
Abstract
The rock bream (Oplegnathus fasciatus) is one of the most economically valuable marine fish in East Asia, and due to various environmental factors, there is substantial revenue loss in the production sector. Therefore, knowledge of its genome is required to uncover the genetic factors and the solutions to these problems. In this study, we constructed the first draft genome of O. fasciatus as a reference for the family Oplegnathidae. The genome size is estimated to be 749 Mb, and it was assembled into 766 Mb by combining Illumina and PacBio sequences. A total of 24,053 transcripts (23,338 genes) are predicted, and among those transcripts, 23,362 (97%), are annotated with functional terms. Finally, the completeness of the genome assembly was assessed by CEGMA, which resulted in the complete mapping of 220 (88.7%) core genes in the genome. To the best of our knowledge, this is the first draft genome for the family Oplegnathidae.
Design Type(s) | sequence analysis objective • sequence assembly objective |
Measurement Type(s) | whole genome sequencing |
Technology Type(s) | DNA sequencing |
Factor Type(s) | |
Sample Characteristic(s) | Oplegnathus fasciatus |
Machine-accessible metadata file describing the reported data (ISA-Tab format)
Similar content being viewed by others

Background & Summary
Oplegnathus fasciatus (commonly known as rock bream, barred knifejaw or striped beakfish), is a fish belonging to the family Oplegnathidae. Those common names are derived from its phenotypic features. Rock bream is a subtropical and carnivorous species and is an economically important teleost fish in East Asia1. Generally, the rock bream inhabits estuaries at various depths according to their growth stage, i.e., as juveniles, they are mostly found in drifting seaweed/algae, and as adults, they are present at depths of 1 to 10 meters1. Moreover, the species growth depends on the photoperiod2. Other factors, such as overfishing and environmental changes, are affecting fish yield and cost, particularly in wild conditions. To overcome these issues, O. fasciatus is propagated via aquaculture to achieve sustainable and cost-effective production. In 2008, the annual production of O. fasciatus in South Korea was 614 tons, and that figure had increased to 909 tons in 20163. However, bacterial and viral diseases cause an enormous economic loss in the Korean aquaculture industry3. As a consequence, the scientific community continues to seek various solutions, including molecular genetic applications, to overcome those problems. Some examples of these applications include genetic breeding4, QTL marker identification5, characterization of immunological pathway genes, proposed sex determination6, sex chromosomal evolution models6, antimicrobial peptides7,8, and vaccine development9.
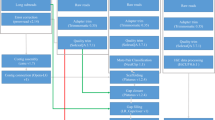
More and more often, advances in molecular sequencing technologies are supporting the scientific community in uncovering the inherited molecular mechanisms of a given species, rather than depending on its model organism10. In this study, we constructed a draft genome for O. fasciatus using next-generation sequencing (NGS) (Fig. 1), which could aid in functional characterization of O. fasciatus-associated problems.

(a) the genome assembly pipeline, (b) the structural and functional annotation pipeline, (c) details of the reference gene sets used for the ab initio and evidence-based gene model predictions.
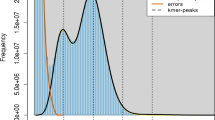
The O. fasciatus genome size is estimated to be ~749 Mb (Fig. 2a) and was assembled into scaffolds with a total size of 762 Mb. Initially, the 224 Gb Illumina library (Table 1) assembled into 108,639 contigs and 31,533 scaffolds. Although the assembled scaffolds are larger than the estimated genome size, it is highly fragmented (Table 2). Therefore, the inclusion of 11.5 Gb of PacBio sequences in the second assembly improved the quality of the overall draft genome when compared to the initial assembly (Fig. 1a). This addition resulted in a 766 Mb draft genome with 4,149 scaffolds, along with improvements to the N50 (0.87 Mb to 1.1 Mb) and to the gaps (5.3% to 5.2%) (Table 2). Furthermore, the repeats were predicted by the de novo method were classified into subclasses (Table 3). In total, 180 Mb (23.56%) of genomic regions consist of repeat sequences, and it is masked in the genome.

