
Abstract
The moon-forming impact caused widespread melting and vaporization of the proto-Earth, leading to the formation of a protolunar disk1,2. Volatile elements trapped in the inner parts of the disk condensed together with heavier elements into a global magma ocean3,4. By subsequent cooling and degassing, volatiles contributed to the formation of the Earth’s secondary atmosphere5. The thermodynamic conditions at the magma ocean-atmosphere interface profoundly impacted the degree of degassing that occurred. Today, our understanding of these conditions remains unclear. Here, we simulate the degassing of carbon and helium from a magma ocean with a bulk silicate Earth composition6 at presumed typical conditions for the early Earth5,7. We employ first-principles molecular dynamics calculations. We find that volatile loss is determined by pressure, temperature, and melt composition. Carbon and helium devolatilize more easily when both elements are present in the melt, leading to pronounced loss from the magma ocean. Our findings suggest that the early Earth atmosphere was carbon-rich, with a high fraction of helium and other noble gases, and thicker and hotter than previously thought. We evaluate the implications for Earth’s composition and thermal evolution, as well as other planets that may undergo similar processes with different chemistries.
Similar content being viewed by others
Introduction
Rocky planetary bodies undergo a similar accretion process, yielding common outcomes. Early on, planetesimals cohere into planetary embryos, which, depending on the formation speed and time, may capture helium and hydrogen gas from the solar nebula8. Some of these gases are trapped in the interior, while the remainder contributes to primordial atmospheres, which might be partially eroded after dissipation of the nebula9. Extensive melting due to accretionary impacts or radiogenic heat results in magma oceans (MO)10. Volatile elements are continuously delivered to planetary embryos11 and are subsequently dissolved in magma bodies or outgassed to form secondary atmospheres. Large impacts also cause extensive melting and vaporization, resulting in resets of the atmosphere12. For Earth, such an impact led to a protolunar disk1,2, from which the Earth and Moon condensed into MO states13. Many volatiles survived the impact14,15 and became well mixed within the silicate liquid in the inner parts of the disk. Heavier volatiles, such as noble gases or carbon, became part of the early Earth, either as dissolved species in the MO or as part of the atmosphere3,4. This volatile and silicate-rich atmosphere was thick and insulating, reaching pressures of several kilobars5,7, delaying MO solidification5,16, and prolonging MO devolatilization5. The devolatilization rate depends on the thermochemical evolution at the MO surface. At first, vaporization was unhindered, but as crystallization advanced, sluggish melt percolation enabled the formation of a surface lid, limiting degassing17. Understanding the factors controlling volatile degassing is paramount to constrain the global budget of primitive volatiles and the conditions and timescales under which Earth crystallized.
Here, we examine carbon’s influence on He degassing from the global MO of the early Earth. We perform ab initio molecular dynamics simulations on pyrolite melt, which reproduces the bulk silicate Earth composition (BSE)6, to which we have added helium and CO2 molecules. Noble gases (particularly He) are used to study degassing18,19 due to their lack of chemical bonding, which makes them excellent tracers of physical processes. Each noble gas has at least one stable and one radiogenically produced isotope, and the ratios of these isotopes allow us to identify relative ages of degassed source material20,21. Carbon is easily devolatilized, and studies show that CO and CO2 dominated the early atmosphere17,22,23. As noble gases were undersaturated in the MO, they could not nucleate bubbles on their own. Instead, they partitioned into bubbles filled with CO2 or other volatiles24,25,26. While previous work examined independent helium27 and carbon22 vaporization from pyrolite, this is the first study to examine their concurrent vaporization.
Results
Nanobubble structure
Above ~ 25 kbar, our melt maintains a continuously polymerized network, indicating that carbon and helium are completely dissolved. With decreasing pressure, nanoscale cavities spontaneously form within the liquid, as previously witnessed in petrology experiments28. These nanobubbles are populated by disconnected, gas-like species, vaporized from the melt (Fig. 1a). Over multiple picoseconds, the nanobubbles change in size, shape, and location. At lower pressures, more vapor is present and the size of the nanobubbles grow, occupying around 64 percent of the total volume in the simulation cell at 3000 K at the largest simulated cell size (Table S1). As the nanobubbles move around over the course of a simulation, the interface between liquid and vapor evolves, encompassing various geometries (Fig. S1). It is important to note that large liquid-vapor interfaces can introduce artifacts into partitioning behavior due to surface effects29. Although the volume of the vapor phase in our simulations is never larger than 65 percent of the total, as liquid-vapor interfaces grow at lower pressures, there is increased uncertainty in the partitioning results. He, CO, and CO2 are common vapor species for this system. Helium and carbon traverse the cavity, exchanging and (for carbon) bonding with melt atoms. Helium diffuses more quickly than carbon and moves ballistically through the nanobubble, as expected from its small mass and lack of bonding. Both atoms span the cell, occupying both the nanobubble and the melt network (Fig. 1b). Quantifying the time each volatile spends in the liquid versus the vapor allows us to determine their solubilities within the melt.

