
Abstract
A challenge in clinical genomics is to predict whether copy number variation (CNV) affecting a gene or multiple genes will manifest as disease. Increasing recognition of gene dosage effects in neurodevelopmental disorders prompted us to develop a computational approach based on critical-exon (highly expressed in brain, highly conserved) examination for potential etiologic effects. Using a large CNV dataset, our updated analyses revealed significant (P < 1.64 × 10−15) enrichment of critical-exons within rare CNVs in cases compared to controls. Separately, we used a weighted gene co-expression network analysis (WGCNA) to construct an unbiased protein module from prenatal and adult tissues and found it significantly enriched for critical exons in prenatal (P < 1.15 × 10−50, OR = 2.11) and adult (P < 6.03 × 10−18, OR = 1.55) tissues. WGCNA yielded 1,206 proteins for which we prioritized the corresponding genes as likely to have a role in neurodevelopmental disorders. We compared the gene lists obtained from critical-exon and WGCNA analysis and found 438 candidate genes associated with CNVs annotated as pathogenic, or as variants of uncertain significance (VOUS), from among 10,619 developmental delay cases. We identified genes containing CNVs previously considered to be VOUS to be new candidate genes for neurodevelopmental disorders (GIT1, MVB12B and PPP1R9A) demonstrating the utility of this strategy to index the clinical effects of CNVs.
Similar content being viewed by others


Introduction
The broad umbrella classification of “developmental disorders” encompasses various conditions characterized by disturbance or delay of developmental milestones that appear in infancy or childhood. The cluster includes diagnostic entities that are themselves collectives, such as autism spectrum disorder (ASD), intellectual disability, learning disability and others. The term may be used rather loosely, but is a common reason for referral to laboratories that offer genomic microarray for diagnostic evaluation. Developmental disorders affect ~3% of the population and reflect a significant genetic contribution1,2,3. In particular, large-scale genome-wide investigations3,4,5,6,7,8,9 have demonstrated the large impact of copy number variation (CNV) on severe pediatric conditions, including various developmental disorders. As a result, whole genome “chromosomal microarray” (CMA) has come to be used as a first tier diagnostic test to elucidate causal CNVs in individuals with developmental disorders and congenital anomalies3,7,10. In a pediatric genetics laboratory, the variants detected by CMA are interpreted with respect to probable clinical significance, based on variant type (deletion or duplication), inheritance (de novo or present in a parent), gene content, gene density and evidence from published literature on association with diseases. For large, rare recurrent deletions and duplications (e.g., 16p11.2, 22q11.2, 15q13.2), the interpretation is rather obvious due to overwhelming genetic and phenotypic evidence. In a large multi-centre clinical data set, 15–20% of cases with developmental delay were associated with diagnostic findings on clinical chromosomal microarray3.
Among these diagnostic cases, many rare CNVs are detected for which the potential functional significance is unknown and are referred to as variants of uncertain (or unknown) significance (VOUS). Variants deemed to be of clinical significance (or pathogenic) are fewer and not all genes impacted by these variants will influence the developmental disability phenotype. Few candidate genes ascertained from clinically significant CNVs have been established as pathogenic through genetic studies; some are supported by model organism experiments6,11,12,13,14,15. The more abundant VOUS remain largely uncharacterized and we need to investigate their expression and regulatory networks to understand their contribution to human cognition, behavior and disease. By virtue of the evaluation criteria, these variants are usually shorter than those classified as significant; they impact fewer exons or genes and are mostly unique observations among cases studied3,7. A typical gene-level case-control association analysis of rare variants reveals thousands of genes to be apparently significant (Coe et al.6 reported ~3800 genes P < 0.01)6 but these include many spurious signals that arise due to physical proximity to truly significant genes, given that large CNVs encompass multiple genes.
Genome-wide molecular data (transcriptomic, proteomic, etc.) facilitate inference of pathogenic mechanisms through information about expression and regulatory networks and pathways at the molecular and cellular level16,17,18,19. Investigations of existing biological networks are biased towards the number of simplified interactions20 or do not take tissue specificity into account21,22, yet these are important factors for the elucidation of phenotypically relevant genes23. The aim of this study was to assess the probable impact of variants in genes from within CNVs–both those established as clinically significant and VOUS–through knowledge of mutational burden and RNA and protein expression in various human brain tissues at different developmental stages (prenatal and adult). Previously, we developed a robust method, coupling information about the burden of exonic mutations with exon-level expression, to quantify the critical nature of an exon in a given tissue at specified times in development (hence, spatio-temporal expression)24. We revealed an inverse correlation between exon expression level in brain and the burden of rare missense mutations found in population controls. Variants in these specific critical exons are significantly enriched among individuals with autism, relative to their unaffected siblings. Building upon this concept, we have now implemented an integrated genomics approach to analyze CMA data from DNA of individuals with developmental delay, to infer biologically relevant genes at the transcriptome and proteome levels. For this analysis, we added RNA-seq data from 388 postmortem brain tissues (prenatal to adult) and re-constructed the exon transcriptome contingency index for 226,845 exons from 19,631 genes. We identified ‘brain critical exons’ with i) a low burden (<75th percentile for the genome) of rare (<5%) missense and loss-of-function (LOF) mutations as identified from the 1,000 Genomes Project25 and ii) high expression (>75th percentile for the genome) in brain tissue. We utilized these data to quantify the enrichment of ‘critical exons’ in 16 brain regions16. Next, we used genome-wide indexing of critical exons to quantify genes impacted by pathogenic or VOUS in developmental delay cases and rare CNVs in controls. We used a comprehensive high resolution proteome dataset from prenatal and adult tissues (16) to analyze and extract biologically relevant gene co-expression networks. New candidate genes from within the pathogenic variants and VOUS were inferred from the aggregate analysis of spatio-temporal mRNA and protein expression data.
Results
Pathogenic variants and variants of uncertain significance
We analyzed 10,619 cases from two Ontario hospitals (The Hospital for Sick Children (SickKids) and Credit Valley Hospital (CVH)) referred for clinical laboratory testing due to developmental delay. We also used 9,692 controls (described in Methods) for whom no known psychiatric condition has been reported (Fig. 1 and Tables S1–S2). From this developmental delay cohort, 10.15% of the samples carried a pathogenic CNV and 50.25% had at least one rare VOUS (Fig. 2A). In the SickKids subset (7,106 cases), which included inheritance information, there were 169 de novo variants (108 deletions and 61 duplications), of which 64% (108/169) were interpreted as pathogenic and 36% (61/169) as VOUS. To reduce platform specific sensitivity and specificity bias, we restricted our analysis to variants of 30 Kb to 5 Mb in length. The developmental delay cohort was 68.53% male and 31.46% female. There were pathogenic deletion variants in 5.26% of females and 3.69% of males and this difference was highly significant (P < 0.0002) (Fig. 2B). This sex difference was not found for duplication CNVs (Fig. S1). The average number of genes with exonic variants differed significantly between pathogenic CNVs and VOUS. Genes per variant averaged 38 for pathogenic duplications, 27 for pathogenic deletions, 4.6 for VOUS duplications and 3.2 for VOUS deletions (Fig. S2). The average number of genes in pathogenic deletions from females was higher than in those from males (Fig. S3). CNVs at loci for known genomic disorders26 were more frequent in our developmental delay cohort relative to controls, for example 16p11.2 (0.86%), 15q13.3 (0.43%), Prader-Willi syndrome (0.52%) and 22q11 deletion/duplication syndrome (1.06%) (Fig. S4).