(a) k-mer based genome size estimation, (b) sequence similarity-based species distribution obtained from BLAST.
A total of 334.3 Gb of mRNA transcriptome sequences from 34 libraries (313.8 Gb of Illumina data and 20.5 Gb of Iso-Seq data) was used for the EVM, and seven genomes were used in the ab initio gene modeler. These analyses predicted 23,338 genes and 24,053 transcripts, and 23,362 (97%) of those transcripts were annotated from biological databases. Moreover, the completeness score produced from CEGMA indicated that 220 (88.7%) eukaryotic core genes are entirely mapped to the genome. Therefore, these results clearly show that the given draft genome could be a near-complete reference genome for O. fasciatus. Moreover, these scaffolds will act as a primary genetic resource for O. fasciatus that can be used to design functional studies, and the annotated transcripts (97%) will aid in detailed characterizations. Finally, based on a literature survey and author knowledge, this is the first draft genome presented to the public from the family of Oplegnathidae; therefore, these data could be a valuable asset for marine researchers.
Methods
Sample collection and genomic DNA extraction
A single rock bream fish (95 ± 5 g) was supplied by the Gyeongsangnam-do Fisheries Resources Research Institute (FRRI) (Tongyeong, Republic of Korea) and was maintained at 22 ± 0.5 °C in aerated seawater. Liver tissue was taken from the fresh rock bream aseptically and stored in liquid nitrogen for the extraction of the genomic DNA. The genomic DNA was extracted using a DNeasy Animal Mini Kit (Qiagen, Hilden, Germany). A total of 24 μg of DNA was quantified using the standard procedure for the Quant-iT PicoGreen ds-DNA Assay Kit (Molecular Probes, Eugene, OR, USA) with a Synergy HTX Multi-Mode Reader (Biotek, Winooski, VT, USA). The quality of the DNA was also checked using an ND-1000 spectrophotometer (Thermo Scientific, Wilmington, DE, USA).
DNA library preparation and sequencing
High-quality high molecular-weight genomic DNA > 100 kb in length was isolated from the given tissues, and two protocols were used to construct the sequencing libraries according to the manufacturer protocols, i.e., Illumina paired-end (PE) and mate pair (MP) libraries, (Illumina, San Diego, CA, USA). Furthermore, these libraries were fragmented and size-selected for Illumina Hi-Seq sequencing (Table 1). To obtain long non-fragmented sequence reads from the libraries, the PacBio manufacturing protocols were used (Pacific Biosciences, CA, USA) with 14 cells, and the sequencing used the P6-C4 chemistry of the PacBio RS II system (Table 1).
Preprocessing and genome size estimation
The entire Illumina DNA sequences were subjected to pre-processing steps, which included adapter trimming, quality trimming (Q20) and contamination removal. The adapter and quality trims were conducted by using Trimmomatic-0.32 functions11, and the microbial contamination of each sample was removed by CLCMapper v4.2.0 (https://www.qiagenbioinformatics.com/products/clc-assembly-cell/) with an in-house database. Here, the in-house database was constructed from the meta-genomes (bacteria (ftp://ftp.ncbi.nlm.nih.gov/genomes/GENOME_REPORTS/prokaryotes.txt), virus (ftp://ftp.ncbi.nlm.nih.gov/genomes/Viruses/) and marine metagenomes (https://www.ncbi.nlm.nih.gov/bioproject/PRJNA13694). Similarly, mate pair sequences were also subjected to adapter and quality trimming, and classification of the mate pairs was performed using the Nextclip v1.1 method12. All the pre-processed sequences (Insert size: 550 bp, 35 Gb) from the paired-end library (Data Citation 1) were subjected to genome size estimation using the k-mer based method (which was used in the panda genome13). The k-mer frequencies (k-mer size = 19) were obtained using the Jellyfish v2.0 method14, and the genome size was calculated from the given formulas: Genome Coverage Depth = (k-mer Coverage Depth × Average Read Length)/(Average Read Length – k-mer size + 1) and Genome size = Total Base Number/Genome Coverage Depth. Alternatively, the PacBio sequences were only subjected to error correction using CLCAssemblyCell v4.2.0 (Fig. 1a).