Vapor and melt coexist within the simulation cell. The nanobubble has been centered in the image. The image contains the entire simulation cell, which is a cubic cell with a length of 21 Å. a A large central cavity containing He, CO, and CO2 vapor species coexists with silicate melt. b Example trajectories of a helium atom (straight red line) and a carbon atom (oscillating blue line) over the length of one simulation. The volatile species traverse the length of the cavity, and exchange and interact with atoms in the melt phase. Helium moves ballistically through the simulation cell, while carbon’s motion is influenced by vibrations, librations, and interactions with other atoms. In natural magmas, the bubbles are not constrained spatially but can merge and migrate, advancing the degassing process.
Helium vaporization
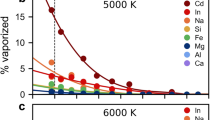
He is devolatilized as isolated atoms (Fig. 1a). With decreasing pressure, larger nanobubbles nucleate within the melt, allowing more helium vapor to congregate inside (Fig. 2). Degassing is very efficient at low pressures. Below 1 kb, He is totally degassed (Fig. 2a). Helium vapor lifetimes are longer at lower pressures (Fig. S2). At 2000 K, the average lifetime at 0.8 kb is 32 fs but only 14 fs at 3.8 kb. Degassing is also temperature-dependent. At higher temperatures, the system has more kinetic energy, and more helium vaporizes. For instance, at 5 kb, ~ 6%, 28%, 44%, and 53% of helium is vaporized at 2000, 3000, 4000, and 5000 K, respectively (Fig. 2a). Variations in vaporization behavior arising from multiple starting configurations are captured in the error bars in Fig. 2. Simulations at higher temperatures tend to be more precise due to the increased movement of atoms, which allows for more configurational space to be sampled during a single simulation. In our volume-constrained simulations, helium vapor lifetimes decrease slightly with temperature, as more kinetic energy facilitates exchange between melt and vapor (Fig. S2). It is possible that in natural systems, temperature enhances He escape due to bubble coalescence.

The data is fit to an exponential function. Error bars on the pressure and vaporization numbers showcase variations arising from multiple starting configurations. There is more uncertainty in the carbon vapor trend compared to helium, because carbon vapor is composed of multiple different species and can bond with atoms in both the melt and gas. a Percentage of He vaporization from this study. He vaporization increases at lower pressures and higher temperatures. b C vaporization percentages from this study. C vaporization follows the same trends as He vaporization. c–f Vaporization of He and C at various temperatures. At lower temperatures, He and C have similar devolatilization numbers, but at higher temperatures, He devolatilizes more readily than C. Comparing this study (circles) to the vaporization numbers of Özgürel and Caracas27 (x’s) and Solomatova and Caracas22 (triangles), we find that the vaporization of He and C is enhanced when both elements are present.
We examine CO2’s effect on helium vaporization by comparing our data to previous work on carbon-free pyrolite melts (Fig. 2d–f). CO2 enhances helium degassing, indicating that additional volatiles stabilize nanobubbles and increase devolatilization. This effect is likely from increased porosity of the melt. Carbon addition causes the melt to become less densely packed, opening channels through which helium can escape. As carbon vaporizes, it creates pathways to the surface that helium can follow. At higher temperatures where more CO2 vapor is formed, helium degassing is further enhanced by the presence of CO2. At 5 kb, helium degassing is increased through CO2 addition by 87% at 5000 K, but only by 23% at 3000 K. CO2 has little effect on helium vapor lifetimes (Fig. S2a).
Our results can also be expressed as solubilities within the melt. Below ~ 10 kb, helium solubility increases linearly with pressure, obeying Henry’s law (Fig. S3a). Our calculated solubilities are slightly higher than experiments and calculations on silicate melts30,31, as the more complex pyrolite melt can incorporate more helium than simpler silicate melts32. At high pressures where no gas phase is exsolved, solubilities may increase if more helium atoms are present in the starting composition.
Carbon vaporization
Carbon vaporization is controlled by pressure, temperature, and composition (Fig. 2). Carbon devolatilizes more readily at lower pressures and higher temperatures (Fig. 2b). At 5000 K and 3.8 kb, 50% of carbon is devolatilized but only 18% at 9.9 kb. At 5 kb, the carbon vapor percent increases with temperature from 19% at 2000 K to 39% at 5000 K. There is more uncertainty in the carbon vapor trend compared to helium, because carbon vapor is composed of multiple different species and can bond with atoms in both the melt and gas. Carbon vapor lifetimes decrease with temperature (Fig. S2b) as carbon exchanges more rapidly between liquid and gas. Lifetimes are almost invariant with pressure. Although more species are vaporized at lower pressures, their exchange with melt atoms does not change. Carbon vaporization is enhanced by the presence of He (Fig. 2c–f). At 5 kb and 3000 K, carbon vaporization increases from 7% to 34% with helium. Below ~ 10 kb, carbon solubility increases linearly, demonstrating that it obeys Henry’s law (Fig. S3b). Our solubilities over a range of pressures at 2000 K (between ~ 0.3 and 1.5 weight %) match well with studies of CO2 solubility in silicate melts and glasses, which report values of 1.48 weight %, 7 weight %, and 0–2 weight % at pressures of 15–30 kb for andesitic melt33, olivine nephelinitic melt33, and sodium aluminosilicate glasses34, respectively. At high pressures, solubilities may further increase with the addition of carbon atoms.
