with a difference of only ~1 kcal mol−1, the barrier for the third methylation is significantly higher (i.e., about 4 kcal mol−1 higher than that of the first methylation) (Fig. 7g, Supplementary Fig. 76). The results support the experimental observations that GLP can only catalyze mono- and dimethylation of H3Koxy9. The free-energy profile for the first methylation reaction involving H3hGln9 in GLP is given in Fig. 7j; the active-site structures for the reactant complex and near-transition state are shown in Fig. 7k, l, respectively. As is evident from Fig. 7j, the free-energy barrier is as high as 42 kcal mol−1, implying that the methylation cannot occur on H3hGln9 in the presence of GLP, in line with our experimental observations.
Discussion
Understanding the molecular origin of enzyme catalysis that plays essential roles in human health and disease is important from a basic molecular perspective as well as from a biomedical perspective. Despite ongoing examinations of basic biomolecular requirements that define the activity of numerous enzymes, an in-depth understanding of the underlying chemical mechanisms that control the enzymatic methylation of lysine and other residues remains incomplete. Members of SAM-dependent methyltransferases represent a widespread and important class of enzymes that catalyze N-, O-, and C-methylation reactions in all kingdoms of life34,35. Our work highlights that cooperative experimental and computational investigations enable the exploration of the chemical foundation for human KMT-catalyzed methylation of histones that possess lysine and its simplest analogs at an unprecedented level of molecular detail. The nucleophilic character and the basicity of lysine and analogous N-, O-, and C-nucleophiles as well as the conformations of the substrates at the active sites appear to define whether the enzymatic methylation takes place or not. In comparison with lysine, protonated forms of Kaza and Koxy are slightly stronger acids that undergo easier deprotonation by KMTs36,37, but their unprotonated forms are somewhat poorer nucleophiles than lysine38,39. The nucleophilic characters and the binding conformations of Kaza and Koxy (as demonstrated from computer simulations) may therefore contribute to the observations that Kaza and Koxy can in general undergo the KMT-catalyzed methylation to a similar degree compared to lysine. The lack of methylation of nArg by KMTs indicates that the Tyr-rich active sites of KMTs may not have an ability to deprotonate the weakly acidic guanidinium cation of nArg and that the electron lone pair of nArg may not be able to align well with the methyl group of SAM for the methyl transfer. Thus, the arginine methylation is catalyzed by functionally related arginine methyltransferases (RMTs) that have different active sites with well aligned methyl donor and acceptor and containing negatively charged Glu residues for deprotonating the weakly acidic guanidinum group of Arg during the methyl transfer processes (Supplementary Fig. 77)40,41. Our experimental observations that KOH does not undergo KMT-catalyzed methylation by SETD8, G9a, and GLP suggest that deprotonation of the very poorly acidic hydroxyl group cannot take place in the KMT active site, thus leading to an inactive substrate (OH is a much poorer nucleophile than O−)42.
The elucidation of the chemical foundation of epigenetics remains one of the great challenges of modern biomolecular sciences. It is envisaged that current chemical biology approaches will contribute to an advanced understanding of biomolecular recognition and enzyme-catalyzed posttranslational modifications on histones and other proteins5,9,43,44,45,46. Toward this aim, our integrated synthetic, enzymatic, and computational studies demonstrate that the biocatalytic scope of biomedically important KMTs is limited to N-methylation, and that the nucleophilic character and related basicity of the functional group importantly contribute to the efficiency of the enzymatic methylation reaction.