De novo Genome Assembly and Scaffolds
The draft genome was built from two type of assemblies, i.e., short-read assemblies and hybrid assemblies. Initially, the complete pre-processed paired-end DNA sequences were subjected to CLCAssemblyCell v4.2.0 to build the contigs. Furthermore, it was scaffolded with mate-pair sequences using the SSPACE v3.0 method15, and the hybrid assembly was built with the SSPACE-LongRead v1.0 method16 from the scaffolds along with the processed PacBio sequences. Next, the hybrid scaffolds were subjected to gap filling with paired-end and mate pair libraries using the GapFiller 1.11 method17. Finally, the gene completeness was assessed using CEGMA18 (Fig. 1a).
De novo repeat region prediction and classification
Initially, repeat regions were predicted using the de novo method and classified into repeat subclasses (Table 3). The de novo repeat prediction for O. fasciatus was conducted using RepeatModeler (http://www.repeatmasker.org/RepeatModeler/), which includes other methods such as RECON19 (http://eddylab.org/software/recon/), RepeatScout20 (https://bix.ucsd.edu/repeatscout/) and TRF21 (https://tandem.bu.edu/trf/trf.html). Furthermore, the repeats were masked using RepeatMasker v4.0.5 (http://www.repeatmasker.org/) with RMBlastn v2.2.27+ and classified into their subclasses using the Repbase22 v20.08 databases for reference (https://www.girinst.org/repbase/).
Gene prediction and annotation
The genes from the O. fasciatus draft genome were predicted using an in-house gene prediction pipeline, which includes three modules: an evidence-based gene modeler (EVM), an ab initio gene modeler and a consensus gene modeler. Finally, the functional annotation processing was conducted for the consensus genes (Fig. 1b). The details of this pipeline were previously explained in articles on the genomes of Capsicum23 and Haliotis24. Initially, the sequenced transcriptomes from two sequencers (Illumina (313.8 GB) and IsoSeq (27.7 GB)) were mapped to the O. fasciatus repeat-masked draft genome using Tophat25, and the transcript/gene structural boundaries were predicted using Cufflink25 and PASA26. To train the ab initio gene modeler and the EVM (which includes Exonerate27, AUGUSTUS28, and GENEID29), several genomes (Gasterosteus aculeatus, Oreochromis niloticus, Tetraodon nigroviridis, Takifugu rubripes, Oryzias latipes, Danio rerio, and Homo sapiens) were used for prediction. Finally, the predicted gene and transcripts models from the EVM and ab initio modeler were subjected to the consensus gene modeler (which includes EVidenceModeler30) to produce the final gene and transcript models. Finally, the consensus transcripts were subjected to functional annotation from biological databases (NCBI - NR databases, Uniprot, Gene Ontologies and KEGG pathways) by using Blast2GO31 (Fig. 1b). From this annotation, 50% of the genes are highly similar to Larimichthys crocea (Fig. 2b).
Code availability
Throughout this study, we were not used any custom specific codes. The command line at each step were executed as instructed in the respective bioinformatics methods.
Data Records
The entire data set used for draft assembly and its corresponding functional and structural annotations were deposited in public repositories. The DNA sequence libraries were deposited in NCBI (Data Citation 1) and see Table 1 for the details. The final assembly super-scaffold were submitted to NCBI Assembly (Data Citation 2) and see Table 2 for details. Moreover, the other files, such as the assembled contigs, scaffolds, and annotation tables, were stored in figshare (Data Citation 3) and see Table 4 for the details.