Helium and carbon have similar devolatilization rates (Fig. 2c–f). At 2000 and 3000 K, the proportions of vaporized carbon and helium are within error. At higher temperatures, helium becomes more volatile than carbon, and this effect increases at low pressures. The differences in spread and slope of the vaporization curves indicate that carbon is less directly affected by changing thermodynamic conditions than helium, due to its complicated chemical bonding. Carbon vapor has much longer lifetimes compared to helium vapor, which diffuses more rapidly in and out of the melt (Fig. S2). For instance, at 0.8 kb, carbon vapor lives an average of 209 fs, while helium vapor only lives 32 fs.
Carbon exists as multiple species in the vapor. Carbon vapor speciation is overwhelmingly dominated by CO2 and CO, although other species like C, NaO2C, and C2O4 are present in small amounts. Uncertainties in carbon vapor speciation are high, especially at low temperatures, because only a subset of the four included carbon atoms are present in the vapor over the course of a simulation (see Methods). At pressures below ~ 5 kb, temperature has little effect on CO2 and CO abundance, with ~ 70% consisting of CO2 and ~ 30% of CO (Fig. S4a, b). The preference for CO2 is unusual at low pressures, as cations typically favor lower coordination numbers35,36,37,38,39. In our coexisting melt-vapor system, however, CO2 abundance is tied to the number of oxygens within the nanobubble. With decreasing pressure, oxygen vaporization increases (see Supplementary Discussion). As more oxygen is available in the vapor, the coordination of carbon by oxygen increases. Above ~ 5 kb, there is a preference for CO with increasing temperature. Favoring lower coordination states at higher temperatures is expected in silicate melts22,35. Our carbon speciation results are in agreement with Solomatova and Caracas22 (Fig. S4d–f), although some variability arises from the limited number of carbon atoms. CO and CO2 lifetimes decrease with temperature (Fig. S5). CO tends to live longer than CO2, although the difference drops significantly with temperature.
Non-vaporized carbon can either be part of the bulk melt network or adsorbed to the melt surface. Which form carbon takes offers insight into its long-term liquid stability and its vaporization mechanism. Carbon in the bulk is deeply embedded and faces physical barriers preventing vaporization. Adsorbed carbon is tethered to the surface and has lifetimes dictated by individual carbon-oxygen bonds. During depressurization, carbon abundance in the bulk decreases exponentially while carbon abundance in the vapor rises (Fig. 3). At mid-pressures (3–8 kb), adsorbed carbon abundances reach a peak. Adsorbed, bulk, and vaporized carbon are about equal, and carbon freely exchanges between all phases. At our lowest pressures, nanobubbles are large and surface area available for adsorption is abundant. Nearly all non-vaporized carbon is adsorbed. The changing abundances in these distinct phases illustrate the transitory nature of carbon’s behavior with pressure. With increasing temperature, non-vaporized carbon prefers to be adsorbed, in line with increased vaporization at similar conditions. With helium, there is a slight increase in adsorbed carbon and a slight decrease in bulk carbon.

Percent of all carbon-bearing species that are vaporized, part of the melt network (bulk), or adsorbed to the melt surface at (a) 2000 K; b 3000 K; c 4000 K; and d 5000 K as a function of pressure. Vaporized and bulk data are fit to exponential functions. Error bars represent uncertainty on the pressure calculations and carbon percentages. At high pressures, carbon is mostly present in the melt network, while vaporized and adsorbed species increase in abundance with decreasing pressure.
Discussion
Due to the size of our simulations, we visualize the formation of bubbles at the nanoscale. In reality, degassing occurs on a much larger scale, and our simulations illustrate the initialization of that process. In the MO, nanobubbles would not only coalesce but continue to grow in size to form larger vapor-filled bubbles, where the size of the bubble is dependent on the concentration of volatiles in the magma40. These bubbles have low density compared to the liquid and rise towards the MO surface over a few hundred meters. However, increased bubble formation does not directly translate to more efficient degassing. Bubble formation induces turbulence in the MO41, which inhibits bubble ascent and traps bubbles in descending liquid. Previous work demonstrated that only small bubbles below 1 mm in size are likely to be entrained in the magma5. Additionally, both vigorous convection and sufficiently long MO lifetimes are required to expose undegassed magma to the lower pressures at the MO surface where degassing occurs42. Due to high temperatures in the MO following the giant impact, the viscosity of the MO was low, allowing for fast convection and the exposure of the majority of the MO to surface conditions5,43. While there is uncertainty both in the efficiency of bubble ascent to the MO surface and in the fraction of the MO that reaches shallow depths, our simulations represent upper bounds on degassing efficiencies based on reasonable estimates of fast convective velocities and large bubble sizes.
Figure 4 presents helium and carbon vaporization as a function of temperature at pressures representing different stages in the Earth’s evolution. Following the giant impact, the Earth’s proto-atmosphere reached pressures and temperatures of several kilobars and thousands of kelvin at the MO surface5,7,44. At these conditions ( ~ 5000 K and 5 kb), vaporization was elevated due to high temperatures; about half of the helium and 40% of the carbon at the MO surface degassed. As the MO cooled, ( ~ 3000–4000 K and 3 kb), degassing was elevated due to lower atmospheric pressures. Slightly more than half the helium and carbon would be lost from the MO surface. In final-stage melts, similar to present-day conditions, ( ~ 2000 K and 1 kb), the vaporization of helium and carbon was efficient ( ~ 65 and 70% loss of helium and carbon, respectively). Our results are in agreement with estimates from present-day degassing of mixed volatile-bearing MORB45 and felsic melts46,47, with vaporization percentages of 65-95% for helium and CO2. These studies concur that carbon and helium vaporize at roughly the same rate.