Methods
Solid-phase synthesis of histone peptides
Histone peptides bearing lysine and its analogs were synthesized on Wang resin using Fmoc solid-phase peptide synthesis (SPPS). Coupling of the amino acids was carried out for 1 h at room temperature with 3.0 equiv. of the desired amino acid, 3.6 equiv. of 1 M N-hydroxybenzotriazole (HOBt) in DMF and 3.3 equiv. of N,N′-diisopropylcarbodiimide (DIPCDI). Fmoc-protected nucleophilic lysine analogs were coupled overnight. Deprotection of the Fmoc-groups was carried out with piperidine in DMF (20%, v/v) for 30 min. After each coupling and deprotection step, a Kaiser test was done to ensure completion of the reaction. After the final Fmoc removal, the peptides were cleaved from the resin with mild cleaving reagents, to ensure that the acid-labile protecting groups remained intact. Cleavage was performed by a mixture of 95% of trifluoroacetic acid (TFA), 2.5% Triisopropylsilane (TIS), and 2.5% water for 4 h at room temperature. Crude peptides were purified by reverse phase HPLC. Fractions containing the pure peptide were collected, frozen, and lyophilized to afford the product as a white-off solid. The purity of histone peptides was examined by analytical HPLC and predicted masses were confirmed by MALDI-TOF MS, LC–MS, and ESI–MS. Results of characterization of histone peptides are presented in Supplementary Figs. 3–12.
HPLC and ESI–MS analyses of histone peptides
Lyophilized crude H3 and H4 peptides were purified by prep-HPLC on a Phenomenex® Gemini-NX 3u C-18 110A reversed-phase column (150 × 21.2 mm) using gradient elution at constant flow rate of 10 mL min−1 and the temperature is 30 °C. A typical run for all histone peptides was performed as follows: C-18 reverse phase column; after 3 mins at 3% B, a gradient of 3–15% over 12 mins was introduced, followed by a gradient of 15–30% over 17 mins and from 30 to 100% B over 19 mins, proceeding with 100 to 100% over 21 mins finalized by 3 mins at 100% CH3CN (total runtime 30 mins). Solvent A is 0.1% TFA in H2O, Solvent B is 0.1% TFA in acetonitrile. The amount of sample applied to the preparative column was 10–15 mg in 1 mL of MilliQ water (100 µL injection per each run). The crude peptides samples were filtered through syringe filters (0.22 µm, Screening Devices B.V, The Netherlands) prior to injection onto the column. H3 peptides were eluted at 8–11 min, whereas H4 peptides were eluted at 15–20 min. Pure fractions containing product were combined, frozen, and freeze-dried overnight to produce pure histone peptides as a white-off solid. Lyophilization was achieved using an ilShin Freeze Dryer (ilShin, Ede, The Netherlands). The purified peptides were characterized by analytical HPLC, MALDI–MS, and LC–MS. Analytical HPLC was performed on a Shimadzu LC-2010A HPLC system (Shimadzu, Kyoto, Japan) using RP C-18 column from Phenomenex, Prodigy ODS3, particle size 5 µm, pore size 110 Å, length 150 mm, and internal diameter 4.60 mm. Linear gradients of acetonitrile (+0.1% TFA) into H2O (+0.1% TFA) were run at 1 mL min−1 flow rate over 50 min. A peptide concentration of 1.0 mg mL−1 in milliQ water offered optimal resolution and separation with the following gradients: After 1 min at 5%, a gradient of 5 to 100% over 30 min was introduced, followed by 5 min at 100 to 100% and followed by a gradient of 100 to 5% in 5 min. Histone peptides were detected at 214 nm wavelength. The retention time of each peptide was shown on the top of the corresponding peak in HPLC chromatogram. The used MilliQ water was purified using a WaterPro PS Polisher (Labconco), set to 18.2 MΩ cm−1. Mass spectrometric analyses of the H3 and H4 peptides were carried out by ESI–MS (Thermo Finnigan LCQ Advantage Max) operating in a positive ionization mode, which was performed on a Thermo Finnigan LCQ-Fleet ESI-ion trap (Thermofischer, Breda, The Netherlands) equipped with a Phenomenex Gemini-NX C-18 column, 50 × 2.0 mm, particle size 3 µM (Phenomenex, Utrecht, The Netherlands). Linear gradients of acetonitrile (+0.1% formic acid) into H2O (+0.1% Formic acid) were run at 0.2 mL min−1 flow rate over 50 min. Ions were scanned in a range of m/z 50–2000 in MS mode. Multiply charged molecular-related ions of each peptide were detected. The observed masses matched the predicted peptide masses which are summarized in Supplementary Table 2.