Technical Validation
Throughout this study, every step was validated with the given metrics. The sampled fish were cultured under controlled conditions in the FRRI. Furthermore, the sequence libraries were quantified with different parameters. For Illumina, the isolated DNA spectrophotometer ratios (SP) were 260/280 ≥ 1.6 and total DNA ≥ 1.1 μg with minimum 20 ng/μl, and for PacBio, the SP was 260/280 ≥ 1.6 and 260/230 ≥ 2.0 and total DNA ≥ 15 μg with minimum 200 ng/μl. Moreover, the default parameters were used in the bioinformatics methods.
Additional information
How to cite this article: Shin, Y. et al. First draft genome sequence of the rock bream in the family Oplegnathidae. Sci. Data. 5:180234 doi: 10.1038/sdata.2018.234 (2018).
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
References
Oh, D. J. et al. Complete mitochondrial genome of the rock bream Oplegnathus fasciatus (Perciformes, Oplegnathidae) with phylogenetic considerations. Gene. 392, 174–180 (2007).
Biswas, A., Takaoka, O., Kumai, H. & Takii, K. Combined effect of photoperiod and self-feeder on the growth performance of striped knifejaw. Oplegnathus fasciatus. Aquaculture. 452, 183–187 (2016).
Statistics Korea. Preliminary Results of the Survey on the Status of Fish Culture in 2017. Korean National Statistical Office (2018).
Kim, J. H., Kim, W. S. & Oh, M. J. Development of Fifteen Novel Microsatellite Markers from Rock Bream (Oplegnathus fasciatus). Open Journal of Genetics 4, 287–291 (2014).
Molecular Ecology Resources Primer Development, C. et al. Permanent genetic resources added to Molecular Ecology Resources Database 1 October 2010-30 November 2010. Mol Ecol Resour. 11, 418–421 (2011).
Xu, D. et al. The testis and ovary transcriptomes of the rock bream (Oplegnathus fasciatus): A bony fish with a unique neo Y chromosome. Genom Data 7, 210–213 (2016).
Bae, J. S. et al. Piscidin: Antimicrobial peptide of rock bream, Oplegnathus fasciatus. Fish Shellfish Immunol 51, 136–142 (2016).
Umasuthan, N., Mothishri, M. S., Thulasitha, W. S., Nam, B. H. & Lee, J. Molecular, genomic, and expressional delineation of a piscidin from rock bream (Oplegnathus fasciatus) with evidence for the potent antimicrobial activities of Of-Pis1 peptide. Fish Shellfish Immunol 48, 154–168 (2016).
Brudeseth, B. E. et al. Status and future perspectives of vaccines for industrialized fin-fish farming. Fish Shellfish Immunol 35, 1759–1768 (2013).
Goodwin, S., McPherson, J. D. & McCombie, W. R. Coming of age: ten years of next-generation sequencing technologies. Nat Rev Genet 17, 333–351 (2016).
Bolger, A. M., Lohse, M. & Usadel, B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics. 30, 2114–2120 (2014).
Leggett, R. M., Clavijo, B. J., Clissold, L., Clark, M. D. & Caccamo, M. NextClip: an analysis and read preparation tool for Nextera Long Mate Pair libraries. Bioinformatics 30, 566–568 (2014).
Li, R. et al. The sequence and de novo assembly of the giant panda genome. Nature. 463, 311–317 (2009).
Marçais, G. & Kingsford, C. A fast, lock-free approach for efficient parallel counting of occurrences of k-mers. Bioinformatics. 27, 764–770 (2011).
Boetzer, M., Henkel, C. V., Jansen, H. J., Butler, D. & Pirovano, W. Scaffolding pre-assembled contigs using SSPACE. Bioinformatics. 27, 578–579 (2011).
Boetzer, M. & Pirovano, W. SSPACE-LongRead: scaffolding bacterial draft genomes using long read sequence information. BMC Bioinformatics. 15, 211 (2014).
Nadalin, F., Vezzi, F. & Policriti, A. GapFiller: a de novo assembly approach to fill the gap within paired reads. BMC Bioinformatics. 13, S8 (2012).