Shaded areas are uncertainty bands on the vaporization numbers and temperatures. a Helium vaporization is enhanced at lower pressure, at higher temperatures, and in the presence of CO2. b Carbon vaporization also increases with lower pressure, higher temperature, and He addition. The trends are not as strong as for helium, due to the more complicated speciation and bonding of carbon-bearing vapor. c Direct comparison of helium and carbon vaporization. Vaporization numbers are quite similar between the two elements, and helium becomes more volatile than carbon at higher temperatures.
CO2 has a strong greenhouse effect, and MO cooling would be influenced by CO2 vapor abundance. As the MO cools and releases heat, that heat is easily stored in the atmosphere by CO2, which creates an insulating blanket that prevents the efficient release of heat16. Our results demonstrate the existence of a feedback loop, where CO2 abundance increases with decreasing pressure and temperature (Fig. S4). As the MO degasses CO2 more efficiently over time, this increases the greenhouse effect of the atmosphere, which prolongs MO cooling. Previous simulations suggest that a steam atmosphere composed of CO2 and H2O prolongs MO solidification by ~ 1 Myr16. Moreover, our simulations predict that the presence of additional volatiles significantly increases devolatilization (Fig. 4a, b). This is particularly true in the case of He, where the addition of CO2 elevates He degassing numbers significantly above the range of error, especially at high temperatures (Fig. 4a). In fact, it has been observed that water and CO2 solubility in basaltic magmas is dependent on the initial volatile concentration, with higher proportions of H2O and CO2 degassing from melts with higher volatile concentrations48. Experiments studying noble gas solubility in the presence of H2O and CO2 find that they are less soluble in silicate melts with CO246,49. The higher degassing flux of volatiles would make the protoatmosphere more insulating than previously thought, futher delaying MO solidification and leading to more degassing.
As carbon is easily devolatilized and relatively abundant in the MO17,22,50, carbon-bearing vapor may have acted as a carrier gas for helium and other volatiles, effectively removing them from the MO. This is supported by experiments46, the strong correlation of CO2 and helium abundances in silicate melts51, and the constancy of their ratio over time52. The enhanced devolatilization of noble gases from the early MO in the presence of carbon needs to be thoroughly considered when assessing the Earth’s noble gas budget. With larger He losses in the MO stage, previous assumptions of accreted He should be revised upwards. This implies more efficient ingassing from the nebula53 and/or more irradiated chondritic material accreted before the Moon-forming impact54,55.
As our vaporization trends cover a wide range of thermodynamic conditions, our results are applicable to secondary atmosphere formation on other planetary bodies. For molten exoplanets around stars with high carbon contents, we would expect elevated degassing due to the prevalence of carbon-bearing vapor. Atmospheres around these exoplanets would be thick, and their interiors would be depleted in volatiles. Around more carbon-poor stars, molten planetary bodies would experience less degassing, and thus have thinner atmospheres and volatile-rich interiors. These planetary bodies would solidify more quickly. Planetary bodies forming a flotation crust early in their cooling histories would store their volatiles within the interior, where they would further affect geochemical processes and planetary evolution.
Methods
We performed ab initio molecular dynamics simulations on a pyrolite6,56 melt with a stoichiometry of NaCa2Fe4Mg30Al3Si24O89, which simulates the composition of the bulk silicate Earth to better than 1 wt.%. To study the vaporization behavior of volatile species in MOs, we added four atoms of helium and four molecules of CO2 to the 153-atom pyrolite supercell. This composition is 1.4 weight % C, which is typical of carbonaceous chondrites. Due to constraints in the size of simulations using ab initio methods, we have added more helium than is observed in the Earth or in chondrites to achieve sufficient statistics to analyze partitioning behavior. Previous work examining partitioning using the two-phase method indicates that simulations with approximately 175 atoms per phase are sufficient to accurately describe partitioning behavior27,57,58. The interatomic forces were computed using the projector-augmented wave method59 of density functional theory as implemented within the Vienna Ab initio Simulation Package60. Electron exchange and correlation were treated in the Perdew-Burke-Ernzerhof form61 of the generalized gradient approximation. We used a kinetic energy cutoff of 550 eV for the plane-wave expansion of the wave functions and of 800 eV for the augmentation charges. We employed the NVT canonical ensemble with temperature controlled by a Nosé-Hoover thermostat62. The Brillouin zone was sampled at the Gamma point, due to the large size of the simulation cell. The mean square displacement as a function of time displays a ballistic regime below about 100 fs, after which the atoms reach a diffusive regime. A timestep of 1-2 fs was used, and simulations generally lasted between 10 to 40 ps, depending on the pressure and temperature conditions. These simulations are sufficiently long to ensure that many different chemical environments are sampled for the speciation analysis. Van der Waals forces were excluded as we found that they did not significantly impact vapor abundance and speciation (i.e. 6% difference in helium vapor abundance at 0.8 kb).