Expression and purification of the KMTs
The expression and purification of SETD8 (residues 186–352), G9a (residues 913–1193), and GLP (residues 951–1235) were carried out as previously described25. Briefly, the WT enzymes were recombinantly expressed in E. coli Rosetta BL21 (DE3)pLysS cells, using the LB broth supplemented with kanamycin and chloramphenicol. The cultures were induced with isopropyl-d-thiogalactopyranoside (IPTG). Cells were harvested by centrifugation and lysed, and the expressed proteins were purified employing Ni-NTA affinity column and size exclusion chromatography using an AKTA system. Protein purity was monitored by SDS-PAGE and the concentrations were determined using the Nanodrop DeNovix DS-11 spectrophotometer.
Methyltransferase activity assays
The standard conditions of methyltransferase activity assays were performed by MALDI-TOF MS in 50 µL final volume for 1 h at 37 °C. Assay conditions for selected KMTs enzymes are described here. For SETD8, the reaction contained enzyme (2 µM), H4 peptide (GGAKRHRK20VLRDNIQ) or any of its unnatural analogs (100 µM), SAM (200 µM) in 50 mM Tris-HCl (pH = 8.0). At high concentration and long incubations, SETD8 was (10 µM) and SAM (1 mM). For G9a and GLP, the reaction contained enzyme (2 µM), H3 peptide (ARTKQTARK9STGGKA) or any of its unnatural analogs (100 µM), excess of SAM (500 µM) in 50 mM Tris-HCl (pH = 8). At longer incubation time and high concentration, G9a and GLP were (10 µM) and SAM (1 mM). Samples were incubated in an Eppendorf vial 1.5 mL in thermomixer. An aliquot of the reaction 5 µL was quenched with 5 µL of MeOH to stop the enzymatic reaction before analysis by MALDI–MS spectra. The spots were placed on a stainless steel MALDI plate (MS 96 target ground steel BC of Bruker, Germany). The mass spectra were measured in the positive reflector mode using α-cyano-4-hydroxycinnamic acid matrix. The mass corresponding to one monomethylation was observed as +14 Da, demethylation was observed as +28 Da, and trimethylation was observed as +42 Da. The MALDI–MS data were annotated employing FlexAnalysis software (Bruker Daltonics, Germany). Enzymatic assays for methylated substrates were carried out in five repeats (distinct samples), whereas for the unmethylated histone peptides in triplicate (distinct samples). The evaluations applied in this work directly measure by mass shifts the substrates activity of SETD8, G9a and GLP. It is noteworthy to mention that in the conditions of MALDI-TOF MS analysis, non-enzyme and non-SAM controls were carried out to ensure that the conditions of MS assay did not affect the observable methylation states. Laser power was adjusted to slightly above the threshold to obtain high resolution and signal/noise ratios. Each measurement was obtained by accumulating three spectra collected at different positions on the plate, 100 shots per position.
The kinetic assays for SETD8-catalyzed methylation of histone peptides was carried out employing a MALDI-TOF MS assay to determine the initial velocity rates for the first methylation reaction47. A solution of histone peptide (0–300 μM), was added to a solution of SAM (3 μM) in assay buffer (50 mM Tris, pH 8.0) at room temperature (the final volume of 100 μL). The reaction was then initiated by the addition of SETD8 (2 µM) and shaken for 10 min. The enzyme activity was quickly neutralized by the addition of methanol:water (1:1). The different reaction mixtures were aliquoted and mixed with α-Cyano-4-hydroxycinnamic acid matrix prior to measurement. All experiments were carried out in replicates (distinct samples). The enzymatic activity was determined by taking the peak areas of each methylation state, including all isotopes and adducts, and is expressed relative to a control reaction in which no monomethylation is present, utilizing the FlexAnalysisTM software. Kinetics values were extrapolated by plotting initial reaction velocities against peptide concentrations, utilizing GraphPad Prism 5.