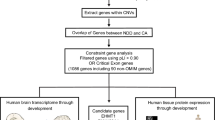
Schematic of the analysis framework to identify candidate genes from copy number variation in developmental delay, using genome-wide human (prenatal and adult) brain transcriptome (RNA sequencing) and proteome data (Fourier-transform mass spectrometry).
The spatio-temporal transcriptome data was used to compute ‘brain critical exon’ analysis for all the genes in the genome. Quantified protein expression for each gene in the genome was used for weighted gene co-expression network analysis. To identify a candidate set of phenotypically relevant genes, integrated (transcriptome and proteome) analysis was conducted for genes impacted by rare CNVs in cases (pathogenic/VOUS) and controls. CVH, Credit Valley Hospital; VOUS, variant of unknown significance; DGV, Database of Genomic Variants.

Ascertainment of pathogenic variants or variants of uncertain significance (VOUS) in 10,619 developmental delay cases.
The rare CNVs of 30 kb to 5 Mb were classified as pathogenic variants or of VOUS. (A) Bars indicate the total number of CNVs in each classification. Of all samples assayed, 4.19% carried a pathogenic deletion, 1.81% a pathogenic duplication, 18.28% a VOUS deletion and 31.97% a VOUS duplication. The green line represents the number of unique genes impacted by the corresponding variants. (B) The percentage of male and female cases in the cohort impacted by pathogenic deletion variants. P value shown is for the one-sided Fisher’s exact test.
Critical Exon Analysis
Genes within pathogenic or VOUS CNVs in this developmental delay cohort had a significantly higher fraction of critical exons (computed over all exons impacted by CNVs), compared with genes in the rare duplications or deletions seen in controls (Table S2). Gene sets from pathogenic CNVs and VOUS were very large and overlapped those from control CNVs; we limited the analysis to genes impacted by pathogenic CNVs or VOUS from the case cohort and not found in unaffected controls (Fig. 3). We observed a striking enrichment of critical exons (computed using prenatal brain transcriptome) within pathogenic deletions (corrected Fisher’s Exact Test (FET) with P < 1.64 × 10−15 for a brain region) and VOUS deletions (P < 1.31 × 10−20 to 6.22 × 10−158) (Fig. 3B). This result strongly suggests that pathogenic and VOUS variants harbor more critical exons than do the rare CNVs in controls. Although deletions showed the consistently highest sensitivity, we observed a similar increased critical exon fraction for pathogenic and VOUS duplications computed using prenatal and adult transcriptome (Fig. 3B and Fig. S5). We then used these exclusively pathogenic and VOUS gene sets to identify candidates for effects on developmental disorders. Of interest, the fraction of critical exons was highest in prenatal neocortical regions (specifically medial prefrontal cortex (MFC) tissues) for genes impacted by either pathogenic or VOUS deletions. This observation strongly supports previous independent reports of multiple neuropsychiatric patients with phenotypes associated with MFC27,28,29.

The fraction of critical exons (over all exons) computed from human prenatal brain regions for the genes impacted by pathogenic, VOUS and rare control deletion and duplication variants.
(A,B) The critical exon fraction was computed using gene expression level quantified from RNA sequencing in 392 brain tissues (controls) from 31 postmortem donors in 2 developmental periods (prenatal and adult) for 16 brain regions (AMY, amygdaloid complex; CBC, cerebellar cortex; V1C, primary visual cortex; STC, posterior (caudal) superior temporal cortex; IPC, posterior inferior parietal cortex; A1C, primary auditory cortex; S1C, primary somatosensory cortex; M1C, primary motor cortex; STR, striatum; DFC, dorsolateral prefrontal cortex; MFC, medial prefrontal cortex; VFC, ventrolateral prefrontal cortex; OFC, orbital frontal cortex; MD, mediodorsal nucleus of thalamus; ITC, inferolateral temporal cortex; HIP, hippocampus). The critical exon fraction was computed using prenatal brain transcriptome for the genes impacted by pathogenic (red dots) or VOUS (orange dots) or rare control deletions (gray dots).
Protein co-expression analysis
Expression of mRNA is not highly correlated with protein expression, even within the same cells under similar conditions, due to many biological processes that impact mRNA prior to protein translation. Integrated analysis of transcriptome and proteome will provide more potent evidence of gene expression within a tissue. To examine the biological relevance of pathogenic and VOUS ‘critical exons’ at the protein level, we applied co-expression analysis using Fourier transformed protein expression data. We used a draft of the human proteome map (HPM) reported by Kim et al.19, obtained from mass spectrometry of 24 different human tissues (each pooled from 3 post-mortem samples) including 17 adult and 7 prenatal19. To our knowledge, ours is the most comprehensive human developmental (prenatal and adult) protein co-expression analysis using this high resolution Fourier transformed dataset. Unlike the biased seeding approach used to construct modules and networks30,31,32, we implemented an unbiased construction of networks, based on spatio-temporal protein expression, by applying a weighted gene co-expression network analysis (WGCNA)33. We applied WGCNA to the 17,294 genes represented by the proteome19 to construct protein co-expression networks. We excluded genes not expressed in at least 90% of the tissues. The analysis revealed networks for 23 independent protein expression modules (Fig. 4A). To identify modules that are relevant to developmental delay, we conducted gene enrichment analysis using 18,826 gene ontology (GO) terms and retained the 20 most significant GO terms. From this analysis, we identified the ‘blue’ module (Fig. 4A), which comprised 2,484 genes (Table S4) and was highly significant (P < 1.0 × 10−39 to 1.0 × 10−81) for pathways involved in synaptic transmission, neuron projection, cell signaling, nervous system development and axon guidance (Fig. 4B).

Genome-wide protein co-expression and enrichment of genes ascertained from pathogenic and VOUS CNVs using human prenatal and adult tissues.
(A) The protein modules generated by weighted gene coexpression network analysis (WGCNA) using high resolution genome-wide Fourier-transform mass spectrometry data from 30 histologically normal human samples (prenatal and adult). Each colour (bars dispersed) represents a module. (B) For the blue module, the 20 most significant results from quantitative association with 18,826 gene sets. (C) Representation of the ‘blue’ module as a functional network, where each node is a gene; the edge between genes represents the weighted Pearson distance. Red nodes represent genes ascertained through CNVs in the developmental delay cohort. (D) The top (25th percentile) critical exon genes in the genome ascertained using prenatal (green) and adult brain (purple) transcriptomes and their corresponding quantified enrichment in the protein module, through Fisher’s exact test (FET) and 100,000 permutations. The orange bar represents the original observation of overlaps between blue module and a gene set. Similarly, (E,F) show the enrichment of genes impacted by pathogenic and VOUS duplications (blue) and deletions (red) within the protein module.