Simulations were conducted at temperatures ranging from 2000 to 5000 K and pressures from 0.5 to 256 kb to represent conditions from the early MO to present-day magmas. To ensure a wide sampling of configurational space, each pressure and temperature condition was simulated using at least four starting configurations, and results were averaged. The length of the simulations and the different initial configurations ensure that many different chemical environments are sampled for the speciation analyses. The pressure range was sufficient to observe the formation of nanobubbles in the pyrolite melt structure which occurs at pressures below ~ 25 kb. These nanobubbles contain small, distinct gas-like species. The species freely enter and exit the void space of the nanobubbles, and thus coexist as both dissolved species in the silicate melt and as vapor species within the nanobubbles. The preference of an element for the vapor phase was determined by measuring the time they spent in the nanobubbles versus the surrounding silicate, which varies based on pressure, temperature, and melt composition. As the nanobubbles change in size, shape, and location over the duration of a simulation run, we utilized the presence of chemical bonds to determine whether a species was dissolved in the melt. Bonding atoms were identified by examining the pair distribution functions of atomic pairs. The first maximum in the pair distribution function corresponds to the most likely bond distance, while the first minimum corresponds to the edge of the first coordination sphere. Fitted values of the first minimums for each bonding pair were taken as maximum acceptable bond distances, which were used to determine the presence of bonding between any two atoms. Speciation analysis was conducted using the UMD package63, and every timestep in the simulation was analyzed to obtain the final speciation results.
To identify vapor species, we examined the bonding matrix for all atoms and identified small atomic clusters coexisting with larger interconnected webs of atoms. Small atomic clusters of 12 atoms or less were classified as vapor species. For these small clusters, no atom in the cluster is close enough to bond with an atom from the melt matrix, indicating that the cluster is spatially separated from the melt phase and exists within the nanobubble. This method of vapor species identification works well for atoms other than helium, as these elements bond with at least one other element. Helium, however, does not exhibit any bonding, making it impossible to distinguish dissolved and vaporized helium species with this method. Thus, to quantify helium vaporization, we analyzed simulations conducted at high pressure where no nanobubbles are formed and the helium atoms are fully dissolved within the silicate melt. We fit the first minimum in the He-O pair distribution function as a proxy for the maximum acceptable distance between helium and oxygen atoms when helium is fully dissolved in the silicate. This number is not a representation of He-O bonding, which does not exist, but rather an expression of the typical distance between helium and oxygen atoms when helium is dissolved in the melt. We then analyzed the coordination of helium to oxygen with this fitted distance and found that helium exhibits a coordination number to oxygen of 3 or more when fully dissolved in the melt phase. Using our average He-O distance from the high pressure simulations, we applied this method to our simulations at lower pressure, and classified helium atoms as vapor species when they exhibited coordination numbers to oxygen of 2 or less. Combining these two methods for vapor phase determination, we examined the speciation of the system at every timestep in each simulation and counted the amount of time each atom spent in the vapor versus the liquid. Because of computational limitations on the size of the simulation cell and the number of atoms, the vaporization values have large uncertainties due to surface effects. However, the vaporization results we present are indicative of trends in volatility behavior with changing thermodynamic parameters, and as such, useful comparisons can be made especially between experiments conducted with similar methods.
To distinguish between carbon atoms dissolved into the bulk melt structure and carbon atoms adsorbed to the surface of the melt, we examined the local bonding environments of non-vaporized carbon atoms. For each timestep, we found the non-vaporized carbon atoms and identified all of the bonding atoms within the first coordination sphere. We then examined the coordination numbers of the bonding anions, and separated them into two categories. Some anions were solely bonded to the central carbon atom while others bonded to cations in addition to the central carbon atom. Anions with more than one bond were classified as atoms embedded in the melt network. Carbon atoms bonded to a single anion embedded in the melt network were classified as adsorbed, while carbon atoms with more than one bond to an anion embedded in the melt network were classified as atoms in the bulk melt structure. In other words, we identify carbon-bearing species tethered to the melt surface by only a singular carbon-anion bond, and classify these atoms as adsorbed carbon atoms. This method, while effective, undercounts the number of the adsorbed atoms, as some adsorbed carbon species may attach via multiple bonds to the melt surface. However, we anticipate that this particular type of adsorbed species is less common than the single tether case, and present our results as a reasonable lower bound for adsorption numbers.
Due to limitations in the number of atoms, ab initio molecular dynamics simulations cannot accurately predict pressure in low density systems64. Thus, the pressure of the gas phase was computed using a modified Van der Waals equation of state65,
where P is the gas pressure, N is the number of atoms in the gas phase, kB is the Boltzmann constant, T is the temperature, V is the volume of the gas phase, a is a constant that takes intermolecular interaction into account, and Na is Avogadro’s number. The constant a is different for each gas species, and values for the common gases found in our simulations (CO2, CO, He, O2) were taken from the Handbook of Chemistry and Physics66. To obtain the volume of the gas phase at each snapshot, we use the Bader charge analysis algorithm67,68,69, which uses minima in the electronic charge density to define Bader volumes. The Bader volume contains the electronic charge that is assigned to a specific atom, and thus, is a useful approximation of individual atom volumes. We run the Bader charge analysis algorithm with the vacuum flag set to auto, which assigns low electronic charge density points to a vacuum volume. The volume of the gas phase is then approximated by adding the volumes of individual gas molecules to the total vacuum volume. The Van der Waals equation calculates the gas pressure at one snapshot during the simulation. To account for fluctuations in gas density, we use a minimum of 6 snapshots and average the results to obtain a representative gas pressure for the entire simulation. When comparing our calculated pressure to the pressure calculated by VASP for a simulation at 5000 K with a density of 1.39 g/cc, we find that the pressures agree within 0.05 kb. Densities calculated from ab initio methods have been shown to systematically deviate from experimental values depending on the approximation used for the exchange correlation functional70,71. When comparing the zero pressure densities of our pure pyrolite liquid to experimentally determined densities of peridotite melt at 2000 K, we find a 7% density decrease, indicating that our calculated pressures are slightly high. However, pressure comparisons made between ab initio studies using the same methods are valid, since pressures are systematically off from experimental values.