Methyltransferase inhibition assays
The inhibition assays were performed in 20 µL in Eppendorf vials in triplicates (distinct samples) as previously described48. Unnatural histone peptide (0–100 µM final concentration) was preincubated with G9a or GLP (100 nM final concentration) for 5 min at 37 °C in 18 µL of 50 mM glycine pH 8.8 containing 2.5% glycerol as assay buffer. The reaction was initiated by the addition of 2 µL of a pre-mixture of SAM (20 µM final concentration of 200 μM stock) and 14-mer histone peptide (5 µM final concentration of 100 μM stock) to afford a final reaction volume of 20 µL. The enzymatic reaction was incubated for an additional 30 min. Then the reaction was quenched with the addition of 20 µL of MeOH. 2 µL of the quenched reaction was mixed with 2 µL of matrix solution (5.0 mg mL−1 of α-CHCA in 50% acetonitrile/H2O, 0.1% TFA) and spotted on the MALDI plate for crystallization. The enzymatic activity was determined by taking the peak areas of each methylation state, including all isotopes and adducts, and is expressed relative to a control reaction in which no unnatural histone peptide is present, utilizing the FlexAnalysisTM software. The half maximal inhibitory concentration (IC50) and the drawing curves, and inhibition studies were calculated using nonlinear regression in GraphPad Prism 5.
NMR experiments
For the NMR experiments of SETD8 with H4K20 peptides, samples (300 µL final volume) were prepared containing SETD8 (8 µM), peptide (400 µM, diluted from a 2 mM stock in 50 mM Tris-D11.HCl at pD 8.0, supplemented with D2O), SAM (2 mM, diluted from a 10 mM stock in 50 mM Tris-D11.HCl at pD 8.0, supplemented with D2O). After incubation for 1 h at 37 °C in an Eppendorf vial using a thermomixer, the reaction mixture was transferred into the NMR tube and then diluted to 550 μL with Tris-D11.HCl buffer and recorded by 1H NMR at 298 K. For the NMR experiments of G9a with H3K9 peptides, similar conditions were applied. Per each NMR experiment, identical incubation was run in parallel but without enzyme as a control. NMR spectra were recorded using a Bruker Avance III-500 MHz magnet equipped with the Prodigy BB cryoprobe. Water suppression was performed by presaturation and the 1D spectra were acquired with 128 or 256 transients and a relaxation delay of 4 s. 2D TOCSY spectra were acquired with presaturation of the water resonance using 1k points per transient, 8.3 kHz spin-lock for 100 ms, 56 transients per increment with a relaxation delay of 2 s and 512 increments with a sweep width of 10 ppm in each dimension. 2D 1H-13C multiplicity-edited HSQC spectra were acquired using 1k points per transient, 64 transients per increment, a relaxation delay of 2 s, and 512 increments. The 13C sweep width spanned from −10 to 130 ppm. 1H NMR characterization of substrates prior to enzymatic catalysis was performed using a 30° excitation pulse, 16–128 transients per compound, and a relaxation delay of 8 s. 1H-13C spectra of the substrates were recorded using a 30° excitation pulse, 512–4096 transients per compound and a relaxation delay of 2 s. 1H and 13C chemical shifts were externally referenced to TMS based on the lock frequency of solvent. NMR enzymatic experiments were conducted at 310 K. MestreNova was used to process the 1D and 2D NMR data.