Identification of candidate developmental delay genes from integrated analysis
To develop a list of candidate genes related to developmental delay, we combined the two approaches - critical exon and WGCNA core protein–analyzing genes from the protein-derived ‘blue’ module for critical exon enrichment. For each gene in the genome, we computed the critical exons for prenatal and for adult tissues. To control for the genes with a large number of exons, for each gene in the genome, we computed the fraction of critical exons (over all exons) for a gene in each brain region. For each developmental period (prenatal or adult), a gene was deemed to be significant if its critical exon fraction fell within the genome’s top 25th percentile for at least 50% of the brain samples (Table S6). We found 48.5% (1,206/2,484) of ‘blue’ module proteins to be within the top 25th percentile of genes enriched for critical exons, both for prenatal (FET, P < 1.15 × 10−50, OR = 2.11) and adult brain (FET, P < 6.03 × 10−18, OR = 1.55) (Table S5). This included genes (SCN2A, NRXN1, NRXN2, NRXN3, SHANK2, NLGN3, STXBP1, NLGN4) known to be associated with neuropsychiatric conditions34,35. Inference of these 1,206 candidate genes, as described, was independent of the knowledge of gene-disease association. The overrepresentation of critical exon genes within the ‘blue’ module was tested by random permutation 100,000 times to obtain an empirical significance (for both periods, P < 1.0 × 10−05) (Fig. 4C,D) using the appropriate background (please see methods).
We then tested from within the ‘blue’ module for overrepresentation in tiers of genes that are associated with certain neurodevelopmental disorders. There was significant enrichment of the ‘blue’ module with 840 fragile X mental retardation protein (FMRP) target genes36 (P < 1.36 × 10−128, OR = 6.25; empirical P < 1.0 × 10−05), 246 genes impacted with de novo LOF mutations in ASD cases35,37,38,39,40 (P < 5.9 × 10−04, OR = 1.79; empirical P < 2.1 × 10−04) and 141 genes that had de novo exonic deleterious mutations in intellectual disability samples41,42 (P < 8.0 × 10−03; OR = 1.80; empirical P < 2.3 × 10−03) from exome and whole genome sequencing (Fig. S6A–C). This result suggested that proteins in this ‘blue’ module are under purifying selection in brain and that perturbing them may lead to neurodevelopmental phenotypic manifestations.
Next, we looked for ‘blue’ module genes among those ascertained from CNVs in the developmental delay cohort. ‘Blue’ module genes were significantly overrepresented among genes within pathogenic deletions (P < 2.97 × 10−09; OR = 1.58; empirical P < 0.024) or duplications (P < 0.002; OR = 1.24; empirical P < 0.006) (1-sided FET and permutation test). They were similarly overrepresented among genes within VOUS deletions (P < 7.44 × 10–07; OR = 1.48; empirical P < 0.002) and duplications (P < 0.004; OR = 1.14; empirical P < 0.023) (Fig. 4E,F). In contrast, the genes within deletions and duplications in controls were not overrepresented within the ‘blue’ module. We found 438 candidate genes (of 1,206 genes) that were included in the ‘blue’ module and also highly enriched for critical exons (top 25th percentile), that had been ascertained by at least one pathogenic variant or VOUS in the developmental delay cohort (Table S5). Of these, 65 (14.84%) were seen in at least 2 VOUS deletions in cases but not controls. Another 37 of these 438 genes were impacted by at least one de novo VOUS in our dataset (Table S5). Genes from large recurrent CNVs known to be associated with neuropsychiatric syndromes (e.g., 16p11.2, 22q11 and 3q29)43 were represented in the ‘blue’ protein module and were highly enriched for ‘critical exons’ in prenatal or adult brain (Table S6).
New candidate gene: GIT1
Through unbiased critical exon analysis coupled with WGCNA we identified 1,206 candidate genes whose disruption is likely to contribute to neurodevelopmental delay (Table S5). For example, in our cohort, VOUS deletions and duplications within chromosome region17q11.2 affect the gene for G protein-coupled receptor kinase interacting ArfGAP1 (GIT1). Within the VOUS in this region, GIT1 was the only gene enriched with critical exons in prenatal brain and was highly clustered with genes (highly connected first degree neighbors) from within the ‘blue’ protein module network that were reported to have de novo mutations in ASD. The GIT1 protein is involved in cell migration44, localizes in pre- and post-synaptic terminals and regulates synapse formation45. Recent studies of knock-out Git1−/− mice showed a decreased brain size, with impaired motor coordination and deficits in learning and memory46. From published data6 and on additional clinical cohorts (Mayo clinic, DatabasE of genomiC varIation and Phenotype in Humans using Ensembl Resources (DECIPHER) and Centre Hospitalier Universitaire Sainte-Justine clinic) we found enrichment of CNVs affecting GIT1 among cases (12 deletions and duplications, including 5 de novo; none in controls). The smallest focal deletion of 180 kb was found in an 11-year-old child (Fig. 5A, Table S7 case 6D) referred for developmental delay, with epilepsy and attention deficit hyperactivity disorder as comorbid conditions. Another focal deletion was found as a de novo event of 299 kb in a 10-year-old child referred for developmental delay (case 1D-DN). The DECIPHER database showed a similar de novo focal 282 kb deletion affecting a child referred for learning disabilities, dysphasia and poor motor coordination (see phenotype Table S7 case 5D-DN). This source also identified two de novo duplications affecting GIT1: a 1.4 Mb de novo duplication in a patient referred for intellectual disability and a focal 466 kb duplication in a patient with broader developmental delay. A de novo damaging missense mutation was also reported in a schizophrenia case47. We conducted quantitative real-time PCR (rt-PCR) analysis to quantify relative mRNA expression (Supplementary text) of GIT1 on 11 human tissues (including prenatal and adult brain) by targeting a critical exon of the gene. GIT1 expression relative to that of the MED13 gene (or of the ACTB gene) showed brain specific expression in prenatal and adult tissue (Fig. 5C).

Deletions within the GIT1 gene identified in developmental disorder cases or controls.
(A) The breakpoints of 12 VOUS deletions (red) and duplications (blue) impacting GIT1 and nearby genes. The dataset includes 5 de novo deletions/duplications (denoted as DN) reported from developmental cases; no rare CNV was found in controls. (B) The analysis of the human protein co-expression network revealed that GIT1 is within the blue module and is highly connected (1-degree neighbors) with genes enriched for ‘critical exons’ (red nodes) and putative ASD genes reported to have de novo mutations (red node with black outline). (C) Expression of GIT1 (primer targeting critical exons) from quantitative real-time PCR (qRT-PCR) relative to housekeeping gene, MED13 (replicated with another housekeeping gene ACTB) in 11 different tissues.