Data availability
The simulation output files can be accessed at https://zenodo.org/records/1004507472. Any additional data can be requested by emailing the corresponding author.
Code availability
The Vienna Ab Initio Simulation Package (VASP) is a proprietary software available for purchase. Parameters for the input files are described in computational methods.
References
Canup, R. M. Simulations of a late lunar-forming impact. Icarus 168, 433–456 (2004).
Asphaug, E. Impact origin of the moon? Annu. Rev. Earth Planet. Sci. 42, 551–578 (2014).
Canup, R. M., Visscher, C., Salmon, J. & Fegley Jr, B. Lunar volatile depletion due to incomplete accretion within an impact-generated disk. Nat. Geosci. 8, 918–921 (2015).
Charnoz, S. & Michaut, C. Evolution of the protolunar disk: Dynamics, cooling timescale and implantation of volatiles onto the Earth. Icarus 260, 440–463 (2015).
Elkins-Tanton, L. Linked magma ocean solidification and atmospheric growth for Earth and Mars. Earth Planet. Sci. Lett. 271, 181–191 (2008).
McDonough, W. & Sun, S.-s The composition of the Earth. Chem. Geol. 120, 223–253 (1995).
Liu, L.-g The inception of the oceans and CO2-atmosphere in the early history of the Earth. Earth Planet. Sci. Lett. 227, 179–184 (2004).
Sasaki, S. & Nakazawa, K. Did a primary solar-type atmosphere exist around the proto-earth? Icarus 85, 21–42 (1990).
Sekiya, M., Hayashi, C. & Nakazawa, K. Dissipation of the primordial terrestrial atmosphere due to irradiation of the solar far-UV during T Tauri stage. Prog. Theor. Phys. 66, 1301–1316 (1981).
Elkins-Tanton, L. T. Magma oceans in the inner solar system. Annu. Rev. Earth Planet. Sci. 40, 113–139 (2012).
Marty, B. The origins and concentrations of water, carbon, nitrogen and noble gases on Earth. Earth Planet. Sci. Lett. 313-314, 56–66 (2012).
Ali-Dib, M., Cumming, A. & Lin, D. N. C. The effect of late giant collisions on the atmospheres of protoplanets and the formation of cold sub-Saturns. Monthly Not. R. Astronomical Soc. 509, 1413–1431 (2021).
Caracas, R. & Stewart, S. T. No magma ocean surface after giant impacts between rocky planets. Earth Planet. Sci. Lett. 608, 118014 (2023).
Pepin, R. O. Evolution of Earth’s noble gases: Consequences of assuming hydrodynamic loss driven by giant impact. Icarus 126, 148–156 (1997).
Abe, Y. Protoatmospheres and surface environment of protoplanets. Earth Moon Planets 108, 9–14 (2011).
Lebrun, T. et al. Thermal evolution of an early magma ocean in interaction with the atmosphere. J. Geophys. Res.: Planets 118, 1155–1176 (2013).
Bower, D. J., Hakim, K., Sossi, P. A. & Sanan, P. Retention of water in terrestrial magma oceans and carbon-rich early atmospheres. Planet. Sci. J. 3, 93 (2022).
Gonnermann, H. M. & Mukhopadhyay, S. Non-equilibrium degassing and a primordial source for helium in ocean-island volcanism. Nature 449, 1037–1040 (2007).
Torgersen, T. Terrestrial helium degassing fluxes and the atmospheric helium budget: Implications with respect to the degassing processes of continental crust. Chem. Geol.: Isot. Geosci. Sect. 79, 1–14 (1989).
Moreira, M. & Sarda, P. Noble gas constraints on degassing processes. Earth Planet. Sci. Lett. 176, 375–386 (2000).
Tolstikhin, I. Helium isotopes in the Earth’s interior and in the atmosphere: A degassing model of the Earth. Earth Planet. Sci. Lett. 26, 88–96 (1975).
Solomatova, N. V. & Caracas, R. Genesis of a CO2-rich and H2O-depleted atmosphere from Earth’s early global magma ocean. Sci. Adv. 7, eabj0406 (2021).
Walker, J. C. G. Carbon dioxide on the early Earth. Orig. Life Evol. Biosphere 16, 117–127 (1985).
Gerlach, T. M. Degassing of carbon dioxide from basaltic magma at spreading centers: II. Mid-oceanic ridge basalts. J. Volcanol. Geotherm. Res. 39, 221–232 (1989).
Jambon, A., Weber, H. & Begemann, F. Helium and argon from an Atlantic MORB glass: concentration, distribution and isotopic composition. Earth Planet. Sci. Lett. 73, 255–268 (1985).