QM/MM computations
QM/MM-free energy (potential of mean force) and MD simulations were performed to study the active-site dynamics of SETD8 and GLP and to calculate the free-energy profiles of the methyl transfers from SAM to the target lysine and lysine analogs containing the N-nucleophiles using the CHARMM program49. The –CH2–CH2–S+(Me) –CH2– part of SAM and lysine/lysine analog chain were treated by QM and the rest of the system by MM. The link-atom approach50 was applied to separate the QM and MM regions. A modified TIP3P water model51 was employed for the solvent, and the stochastic boundary molecular dynamics method52 was used for the QM/MM simulations. The reaction region was a sphere with radius r of 20 Å, and the buffer region extended over 20 Å ≤ r ≤ 22 Å. The reference center for partitioning the system was chosen to be the Nζ atom of the target lysine or the corresponding atoms in the lysine analogs. The resulting systems contained around 5800 atoms, including about 700–800 water molecules. The DFTB3 method52,53 implemented in CHARMM was used for the QM atoms. The semi-empirical approach adopted here has been used previously on a number of systems, and the results seem to be quite reasonable54,55. The all-hydrogen CHARMM potential function (PARAM27)56 was used for the MM atoms.
The initial coordinates for the reactant complexes of the methylation were based on the crystallographic complexes (PDB codes: 2BQZ and 3HNA for SETD8 and GLP, respectively) containing, SAH and methyl lysine (i.e., the product complexes). In each of the cases, a methyl group was manually added to SAH to change it to SAM and the methyl group(s) on the methyl lysine were manually deleted to generate the target lysine. For the models with the lysine analogs, the –CH2–NH3 group on lysine was modified to change to the corresponding groups on the analogs. The initial structures for the entire stochastic boundary systems were optimized using the steepest descent (SD) and adopted-basis Newton–Raphson (ABNR) methods. The systems were gradually heated from 50.0 to 298.15 K in 50 ps. A 1-fs time step was used for integration of the equation of motion, and the coordinates were saved every 50 fs for analyses. 1.5 ns QM/MM MD simulations were carried out for each of the reactant complexes, and the similar approaches have been used previously17,31,32,33,57.
The umbrella sampling method58 implemented in the CHARMM program along with the Weighted Histogram Analysis Method (WHAM)59 was applied to determine the change of the free energy (potential of mean force) as a function of the reaction coordinate for the methyl transfer from SAM to the target lysine or lysine analog in each enzyme. The reaction coordinate was defined as a linear combination of r(CM-Nε) and r(CM-Sδ) [R = r(CM-Sδ)- r(CM-Nε)] (see Fig. 6b for the atom designation). Thirty windows were used, and for each window 50 ps production runs were performed after 50 ps equilibration. The force constants of the harmonic biasing potentials used in the PMF simulations were 50–400 kcal mol–1 Å–2.
Reporting summary
Further information on research design is available in the Nature Research Reporting Summary linked to this article.
Data availability
The authors declare that the main data supporting the findings of this study are available within the paper and its Supplementary Information file. Other relevant data are available from the corresponding authors upon reasonable request.
References
Bannister, A. J. & Kouzarides, T. Regulation of chromatin by histone modifications. Cell Res. 21, 381–395 (2011).
Kouzarides, T. Chromatin modifications and their function. Cell 128, 693–705 (2007).
Strahl, B. D. & Allis, C. D. The language of covalent histone modifications. Nature 403, 41–45 (2000).
Black, J. C., Van Rechem, C. & Whetstine, J. R. Histone lysine methylation dynamics: establishment, regulation, and biological impact. Mol. Cell 48, 491–507 (2012).
Luo, M. Chemical and biochemical perspectives of protein lysine methylation. Chem. Rev. 118, 6656–6705 (2018).
Rea, S. et al. Regulation of chromatin structure by site-specific histone H3 methyltransferases. Nature 406, 593–599 (2000).
Martin, C. & Zhang, Y. The diverse functions of histone lysine methylation. Nat. Rev. Mol. Cell Biol. 6, 838–849 (2005).
Qian, C. & Zhou, M.-M. SET domain protein lysine methyltransferases: structure, specificity and catalysis. Cell. Mol. Life Sci. 63, 2755–2763 (2006).
Luo, M. Current chemical biology approaches to interrogate protein methyltransferases. ACS Chem. Biol. 7, 443–463 (2012).