New candidate gene: MVB12B
We ascertained another candidate gene, multivesicular body subunit 12B (MVB12B), with enrichment of critical exons and also clustered with genes in the ‘blue’ network that were reported to be impacted by de novo exonic mutations in individuals with neuropsychiatric conditions. Initially we observed a single de novo duplication in our cohort, but subsequently found 18 VOUS involving this gene, including 12 that were de novo in origin (8 deletions and 4 duplications) (Fig. 6). This included 3 recurrent de novo CNVs ascertained from developmental delay cases impacting only the MVB12B gene (Fig. 6 and Table S7 cases 6D-DN,4G-DN and 3G). The protein encoded by this gene is an endosomal sorting complex required for transport (ESCRT-I)48 which is involved in sorting of ubiquitinated cargo protein from the plasma membrane. Mutations in the ESCRT complex family have been implicated in frontotemporal dementia, a neurodegenerative disease49. Our data included a de novo 839 kb duplication in a child (case 5G-DN) with Tourette syndrome, attention deficit hyperactivity disorder and learning disability. Another case had an adjacent de novo 700 kb duplication and was described as having severe intellectual disability, whereas an individual (Fig. 6 case 3G) with a recurrent duplication was reported to be normal, thereby demonstrating variable expression (or reduced penetrance) of the duplication. Deletions were found in cases referred for developmental delay and reported to have other comorbid conditions (Table S7). The 73 kb smaller transcript of this gene (NM_001011703.2) containing the ‘critical exons’ in our analysis was impacted by 12 of our reported de novo VOUS and no CNVs in controls. Quantitative PCR analysis of 11 tissues for both MVB12B and PPP1R9A showed prenatal and adult brain-specific mRNA expression relative to that of the MED13 gene (or the ACTB gene).

Deletions within MVB12B gene identified in developmental disorder cases and controls.
(A) The breakpoints of 18 VOUS deletions (red) and duplications (blue) impacting MVB12B and nearby genes. The breakpoints include 12 de novo VOUS reported from developmental delay cases, including 3 recurrent breakpoints (6D-DN, 4G-DN, 3G). All de novo VOUS impacted the smaller isoform (highlighted by vertical dashed lines) of the gene (NM_001011703.2) and this was not impacted by CNV in controls. (B) The human protein co-expression network revealed that the MBV12B gene is the within the blue protein module and enriched for ‘critical exons’ (red nodes) and putative ASD genes reported to have de novo mutations (red node with black outline). (C) Expression of MVB12B (primer targeting critical exons) from quantitative real-time PCR (qRT-PCR) relative to housekeeping gene, MED13 (replicated with another housekeeping gene ACTB) in 11 different tissues.
New candidate gene: PPP1R9A
Our data set showed enrichment of deletion variants (4 VOUS, 1 de novo) containing another gene from our candidate list: the protein phosphatase 1 regulatory subunit 9A (PPP1R9A) gene (Fig. S7). Additional data sets revealed 18 CNVs (13 deletions and 5 duplications), including 3 additional de novo deletions impacting exons of the gene. PPP1R9A is the only gene from within the de novo variants that is enriched with critical exons and clustered within the blue protein module (Fig. S7B). The nearby genes are impacted by polymorphic deletions in controls, whereas no deletions encompassing PPP1R9A were identified from the control data set. A de novo missense mutation of this gene was recently reported in an ASD proband40. This gene shows tissue-specific imprinting; it is maternally expressed in skeletal muscle, but both alleles are expressed in other embryonic tissues, including the brain50. The protein encoded by this gene, Neurabin I, is a key candidate molecule in synaptic formation and function50. PPP1R9A also has a role in synaptic structure and function, spine motility and neurite formation51. We observed enrichment of critical exons of this gene within adult brain regions (Fig. S7) and it is part of the ‘blue’ module. An individual with autism was found to have a de novo missense variant of PPP1R9A40 and a rare LOF mutation was reported in a schizophrenia case52. This gene also shows increased expression in brain from individuals with bipolar disorder compared with controls53. Upon further investigation of the ‘blue’ protein co-expression network, highly connected neighboring (first degree) genes of PPP1R9A were reported to have de novo mutations in individuals with neuropsychiatric conditions32,35,37,42. In our cohort, a focal de novo 201 kb deletion affecting this gene was found in a child (Fig. S7 and Table S7 case 11D-DN) referred for language delay, repetitive behaviors and sensory sensitivities, consistent with her diagnosis of ASD. A 2 Mb deletion impacting 15 genes–including PPP1R9A and the sarcoglycan epsilon (SGCE) gene - was found in a 12 year old girl (case 7D) referred for myoclonus dystonia, short stature, failure to thrive, severe anxiety and obsessive-compulsive behavior. She also had mild dysmorphic features (triangular facies, broad forehead, thin lips) but no cognitive concerns were reported.
Discussion
The brain transcriptome and proteomic method implemented here can infer candidate genes from within the boundaries of a pathogenic or VOUS CNV that are likely to impact brain-related conditions. Such lists of candidate genes can then be used to index the potential effect of a particular CNV or a set of CNVs in neurodevelopmental disorders.
The detection of genes critical for neurodevelopmental disorders is highly dependent on the breakpoints of the CNVs. Compared with sequencing technologies, clinical microarray tends to extend CNV breakpoints due to low probe density for the genome54. The low resolution impacts the precision of apparent breakpoints55 and CNVs may appear to encompass genes that may not be impacted by the real variant. Such false positives are reduced by large cohorts and high-resolution breakpoint detection through sequencing. Although HPM is one of the most comprehensive protein expression datasets, it may not be perfect in the quantification of peptides across all tissues partly due to the peptide fractionation techniques. Further improvement on fractionation technique will provide more concrete protein expression profile of genes across different tissue types.
We have demonstrated a quantifiable approach to screen for genes that are candidates to be involved in neurodevelopmental disorders, through the coordinated application of multiple genome-scale data sets. The critical exon approach reveals a negative selective pressure, whereas the protein expression analysis brings out networks that are in pathways biologically relevant to neurodevelopmental disorders31,56. This approach using CNVs could be augmented, through sequence analysis, to identify new genes, pathways and regulatory elements for a wide spectrum of neurodevelopmental phenotypes. Our recent work57,58 has shown that CNVs and smaller sequence-level variants (indels/single nucleotide variants (SNVs)) contribute - to autism spectrum disorder and that whole genome sequencing, (which is capable of detecting both SNVs and CNVs) will become the ultimate standard for clinical genetic testing35,59. An approach such as ours that can assess the effects of mutations on genes in phenotypically relevant tissues will be important to reveal candidates for other classes of disorders. As large CNV and sequence level mutation datasets become available, large tissue specific RNA60 and protein expression dataset can be utilized for additional disease associations.