Sarda, P. & Graham, D. Mid-ocean ridge popping rocks: Implications for degassing at ridge crests. Earth Planet. Sci. Lett. 97, 268–289 (1990).
Ozgurel, O. & Caracas, R. The magma ocean was a huge helium reservoir in the early Earth. Geochem. Persp. Lett. 25, 46–50 (2023).
Gudfinnsson, G. H. & Presnall, D. C. Continuous gradations among primary carbonatitic, kimberlitic, melilititic, basaltic, picritic, and komatiitic melts in equilibrium with garnet lherzolite at 3-8 GPa. J. Petrol. 46, 1645–1659 (2005).
Hong, Q.-J. & Van De Walle, A. Solid-liquid coexistence in small systems: A statistical method to calculate melting temperatures. J. Chem. Phys. 139, 094114 (2013).
Bouhifd, M. A., Jephcoat, A. P., Heber, V. S. & Kelley, S. P. Helium in Earth’s early core. Nat. Geosci. 6, 982–986 (2013).
Wang, K., Lu, X., Liu, X. & Yin, K. Noble gas (He, Ne, and Ar) solubilities in high-pressure silicate melts calculated based on deep-potential modeling. Geochimica et. Cosmochim. Acta 350, 57–68 (2023).
Carroll, M. R. & Stolper, E. M. Noble gas solubilities in silicate melts and glasses: New experimental results for argon and the relationship between solubility and ionic porosity. Geochimica et. Cosmochim. Acta 57, 5039–5051 (1993).
Mysen, B. O., Arculus, R. J. & Eggler, D. H. Solubility of carbon dioxide in melts of andesite, tholeiite, and olivine nephelinite composition to 30 kbar pressure. Contr. Mineral. Petrol. 53, 227–239 (1975).
Fine, G. & Stolper, E. The speciation of carbon dioxide in sodium aluminosilicate glasses. Contr. Mineral. Petrol. 91, 105–121 (1985).
Solomatova, N. & Caracas, R. Pressure-induced coordination changes in a pyrolitic silicate melt from ab initio molecular dynamics simulations. J. Geophys. Res.: Solid Earth 124, 11232–11250 (2019).
Davis, A. H., Solomatova, N. V., Campbell, A. J. & Caracas, R. The speciation and coordination of a deep Earth carbonate-silicate-metal melt. J. Geophys. Res.: Solid Earth 127, e2021JB023314 (2022).
Paris, E., Dingwell, D. B., Seifert, F. A., Mottana, A. & Romano, C. Pressure-induced coordination change of Ti in silicate glass: A XANES study. Phys. Chem. Miner. 21, 510–515 (1994).
Mookherjee, M., Stixrude, L. & Karki, B. Hydrous silicate melt at high pressure. Nature 452, 983–986 (2008).
Williams, Q. & Jeanloz, R. Spectroscopic evidence for pressure-induced coordination changes in silicate glasses and melts. Science 239, 902–905 (1988).
Aubry, G. J., Sator, N. & Guillot, B. Vesicularity, bubble formation and noble gas fractionation during morb degassing. Chem. Geol. 343, 85–98 (2013).
Massol, H. & Koyaguchi, T. The effect of magma flow on nucleation of gas bubbles in a volcanic conduit. J. Volcanol. Geotherm. Res. 143, 69–88 (2005).
Salvador, A. & Samuel, H. Convective outgassing efficiency in planetary magma oceans: Insights from computational fluid dynamics. Icarus 390, 115265 (2023).
Liebske,C. et al. Viscosity of peridotite liquid up to 13 gpa: Implications for magma ocean viscosities. Earth Planet. Sci. Lett. 240, 589–604 (2005).
Zhang, J., Dauphas, N., Davis, A. M., Leya, I. & Fedkin, A. The proto-earth as a significant source of lunar material. Nat. Geosci. 5, 251–255 (2012).
Tucker, J. M., Mukhopadhyay, S. & Gonnermann, H. M. Reconstructing mantle carbon and noble gas contents from degassed mid-ocean ridge basalts. Earth Planet. Sci. Lett. 496, 108–119 (2018).
Paonita, A., Gigli, G., Gozzi, D., Nuccio, P. & Trigila, R. Investigation of the He solubility in H2O-CO2 bearing silicate liquids at moderate pressure: A new experimental method. Earth Planet. Sci. Lett. 181, 595–604 (2000).
Nuccio, P. & Paonita, A. Magmatic degassing of multicomponent vapors and assessment of magma depth: Application to Vulcano Island (Italy). Earth Planet. Sci. Lett. 193, 467–481 (2001).
Dixon, J. E. & Stolper, E. M. An experimental study of water and carbon dioxide solubilities in mid-ocean ridge basaltic liquids. Part II: Applications to degassing. J. Petrol. 36, 1633–1646 (1995).
Nuccio, P. & Paonita, A. Investigation of the noble gas solubility in H2O-CO2 bearing silicate liquids at moderate pressure II: The extended ionic porosity (EIP) model. Earth Planet. Sci. Lett. 183, 499–512 (2000).
Kuwahara, H., Itoh, S., Nakada, R. & Irifune, T. The effects of carbon concentration and silicate composition on the metal-silicate partitioning of carbon in a shallow magma ocean. Geophys. Res. Lett. 46, 9422–9429 (2019).