Smith, B. C. & Denu, J. M. Chemical mechanisms of histone lysine and arginine modifications. Biochim. Biophys. Acta 1789, 45–57 (2009).
Dillon, S. C., Zhang, X., Trievel, R. C. & Cheng, X. The SET-domain protein superfamily: protein lysine methyltransferases. Genome Biol. 6, 227–227 (2005).
Couture, J.-F., Dirk, L. M. A., Brunzelle, J. S., Houtz, R. L. & Trievel, R. C. Structural origins for the product specificity of SET domain protein methyltransferases. Proc. Natl Acad. Sci. USA 105, 20659–20664 (2008).
Del Rizzo, P. A. & Trievel, R. C. Substrate and product specificities of SET domain methyltransferases. Epigenetics 6, 1059–1067 (2011).
Herz, H.-M., Garruss, A. & Shilatifard, A. SET for life: biochemical activities and biological functions of SET domain-containing proteins. Trends Biochem. Sci. 38, 621–639 (2013).
Schapira, M. Structural chemistry of human SET domain protein methyltransferases. Curr. Chem. Genom. 5, 85–94 (2011).
Wu, H. et al. Structural biology of human H3K9 methyltransferases. PLoS ONE 5, e8570 (2010).
Guo, H.-B. & Guo, H. Mechanism of histone methylation catalyzed by protein lysine methyltransferase SET7/9 and origin of product specificity. Proc. Natl Acad. Sci. USA 104, 8797–8802 (2007).
Zhang, X. & Bruice, T. C. Enzymatic mechanism and product specificity of SET-domain protein lysine methyltransferases. Proc. Natl Acad. Sci. USA 105, 5728–5732 (2008).
Linscott, J. A. et al. Kinetic isotope effects reveal early transition state of protein lysine methyltransferase SET8. Proc. Natl Acad. Sci. USA 113, E8369–E8378 (2016).
Poulin, M. B. et al. Transition state for the NSD2-catalyzed methylation of histone H3 lysine 36. Proc. Natl Acad. Sci. USA 113, 1197–1201 (2016).
Huber, T. D., Johnson, B. R., Zhang, J. & Thorson, J. S. AdoMet analog synthesis and utilization: current state of the art. Curr. Opin. Biotechnol. 42, 189–197 (2016).
Klimašauskas, S. & Weinhold, E. A new tool for biotechnology: AdoMet-dependent methyltransferases. Trends Biotechnol. 25, 99–104 (2007).
Zhang, J. & Zheng, Y. G. SAM/SAH analogs as versatile tools for SAM-dependent methyltransferases. ACS Chem. Biol. 11, 583–597 (2016).
Belle, R. et al. Investigating d-lysine stereochemistry for epigenetic methylation, demethylation and recognition. Chem. Commun. 53, 13264–13267 (2017).
Al Temimi, A. H. K. et al. Lysine possesses the optimal chain length for histone lysine methyltransferase. Sci. Rep. 7, 16148 (2017).
Al Temimi, A. H. K. et al. Importance of the main chain of lysine for histone lysine methyltransferase catalysis. Org. Biomol. Chem. 17, 5693–5697 (2019).
Al Temimi, A. H. K. et al. γ-Thialysine versus lysine: an insight into the epigenetic methylation of histones. Bioconjugate Chem. 30, 1798–1804 (2019).
Dancy, B. C. R. et al. Azalysine analogues as probes for protein lysine deacetylation and demethylation. J. Am. Chem. Soc. 134, 5138–5148 (2012).
Liu, F., Thomas, J. & Burke, T. R. Jr Synthesis of a homologous series of side-chain-extended orthogonally protected aminooxy-containing amino acids. Synthesis 15, 2432–2438 (2008).
Theillet, F.-X. et al. Site-specific mapping and time-resolved monitoring of lysine methylation by high-resolution NMR spectroscopy. J. Am. Chem. Soc. 134, 7616–7619 (2012).