Through the approach described in this study, we have demonstrated the combined use of different types of molecular data from the human brain to interpret and identify candidate genes for developmental disorders, from pathogenic variants and VOUS. This quantifiable approach begins to enable the indexing of genes affected by CNVs for their potential role in neurodevelopmental disorders. Further functional characterization of these candidate genes and their products will allow us to define their regulation among tissues and throughout development. This, in turn, may aid in steps for timely interventions to mitigate untoward effects of various genomic alterations.
Materials and Methods
Clinical microarray datasets
The clinical microarray (CMA) data were obtained from two independent sites: The Hospital for Sick Children (SickKids) (7.106 cases) and Credit Valley Hospital (CVH) (3.513 cases) from individuals referred for investigation of developmental delay (Table S1). In both sites, a International Standards for Cytogenomic Arrays ISCA 180 K comparative genomic hybridization array (aCGH) was used to detect large CNVs by applying a circular binary segmentation algorithm61. For reference, we used a pool of 10 samples to compare individual probe intensities. The clinical annotation for each sample variant was conducted by the clinical laboratory geneticist in each site.
Briefly, DNA from each case and pooled same-sex reference DNA (Promega, Madison, WI) were differentially labeled with Cy3-dCTP or Cy5-dCTP, respectively and hybridized to the array slide according to the manufacturer’s protocol (Oxford Gene Technology). We scanned arrays using the Agilent G2505Bmicroarray scanner and analyzed data using the Agilent Feature Extraction software (10.7.11) and CytoSure Interpret Software version 3.4.3 (OGT). Clinical interpretation of copy number variants was consistent with the American College of Genetics and Genomics guidelines62. When needed, we performed fluorescence in situ hybridization) analysis on cultured lymphocytes of parents, using standard protocols. Metaphase chromosomes were counter-stained with 4’,6-diamidino-2-phenylindole and inverted grey scale imaging was used to visualize chromosome banding patterns for chromosome identification, using the ISIS Metasytems imaging software version 5.5.4 (Newton, MA, USA). Deletions of less than 200 kb and duplications less than 700 kb were followed up by aCGH in parental samples.
As controls, we used data from 9,692 unrelated samples from individuals with no obvious psychiatric history, from multiple major population-scale studies that used high-resolution microarray platforms. These included 4,347 control samples assayed by Illumina 1 M from the Study of Addiction Genetics and Environment (SAGE)63 and the Health, Aging and Body Composition (HABC)64; 2,988 control samples assayed by Illumina Omni 2.5 M from the Collaborative Genetic Study of Nicotine Dependence (COGEND)]65 and Cooperative Health Research in the Region of Augsburg KORA projects66; 2,357 control samples assayed by Affymetrix 6.0 from the Ottawa Heart Institute67 and the PopGen project68. In addition, we incorporated 11,255 control datasets assayed on Illumina platforms from ARIC and Wellcome Trust case control consortium (WTCCC2) projects6.
Critical exon classification
Burden of rare missense mutations
We used whole genome sequence data from the 1000 genomes project25 initiated by the US National Health Heart, Lung and Blood Institute (NHLBI) to calculate the burden of rare missense mutations in humans (495 males and 544 females). Exonic regions had mean sequenced coverage of at least 20X. We used the RefSeq gene annotation model (which includes all exons from annotated isoforms) for our analysis. Genes with no variant calls were excluded. As described previously24, we annotated the variants using Annovar and considered rare missense and LOF variants as strong proxies for recent (mostly within the last 5,000–10,000 years) rare deleterious mutation events in humans.
Spatio-temporal expression in human brain
We downloaded normalized RNA-seq data for spatio-temporal expression profiles of human brains from the BrainSpan database (http://www.brainspan.org/static/download.html). We analyzed 388 tissue samples from 32 post-mortem brain donors (prenatal and adult). The expression measures for exons were provided as reads per kilobase per million (RPKM) from mapped reads. Method details for sequencing, alignment, quality control and expression quantification can be found in the BrainSpan Technical White Paper (http://www.brainspan.org/). We conducted our spatio-temporal (prenatal and adult) analysis on 16 brain regions, including 11 neocortex regions (V1C, primary visual cortex; STC, posterior (caudal) superior temporal cortex; IPC, posterior inferior parietal cortex; A1C, primary auditory cortex; S1C, primary somatosensory cortex; M1C, primary motor cortex; DFC, dorsolateral prefrontal cortex; MFC, medial prefrontal cortex; VFC, ventrolateral prefrontal cortex; OFC, orbital frontal cortex; ITC, inferolateral temporal cortex) and AMY, amygdaloid complex; CBC, cerebellar cortex; HIP, hippocampus; MD, mediodorsal nucleus of thalamus; and STR, striatum. We classified critical exons as described previously24.
Data from human protein expression at developmental stages
We used high-resolution genome-wide Fourier-transform mass spectrometry data (downloaded from the Human Proteome Map)19 to analyze protein expression levels in human tissues at two developmental stages. This included in-depth proteomic profiling of 30 histologically normal human samples, including 17 adult tissues (lung, heart, liver, gall bladder, adrenal gland, kidney, urinary bladder, prostate, testis, ovary, rectum, colon, pancreas, oesophagus, retina, frontal cortex and spinal cord) and 7 fetal tissues (liver, heart, brain, placenta, gut, ovary, testis)19. High-resolution Fourier transform mass spectrometers had been used for fragmentation (high-high mode) to process the data. This resulted in the identification of proteins encoded by 17,294 genes, accounting for approximately 84% of annotated protein-coding human genes19. We used average spectral counts per gene per sample as the measure for protein expression.
Weighted gene coexpression network analysis (WGCNA)
We used the R WGCNA package69,70 to analyze the human protein expression. The use of weighted networks represents an improvement over unweighted networks because it preserves continuity of the co-expression information and it is biologically robust with respect to parameter ß33. We excluded proteins that are rarely expressed (expression = 0 in at least 90% of the samples) because such low-expressed features tend to reflect noise and correlations based on such counts are not really meaningful. We calculated the absolute value of the Pearson correlation coefficient for all pair-wise comparisons of protein expression values across all developmental tissue samples into a similarity matrix. We used blockwise network construction and a module detection method, where a block cluster consists of a maximum of 20,000 proteins. We constructed a signed adjacency matrix using a “soft” power adjacency function aij = |0.5 + 0.5 * cor(xi, xj)|β , where the absolute value of the Pearson correlation measures protein co-expression similarity and aij represents the resulting adjacency–reflecting the connection strength. We chose for our analysis the soft threshold beta = 18, based on the scale-free topology33 criterion ß (63). Next, to compute modules, where the proteins have high “topological overlap”, we compared connection strength between proteins in the network. The parameters for module detection were: minimum 30 proteins per module and a medium sensitivity deepsplit = 2 was applied to cluster splitting. The clustering of genes for modules used average linkage hierarchical clustering; modules were identified in the resulting dendrogram by the dynamic hybrid tree cut. Such modules were trimmed of genes whose correlation with module eigengene (KME) was less than a threshold defined by the function minKMEtoStay; for merging similar modules, we used 0.35 as a threshold. The connectivity of each node i is the sum of connections to other nodes.