Trull, T., Nadeau, S., Pineau, F., Polve, M. & Javoy, M. C-He systematics in hotspot xenoliths: Implications for mantle carbon contents and carbon recycling. Earth Planet. Sci. Lett. 118, 43–64 (1993).
Hilton, D. R., McMurtry, G. M. & Kreulen, R. Evidence for extensive degassing of the Hawaiian Mantle Plume from helium-carbon relationships at Kilauea Volcano. Geophys. Res. Lett. 24, 3065–3068 (1997).
Olson, P. L. & Sharp, Z. D. Primordial helium-3 exchange between Earth’s core and mantle. Geochem. Geophys. Geosyst. 23, e2021GC009985 (2022).
Péron, S., Moreira, M. & Agranier, A. Origin of light noble gases (He, Ne, and Ar) on Earth: A review. Geochem. Geophys. Geosyst. 19, 979–996 (2018).
Moreira, M. & Charnoz, S. The origin of the neon isotopes in chondrites and on Earth. Earth Planet. Sci. Lett. 433, 249–256 (2016).
Caracas, R., Hirose, K., Nomura, R. & Ballmer, M. D. Melt-crystal density crossover in a deep magma ocean. Earth Planet. Sci. Lett. 516, 202–211 (2019).
Yuan, L. & Steinle-Neumann, G. Earth’s “missing” chlorine may be in the core. J. Geophys. Res.: Solid Earth 129, e2023JB027731 (2024).
Zhang, Y. & Guo, G. Partitioning of si and o between liquid iron and silicate melt: A two-phase ab-initio molecular dynamics study. Geophys. Res. Lett. 36, 18305 (2009).
Blöchl, P. E. Projector augmented-wave method. Phys. Rev. B 50, 17953–17979 (1994).
Kresse, G. & Furthmüller, J. Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set. Phys. Rev. B 54, 11169–11186 (1996).
Perdew, J. P., Burke, K. & Ernzerhof, M. Generalized gradient approximation made simple. Phys. Rev. Lett. 77, 3865–3868 (1996).
Nosé, S. A unified formulation of the constant temperature molecular dynamics methods. J. Chem. Phys. 81, 511–519 (1984).
Caracas, R. et al. Analyzing melts and fluids from ab initio molecular dynamics simulations with the UMD package. J. Vis. Exper. (2021).
Binder, K., Block, B. J., Virnau, P. & Tröster, A. Beyond the Van Der Waals loop: What can be learned from simulating Lennard-Jones fluids inside the region of phase coexistence. Am. J. Phys. 80, 1099–1109 (2012).
Van der Waals, J. D. On the Continuity of the Gaseous and Liquid States (Leiden, 1873).
Weast, R. Handbook of Chemistry and Physics 53rd edn (Chemical Rubber Pub., 1972). https://books.google.no/books?id=aXHKtAEACAAJ.
Tang, W., Sanville, E. & Henkelman, G. A grid-based bader analysis algorithm without lattice bias. J. Phys.: Condens. Matter 21, 084204 (2009).
Sanville, E., Kenny, S. D., Smith, R. & Henkelman, G. Improved grid-based algorithm for bader charge allocation. J. Comput. Chem. 28, 899–908 (2007).
Henkelman, G., Arnaldsson, A. & Jónsson, H. A fast and robust algorithm for Bader decomposition of charge density. Comput. Mater. Sci. 36, 354–360 (2006).
Zhang, Z., Stixrude, L. & Brodholt, J. Elastic properties of mgsio3-perovskite under lower mantle conditions and the composition of the deep earth. Earth Planet. Sci. Lett. 379, 1–12 (2013).
Zhao, G., Mu, H., Tan, X., Wang, D. & Yang, C. Structural and dynamical properties of mgsio3 melt over the pressure range 200–500gpa: Ab initio molecular dynamics. J. Non-Crystalline Solids 385, 169–174 (2014).
Davis, A. H. & Caracas, R. Output Files for Degassing of CO2 Triggers Large-scale Loss of Helium from Magma Oceans (Zenodo, 2023). https://zenodo.org/records/10045074.
Acknowledgements
This research was supported by the Research Council of Norway (NFR) (Project number 325567-HIDDEN to R.C) and the European Research Council under EU Horizon 2020 research and innovation program (grant agreement 681818 - IMPACT to RC). This project has received funding from the Research Council of Norway through the Centres of Excellence funding scheme, project number 332523 (PHAB). The simulations were performed on resources provided by Sigma2 - the National Infrastructure for High-Performance Computing and Data Storage in Norway (Uninet2 NN9697K grant) and supercomputing facilities in France via eDARI stl2816 grant. We would also like to acknowledge two anonymous reviewers for their helpful insights which improved the manuscript.
Author information
Authors and Affiliations
Contributions
Anne H. Davis designed the study, performed calculations, analyzed the data, interpreted the results, discussed the implications, and wrote the manuscript. Razvan Caracas designed the study, performed calculations, interpreted the results, discussed the implications, and contributed to writing the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Communications Earth & Environment thanks Zhongqing Wu and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. Primary Handling Editor: Carolina Ortiz Guerrero. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Davis, A.H., Caracas, R. Degassing of CO2 triggers large-scale loss of helium from magma oceans. Commun Earth Environ 5, 344 (2024). https://doi.org/10.1038/s43247-024-01509-1
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s43247-024-01509-1
- Springer Nature Limited