Chu, Y., Xu, Q. & Guo, H. Understanding energetic origins of product specificity of SET8 from QM/MM free energy simulations: what causes the stop of methyl addition during histone lysine methylation? J. Chem. Theory Comput. 6, 1380–1389 (2010).
Chu, Y., Yao, J. & Guo, H. QM/MM MD and free energy simulations of G9a-like protein (GLP) and its mutants: understanding the factors that determine the product specificity. PLoS ONE 7, e37674 (2012).
Qian, P., Guo, H., Wang, L. & Guo, H. QM/MM investigation of substrate and product specificities of Suv4-20h2: how does this enzyme generate dimethylated H4K20 from monomethylated substrate? J. Chem. Theory Comput. 13, 2977–2986 (2017).
Bennett, M. R., Shepherd, S. A., Cronin, V. A. & Micklefield, J. Recent advances in methyltransferase biocatalysis. Curr. Opin. Chem. Biol. 37, 97–106 (2017).
Struck, A.-W., Thompson, M. L., Wong, L. S. & Micklefield, J. S-adenosyl-methionine-dependent methyltransferases: highly versatile enzymes in biocatalysis, biosynthesis and other biotechnological applications. ChemBioChem 13, 2642–2655 (2012).
Bissot, T. C., Parry, R. W. & Campbell, D. H. The physical and chemical properties of the methylhydroxylamines1. J. Am. Chem. Soc. 79, 796–800 (1957).
Hinman, R. Notes: base strengths of some alkylhydrazines. J. Org. Chem. 23, 1587–1588 (1958).
Nigst, T. A., Ammer, J. & Mayr, H. Ambident reactivities of methylhydrazines. Angew. Chem. Int. Ed. 51, 1353–1356 (2012).
Nigst, T. A., Antipova, A. & Mayr, H. Nucleophilic reactivities of hydrazines and amines: the futile search for the α-effect in hydrazine reactivities. J. Org. Chem. 77, 8142–8155 (2012).
Schapira, M. & Ferreira de Freitas, R. Structural biology and chemistry of protein arginine methyltransferases. MedChemComm 5, 1779–1788 (2014).
Yue, Y., Chu, Y. & Guo, H. Computational study of symmetric methylation on histone arginine catalyzed by protein arginine methyltransferase PRMT5 through QM/MM MD and free energy simulations. Molecules 20, 10032 (2015).
Minegishi, S. & Mayr, H. How constant are Ritchie’s “Constant Selectivity Relationships”? A general reactivity scale for n-, π-, and σ-nucleophiles. J. Am. Chem. Soc. 125, 286–295 (2003).
David, Y. & Muir, T. W. Emerging chemistry strategies for engineering native chromatin. J. Am. Chem. Soc. 139, 9090–9096 (2017).
Fierz, B. & Muir, T. W. Chromatin as an expansive canvas for chemical biology. Nat. Chem. Biol. 8, 417–427 (2012).
Müller, M. M. & Muir, T. W. Histones: at the crossroads of peptide and protein chemistry. Chem. Rev. 115, 2296–2349 (2015).
Nadal, S., Raj, R., Mohammed, S. & Davis, B. G. Synthetic post-translational modification of histones. Curr. Opin. Chem. Biol. 45, 35–47 (2018).
Guitot, K. et al. Label-free measurement of histone lysine methyltransferases activity by matrix-assisted laser desorption/ionization time-of-flight mass spectrometry. Anal. Biochem. 456, 25–31 (2014).
Lenstra, D. C. et al. Structure–activity relationship studies on (R)-PFI-2 analogues as inhibitors of histone lysine methyltransferase SETD7. ChemMedChem 13, 1405–1413 (2018).
Brooks, B. R. et al. CHARMM: a program for macromolecular energy, minimization, and dynamics calculations. J. Comput. Chem. 4, 187–217 (1983).
Field, M. J., Bash, P. A. & Karplus, M. A combined quantum mechanical and molecular mechanical potential for molecular dynamics simulations. J. Comput. Chem. 11, 700–733 (1990).