For visualizing the protein co-expression network, we used Cytoscape network software v.2.8.3. A node is represented by a circle and the edge represented by a line between the nodes implies the co-expression weighted Pearson distance. The color of the node represents membership to a phenotype.
Significant test analysis and permutation test
We used Fisher’s exact test (FET) for all count data and g p-value <0.05 (after Bonferroni multiple test correction) as the threshold for significance in tests of gene enrichment. To reveal the strength of enrichment association with the gene lists, we undertook a permutation test by randomly drawing equal numbers of genes and re-analyzing the data under the null-hypothesis. The random draw was conducted from a background appropriate for the test. To analyze enrichment of critical exon genes (top 25th percentile) within the ‘blue’ protein module (Fig. 4D), the background included all genes for which we had both protein expression and mRNA expression from RNA-seq data. For the analysis of pathogenic and VOUS deletion/duplication gene enrichment within the ‘blue’ protein module, we used all genes with protein expression as the background. In each iteration of the random draw, an equal (to the original set) number of genes was drawn. With sufficient iterations (100,000 times), the resulting sets of p-values are presumed to be a reasonable approximation of the null distribution of the p-values.
Reverse transcription polymerase chain reaction (RT-PCR) and quantitative RT-PCR (qRT-PCR)
To quantitate ‘critical exons’ by qRT-PCR, primers were designed to prime from within the specific exon (Supplementary Table S8). We tested PCR efficiency with a dilution standard curve and for specificity with melting curve analysis using adult whole brain cDNA. To quantify the ‘critical exon’ expression from selected genes, we used RNA from a panel of 11 human tissues: liver (BD Biosciences), kidney (Stratagene), mammary gland (BD Biosciences), cerebellum (Clonetech), skeletal muscle (Stratagene), prostate (Clonetech), spleen (Stratagene), thyroid (Stratagene) and testis (Clonetech). Reverse transcription was performed using the Superscript III First strand Synthesis Supermix (Invitrogen). We used 10 ng of cDNA as template for RT-PCR under standard PCR conditions, using Brilliant III SYBR® Green PCR Master Mix (Agilent) and the MX300 software (Agilent). Gene expression was normalized using MED13 or ACTB (dCt) and quantified as relative expression (2^(-dCt)).
Additional Information
Accession codes: The dataset used for the analyses described in this manuscript was obtained from the database of Genotype and Phenotype (dbGaP) found at Accession Number: phs001154.v1.p1.
How to cite this article: Uddin, M. et al. Indexing Effects of Copy Number Variation on Genes Involved in Developmental Delay. Sci. Rep. 6, 28663; doi: 10.1038/srep28663 (2016).
References
Flore, L. A. & Milunsky, J. M. Updates in the genetic evaluation of the child with global developmental delay or intellectual disability. Semin Pediatr Neurol 19, 173–180, doi: 10.1016/j.spen.2012.09.004 (2012).
Michelson, D. J. et al. Evidence report: Genetic and metabolic testing on children with global developmental delay: report of the Quality Standards Subcommittee of the American Academy of Neurology and the Practice Committee of the Child Neurology Society. Neurology 77, 1629–1635, doi: 10.1212/WNL.0b013e3182345896 (2011).
Miller, D. T. et al. Consensus statement: chromosomal microarray is a first-tier clinical diagnostic test for individuals with developmental disabilities or congenital anomalies. Am J Hum Genet 86, 749–764, doi: 10.1016/j.ajhg.2010.04.006 (2010).
Buchanan, J. A. & Scherer, S. W. Contemplating effects of genomic structural variation. Genetics in Medicine: Official Journal of the American College of Medical Genetics 10, 639–647, doi: 10.1097GIM.0b013e318183f848 (2008).
Oskoui, M. et al. Clinically relevant copy number variations detected in cerebral palsy. Nature Communications 6, 7949, doi: 10.1038/ncomms8949 (2015).
Coe, B. P. et al. Refining analyses of copy number variation identifies specific genes associated with developmental delay. Nat Genet 46, 1063–1071, doi: 10.1038/ng.3092 (2014).
Kaminsky, E. B. et al. An evidence-based approach to establish the functional and clinical significance of copy number variants in intellectual and developmental disabilities. Genetics in Medicine: Official Journal of the American College of Medical Genetics 13, 777–784, doi: 10.1097/GIM.0b013e31822c79f9 (2011).
Zarrei, M., MacDonald, J. R., Merico, D. & Scherer, S. W. A copy number variation map of the human genome. Nat Rev Genet 16, 172–183, doi: 10.1038/nrg3871 (2015).
Stavropoulos, D. J. et al. Whole-genome sequencing expands diagnostic utility and improves clinical management in paediatric medicine. npj Genomic Medicine, doi: 10.1038/npjgenmed.2016.8 (2016).
Duncan, A. M. & Chodirker, B. Use of array genomic hybridization technology for constitutional genetic diagnosis in Canada. Paediatr Child Health 16, 211–212 (2011).
Talkowski, M. E. et al. Assessment of 2q23.1 microdeletion syndrome implicates MBD5 as a single causal locus of intellectual disability, epilepsy and autism spectrum disorder. Am J Hum Genet 89, 551–563, doi: 10.1016/j.ajhg.2011.09.011 (2011).
Golzio, C. et al. KCTD13 is a major driver of mirrored neuroanatomical phenotypes of the 16p11.2 copy number variant. Nature 485, 363–367, doi: 10.1038/nature11091 (2012).
Cooper, G. M. et al. A copy number variation morbidity map of developmental delay. Nat Genet 43, 838–846, doi: 10.1038/ng.909 (2011).
Beunders, G. et al. Exonic deletions in AUTS2 cause a syndromic form of intellectual disability and suggest a critical role for the C terminus. Am J Hum Genet 92, 210–220, doi: 10.1016/j.ajhg.2012.12.011 (2013).
Lionel, A. C. et al. Disruption of the ASTN2/TRIM32 locus at 9q33.1 is a risk factor in males for autism spectrum disorders, ADHD and other neurodevelopmental phenotypes. Hum Mol Genet 23, 2752–2768, doi: 10.1093/hmg/ddt669 (2014).
Kang, H. J. et al. Spatio-temporal transcriptome of the human brain. Nature 478, 483–489, doi: 10.1038/nature10523 (2011).
Andersson, R. et al. An atlas of active enhancers across human cell types and tissues. Nature 507, 455–461, doi: 10.1038/nature12787 (2014).
Consortium, E. P. An integrated encyclopedia of DNA elements in the human genome. Nature 489, 57–74, doi: 10.1038/nature11247 (2012).
Kim, M. S. et al. A draft map of the human proteome. Nature 509, 575–581, doi: 10.1038/nature13302 (2014).
Khurana, E., Fu, Y., Chen, J. & Gerstein, M. Interpretation of genomic variants using a unified biological network approach. PLoS Comput Biol 9, e1002886, doi: 10.1371/journal.pcbi.1002886 (2013).