Parra, G., Bradnam, K. & Korf, I. CEGMA: a pipeline to accurately annotate core genes in eukaryotic genomes. Bioinformatics. 23, 1061–1067 (2007).
Bao, Z. & Eddy, S. R. Automated denovo identification of repeat sequence families in sequenced genomes. Genome Res 12, 1269–1276 (2002).
Price, A. L., Jones, N. C. & Pevzner, P. A. Denovo identification of repeat families in large genomes. Bioinformatics. 21, 351–358 (2005).
Benson, G. Tandem repeats finder: a program to analyze DNA sequences. Nucleic Acids Res 27, 573–580 (1999).
Bao, W., Kojima, K. K. & Kohany, O. Repbase Update, a database of repetitive elements in eukaryotic genomes. Mobile DNA 6, 11 (2015).
Kim, S. et al. Genome sequence of the hot pepper provides insights into the evolution of pungency in Capsicum species. Nat Genet. 46, 270–278 (2014).
Nam, B. H. et al. Genome sequence of Pacific abalone (Haliotis discus hannai): the first draft genome in family Haliotidae. Gigascience. 6, 1–8 (2017).
Trapnell, C. et al. Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks. Nat. Protocols 7, 562–578 (2012).
Haas, B. J. et al. Improving the Arabidopsis genome annotation using maximal transcript alignment assemblies. Nucleic Acids Research 31, 5654–5666 (2003).
Slater, G. S. & Birney, E. Automated generation of heuristics for biological sequence comparison. BMC Bioinformatics. 6, 31 (2005).
Stanke, M., Schoffmann, O., Morgenstern, B. & Waack, S. Gene prediction in eukaryotes with a generalized hidden Markov model that uses hints from external sources. BMC Bioinformatics. 7, 62 (2006).
Blanco, E, Parra, G . & Guigó, R. in Current Protocols in Bioinformatics Vol. 00 Ch.4. John Wiley & Sons, Inc., (2002).
Haas, B. J. et al. Automated eukaryotic gene structure annotation using EVidenceModeler and the Program to Assemble Spliced Alignments. Genome Biol. 9, R7 (2008).
Gotz, S. et al. High-throughput functional annotation and data mining with the Blast2GO suite. Nucleic Acids Res 36, 3420–3435 (2008).
Data Citations
NCBI Sequence Read Archive SRP113430 (2017)
NCBI Assembly GCA_003416845.1 (2018)
Shin, Y. Figshare https://doi.org/10.6084/m9.figshare.5705269.v1 (2017)
Acknowledgements
This research was supported by the collaborative Grant Program of the Korea Institute of Marine Science and Technology Promotion (KIMST) funded by the Ministry of Oceans and Fisheries (MOF) (No. 20180430). And also this study was supported by a grant from the National Institute of Fisheries Science (NIFS) (No. R2018021).
Author information
Authors and Affiliations
Contributions
Sampling – Bo-Hye Nam, Jae Koo Noh, Young-Ok Kim, Jung Youn Park, Cheul Min An, Ju-Won Kim. Sequencing – Chan-Il Park, Bo-Hye Nam, Ga-hee Shin. Genome assembly – Younhee Shin, Myunghee Jung, Su-Jin Baek, Ji-man Hong. Gene prediction – Younhee Shin, Myunghee Jung, Ga-hee Shin, Ho-Jin Jung, Gi-young Lee. Funding and experimental design – Bo-Hye Nam, Chan-Il Park, Ga-hee Shin, Hojin Jung.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
ISA-Tab metadata
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/ The Creative Commons Public Domain Dedication waiver http://creativecommons.org/publicdomain/zero/1.0/ applies to the metadata files made available in this article.
About this article

Cite this article
Shin, Y., Jung, M., Shin, Gh. et al. First draft genome sequence of the rock bream in the family Oplegnathidae. Sci Data 5, 180234 (2018). https://doi.org/10.1038/sdata.2018.234
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/sdata.2018.234
- Springer Nature Limited