Jorgensen, W. L., Chandrasekhar, J., Madura, J. D., Impey, R. W. & Klein, M. L. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 79, 926–935 (1983).
Brooks, C. L., Brünger, A. & Karplus, M. Active site dynamics in protein molecules: a stochastic boundary molecular-dynamics approach. Biopolymers 24, 843–865 (1985).
Elstner, M. et al. Self-consistent-charge density-functional tight-binding method for simulations of complex materials properties. Phys. Rev. B 58, 7260–7268 (1998).
Christensen, A. S., Kubař, T., Cui, Q. & Elstner, M. Semiempirical quantum mechanical methods for noncovalent interactions for chemical and biochemical applications. Chem. Rev. 116, 5301–5337 (2016).
Cui, Q., Elstner, M., Kaxiras, E., Frauenheim, T. & Karplus, M. A QM/MM implementation of the self-consistent charge density functional tight binding (SCC-DFTB) method. J. Phys. Chem. B 105, 569–585 (2001).
MacKerell, A. D. et al. All-atom empirical potential for molecular modeling and dynamics studies of proteins. J. Phys. Chem. B 102, 3586–3616 (1998).
Xu, Q., Chu, Y.-z, Guo, H.-B., Smith, J. C. & Guo, H. Energy triplets for writing epigenetic marks: insights from QM/MM free-energy simulations of protein lysine methyltransferases. Chem. Eur. J. 15, 12596–12599 (2009).
Torrie, G. M. & Valleau, J. P. Monte Carlo free energy estimates using non-Boltzmann sampling: application to the sub-critical Lennard–Jones fluid. Chem. Phys. Lett. 28, 578–581 (1974).
Kumar, S., Rosenberg, J. M., Bouzida, D., Swendsen, R. H. & Kollman, P. A. THE weighted histogram analysis method for free-energy calculations on biomolecules. I. The method. J. Comput. Chem. 13, 1011–1021 (1992).
Acknowledgements
We gratefully acknowledge the financial support from the ERC Starting Grant to J.M. (ChemEpigen-715691) and the Netherlands Organization for Scientific Research (NWO, NCI-TA grant 731.015.202). The research has been aided by the Natural Science Foundation of Shandong Province (No. ZR2017MB048 to P.Q.). We thank Professor Martin Karplus for a gift of the CHARMM program and Giordano Proietti for help with kinetics analyses.
Author information
Authors and Affiliations
Contributions
J.M. conceived and supervised the project. A.H.K.A.T. synthesized building blocks and histone peptides, produced proteins, and carried out enzymatic assays. H.I.V.A.G. contributed to the synthesis of building blocks 1 and 2. Y.V.R and A.H.K.A.T. carried out NMR experiments and analyzed results. R.H.B. provided expertise with unnatural amino acids. H.G. and P.Q. carried out computational studies and analyzed results. A.H.K.A.T., H.G., P.Q. and J.M. wrote the manuscript. All authors contributed in editing the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Al Temimi, A.H.K., Amatdjais-Groenen, H.I.V., Reddy, Y.V. et al. The nucleophilic amino group of lysine is central for histone lysine methyltransferase catalysis. Commun Chem 2, 112 (2019). https://doi.org/10.1038/s42004-019-0210-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s42004-019-0210-8
- Springer Nature Limited
This article is cited by
-
Roles for the methyltransferase SETD8 in DNA damage repair
Clinical Epigenetics (2022)
-
Investigation of in vitro histone H3 glycosylation using H3 tail peptides
Scientific Reports (2022)
-
Examining sterically demanding lysine analogs for histone lysine methyltransferase catalysis
Scientific Reports (2020)
-
Fine-tuning of lysine side chain modulates the activity of histone lysine methyltransferases
Scientific Reports (2020)
-
Effect of lysine side chain length on histone lysine acetyltransferase catalysis
Scientific Reports (2020)