Huang, N., Lee, I., Marcotte, E. M. & Hurles, M. E. Characterising and predicting haploinsufficiency in the human genome. PLoS Genet 6, e1001154, doi: 10.1371/journal.pgen.1001154 (2010).
Steinberg, J., Honti, F., Meader, S. & Webber, C. Haploinsufficiency predictions without study bias. Nucleic Acids Res 43, e101, doi: 10.1093/nar/gkv474 (2015).
Greene, C. S. et al. Understanding multicellular function and disease with human tissue-specific networks. Nat Genet 47, 569–576, doi: 10.1038/ng.3259 (2015).
Uddin, M. et al. Brain-expressed exons under purifying selection are enriched for de novo mutations in autism spectrum disorder. Nat Genet 46, 742–747, doi: 10.1038/ng.2980 (2014).
Genomes Project, C. et al. An integrated map of genetic variation from 1,092 human genomes. Nature 491, 56–65, doi: 10.1038/nature11632 (2012).
Firth, H. V. et al. DECIPHER: Database of Chromosomal Imbalance and Phenotype in Humans Using Ensembl Resources. Am J Hum Genet 84, 524–533, doi: 10.1016/j.ajhg.2009.03.010 (2009).
Lindstrom, L. H. et al. Increased dopamine synthesis rate in medial prefrontal cortex and striatum in schizophrenia indicated by L-(beta-11C) DOPA and PET. Biol Psychiatry 46, 681–688 (1999).
Gilbert, S. J., Meuwese, J. D., Towgood, K. J., Frith, C. D. & Burgess, P. W. Abnormal functional specialization within medial prefrontal cortex in high-functioning autism: a multi-voxel similarity analysis. Brain 132, 869–878, doi: 10.1093/brain/awn365 (2009).
Testa-Silva, G. et al. Hyperconnectivity and slow synapses during early development of medial prefrontal cortex in a mouse model for mental retardation and autism. Cereb Cortex 22, 1333–1342, doi: 10.1093/cercor/bhr224 (2012).
Willsey, A. J. et al. Coexpression networks implicate human midfetal deep cortical projection neurons in the pathogenesis of autism. Cell 155, 997–1007, doi: 10.1016/j.cell.2013.10.020 (2013).
Gulsuner, S. et al. Spatial and temporal mapping of de novo mutations in schizophrenia to a fetal prefrontal cortical network. Cell 154, 518–529, doi: 10.1016/j.cell.2013.06.049 (2013).
O’Roak, B. J. et al. Sporadic autism exomes reveal a highly interconnected protein network of de novo mutations. Nature 485, 246–250, doi: 10.1038/nature10989 (2012).
Zhang, B. & Horvath, S. A general framework for weighted gene co-expression network analysis. Stat Appl Genet Mol Biol 4, Article17, doi: 10.2202/1544-6115.1128 (2005).
Lee, C. & Scherer, S. W. The clinical context of copy number variation in the human genome. Expert Rev Mol Med 12, e8, doi: 10.1017/S1462399410001390 (2010).
Yuen, R. K. et al. Whole-genome sequencing of quartet families with autism spectrum disorder. Nat Med 21, 185–191, doi: 10.1038/nm.3792 (2015).
Darnell, J. C. et al. FMRP stalls ribosomal translocation on mRNAs linked to synaptic function and autism. Cell 146, 247–261, doi: 10.1016/j.cell.2011.06.013 (2011).
De Rubeis, S. et al. Synaptic, transcriptional and chromatin genes disrupted in autism. Nature 515, 209–215, doi: 10.1038/nature13772 (2014).
Sanders, S. J. et al. De novo mutations revealed by whole-exome sequencing are strongly associated with autism. Nature 485, 237–241, doi: 10.1038/nature10945 (2012).
Iossifov, I. et al. De novo gene disruptions in children on the autistic spectrum. Neuron 74, 285–299, doi: 10.1016/j.neuron.2012.04.009 (2012).
Iossifov, I. et al. The contribution of de novo coding mutations to autism spectrum disorder. Nature 515, 216–221, doi: 10.1038/nature13908 (2014).
Rauch, A. et al. Range of genetic mutations associated with severe non-syndromic sporadic intellectual disability: an exome sequencing study. Lancet 380, 1674–1682, doi: 10.1016/S0140-6736(12)61480-9 (2012).
Vissers, L. E. et al. A de novo paradigm for mental retardation. Nat Genet 42, 1109–1112, doi: 10.1038/ng.712 (2010).
Cook, E. H. Jr. & Scherer, S. W. Copy-number variations associated with neuropsychiatric conditions. Nature 455, 919–923, doi: 10.1038/nature07458 (2008).
Nishiya, N., Kiosses, W. B., Han, J. & Ginsberg, M. H. An alpha4 integrin-paxillin-Arf-GAP complex restricts Rac activation to the leading edge of migrating cells. Nat Cell Biol 7, 343–352, doi: 10.1038/ncb1234 (2005).
Zhang, H., Webb, D. J., Asmussen, H. & Horwitz, A. F. Synapse formation is regulated by the signaling adaptor GIT1. J Cell Biol 161, 131–142, doi: 10.1083/jcb.200211002 (2003).
Hong, S. T. & Mah, W. A Critical Role of GIT1 in Vertebrate and Invertebrate Brain Development. Exp Neurobiol 24, 8–16, doi: 10.5607/en.2015.24.1.8 (2015).
Fromer, M. et al. De novo mutations in schizophrenia implicate synaptic networks. Nature 506, 179–184, doi: 10.1038/nature12929 (2014).
Oestreich, A. J., Davies, B. A., Payne, J. A. & Katzmann, D. J. Mvb12 is a novel member of ESCRT-I involved in cargo selection by the multivesicular body pathway. Mol Biol Cell 18, 646–657, doi: 10.1091/mbc.E06-07-0601 (2007).
Skibinski, G. et al. Mutations in the endosomal ESCRTIII-complex subunit CHMP2B in frontotemporal dementia. Nat Genet 37, 806–808, doi: 10.1038/ng1609 (2005).
Nakabayashi, K. et al. Genomic imprinting of PPP1R9A encoding neurabin I in skeletal muscle and extra-embryonic tissues. J Med Genet 41, 601–608, doi: 10.1136/jmg.2003.014142 (2004).
Nakanishi, H. et al. Neurabin: a novel neural tissue-specific actin filament-binding protein involved in neurite formation. J Cell Biol 139, 951–961 (1997).
Purcell, S. M. et al. A polygenic burden of rare disruptive mutations in schizophrenia. Nature 506, 185–190, doi: 10.1038/nature12975 (2014).
Konopaske, G. T., Subburaju, S., Coyle, J. T. & Benes, F. M. Altered prefrontal cortical MARCKS and PPP1R9A mRNA expression in schizophrenia and bipolar disorder. Schizophr Res, doi: 10.1016/j.schres.2015.02.005 (2015).
Pinto, D. et al. Comprehensive assessment of array-based platforms and calling algorithms for detection of copy number variants. Nat Biotechnol 29, 512–520, doi: 10.1038/nbt.1852 (2011).
Uddin, M. et al. A high-resolution copy-number variation resource for clinical and population genetics. Genetics in Medicine: Official Journal of the American College of Medical Genetics, doi: 10.1038/gim.2014.178 (2014).
Pinto, D. et al. Convergence of genes and cellular pathways dysregulated in autism spectrum disorders. Am J Hum Genet 94, 677–694, doi: 10.1016/j.ajhg.2014.03.018 (2014).
Tammimies, K. et al. Molecular Diagnostic Yield of Chromosomal Microarray Analysis and Whole-Exome Sequencing in Children With Autism Spectrum Disorder. JAMA 314, 895–903, doi: 10.1001/jama.2015.10078 (2015).
Merikangas, A. K. et al. The phenotypic manifestations of rare genic CNVs in autism spectrum disorder. Molecular psychiatry 20, 1366–1372, doi: 10.1038/mp.2014.150 (2015).
Jiang, Y. H. et al. Detection of clinically relevant genetic variants in autism spectrum disorder by whole-genome sequencing. Am J Hum Genet 93, 249–263, doi: 10.1016/j.ajhg.2013.06.012 (2013).
Consortium, G. T. Human genomics. The Genotype-Tissue Expression (GTEx) pilot analysis: multitissue gene regulation in humans. Science 348, 648–660, doi: 10.1126/science.1262110 (2015).
Olshen, A. B., Venkatraman, E. S., Lucito, R. & Wigler, M. Circular binary segmentation for the analysis of array-based DNA copy number data. Biostatistics 5, 557–572, doi: 10.1093/biostatistics/kxh008 (2004).
Kearney, H. M. et al. American College of Medical Genetics standards and guidelines for interpretation and reporting of postnatal constitutional copy number variants. Genetics in Medicine: Official Journal of the American College of Medical Genetics 13, 680–685, doi: 10.1097/GIM.0b013e3182217a3a (2011).
Bierut, L. J. et al. A genome-wide association study of alcohol dependence. Proc Natl Acad Sci USA 107, 5082–5087, doi: 10.1073/pnas.0911109107 (2010).
Coviello, A. D. et al. A genome-wide association meta-analysis of circulating sex hormone-binding globulin reveals multiple Loci implicated in sex steroid hormone regulation. PLoS Genet 8, e1002805, doi: 10.1371/journal.pgen.1002805 (2012).
Bierut, L. J. et al. Novel genes identified in a high-density genome wide association study for nicotine dependence. Hum Mol Genet 16, 24–35, doi: 10.1093/hmg/ddl441 (2007).
Verhoeven, V. J. et al. Genome-wide meta-analyses of multiancestry cohorts identify multiple new susceptibility loci for refractive error and myopia. Nat Genet 45, 314–318, doi: 10.1038/ng.2554 (2013).
Stewart, A. F. et al. Kinesin family member 6 variant Trp719Arg does not associate with angiographically defined coronary artery disease in the Ottawa Heart Genomics Study. J Am Coll Cardiol 53, 1471–1472, doi: 10.1016/j.jacc.2008.12.051 (2009).
Krawczak, M. et al. PopGen: population-based recruitment of patients and controls for the analysis of complex genotype-phenotype relationships. Community Genet 9, 55–61, doi: 10.1159/000090694 (2006).
Langfelder, P. & Horvath, S. WGCNA: an R package for weighted correlation network analysis. BMC Bioinformatics 9, 559, doi: 10.1186/1471-2105-9-559 (2008).
Langfelder, P. & Horvath, S. Fast R Functions for Robust Correlations and Hierarchical Clustering. J Stat Softw 46 (2012).
Acknowledgements
We thank The Centre for Applied Genomics (TCAG), which is funded by Genome Canada and the Ontario Genomics Institute, Canada Foundation for Innovation (CFI) and the Ontario Research Fund of the Government of Ontario. The project was also supported by funds from the University of Toronto McLaughlin Centre and Genome Canada. M.U. holds a Banting postdoctoral fellowship from the Canadian Institutes of Health Research (CIHR). A.C. holds a Banting and Best CIHR scholarship; R.K.C. holds a CIHR postdoctoral fellowship and National Alliance for Research on Schizophrenia and Depression (NARSAD) Young Investigator Award. S.W.S. holds the GlaxoSmithKline-CIHR Chair in Genome Sciences at the University of Toronto and The Hospital for Sick Children.
Author information
Authors and Affiliations
Contributions
M.U. and S.W.S. conceived the study design and develop the concept. S.W.S. supervised the overall study and M.U. conducted all the analyses. G.P. helped with transcriptome analysis, B.T. helped with transcriptome analysis, D.M. helped with transcriptome analysis, A.C. helped with proteome analysis, M.Z. proteome analysis, K.T. conducted transcriptome analysis, S.W. conducted proteome analysis, M.J.G. helped with transcriptome and proteome analysis, T.N. helped with annotation, R.K.C.Y. and C.R.M. helped with transcriptome, protein expression analysis and result interpretation. M.U. and L.D.A. conducted the quantitative gene expression analysis on multiple tissues. K.D. provided clinical microarray and phenotype data, G.M. provided clinical microarray and phenotype data, E.L. provided clinical microarray and phenotype data, S.N. provided clinical microarray and phenotype data, M.S. provided clinical microarray and phenotype data, M.A.J. provided clinical microarray and phenotype data, A.N. provided clinical microarray and phenotype data, M.T.C. provided clinical microarray and phenotype data, G.Y. provided clinical microarray and phenotype data, P.K. provided clinical microarray and phenotype data, F.T. provided clinical microarray and phenotype data, E.C.T. provided clinical microarray and phenotype data. M.S. and D.J.S. provided clinical microarray and provided detail phenotype data. M.U., J.A.B. and S.W.S. wrote the main manuscript text. All authors reviewed the manuscript.
Ethics declarations
Competing interests
The concept of an exon transcriptome-mutation contingency index for autism diagnosis has been filed under reference H8312944USP (US provisional application number 61/892920) with the US Patent and Trademark Office.
Electronic supplementary material
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Uddin, M., Pellecchia, G., Thiruvahindrapuram, B. et al. Indexing Effects of Copy Number Variation on Genes Involved in Developmental Delay. Sci Rep 6, 28663 (2016). https://doi.org/10.1038/srep28663
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep28663
- Springer Nature Limited
This article is cited by
-
Mutational spectrum and phenotypic variability of Duchenne muscular dystrophy and related disorders in a Bangladeshi population
Scientific Reports (2023)
-
Overlapping pathogenic de novo CNVs in neurodevelopmental disorders and congenital anomalies impacting constraint genes regulating early development
Human Genetics (2023)
-
Chromosomal microarray analysis of 410 Han Chinese patients with autism spectrum disorder or unexplained intellectual disability and developmental delay
npj Genomic Medicine (2022)