
Abstract
We demonstrate the controversial origin of a biological species within an area of a few kilometres in the absence of physical barriers. We employed nuclear rRNA/mitochondrial and genome-wide SNP approaches to infer relationships of four species of net-winged beetles characterised by female neoteny. Three species are distributed at low elevations and a single population colonised a 40 km2 highland plateau and established distinct biological species despite incomplete genetic isolation. The speciation process is extreme in the highly localised spatial scale, due to the low dispersal power of neotenics and provides clear support for a microallopatric model based on ecological conditions. In contrast with neutral evolution in a homogenous environment, as demonstrated by the genetic divergence and morphological similarity of two widely distributed low-mountain species, the environmental characteristics of the high-mountain plateau led to the origin of a species adapted to the local mimetic pattern and characterised by morphologically distinct genitalia. We conclude that the low dispersal propensity promotes neutral genetic differentiation in the first stage, but environmental characteristics play an important role the final phase of the speciation process. The unexpected speciation at such an extreme geographic scale points to the in situ origin and uniqueness of the mountain fauna.
Similar content being viewed by others

Introduction
A large part of insect diversity remains unrecognised especially when genetic diversity is decoupled from morphological differentiation1,2,3 and when species occur in very small ranges and in under-investigated regions4. Studies of speciation processes limited to an extremely small spatial scale are needed to understand the extent and spatial distribution of diversity, species delimitation and to identify the processes which produce endemism in limited areas, e.g., in the tropical mountains5,6.
The common speciation model suggests allopatry over a large geographic scale and with prominent isolation barriers preventing gene flow7. Alternatively, various animal lineages with limited dispersal power readily diversify within much smaller areas without clear geographic or ecological barriers with neutral evolution being an important component generating their geographically structured genetic diversity8,9. The neotenic lineages are an extreme example of animals with very low dispersal propensity as they readily diversify within limited areas and so can be used as a model system to demonstrate the effect of neoteny and flightlessness on the speciation process8. Besides neutral processes, additional factors can support differentiation such as habitat diversity10 or temporal, spatial11 and ecological separation12 in a single ecosystem. Nevertheless, these factors cannot be decoupled from dispersal propensity which determines the intensity of gene flow between populations and consequently at what scale the deep genetic divergence leading to speciation can occur. Speciation across a small geographical range has been considered as an uncommon scenario7, but detailed studies can challenge this expectation. We propose that extremely low dispersal propensity can be the principal factor rescaling the meaning of the geographical distance and might be a primary cause for the observed high speciation rate in poor dispersers8.
The divergence caused solely by neutral processes is slow and only a stable and continuous environment can provide sufficient space and time for high levels of genetic differentiation. The optimal areas for studies dealing with poor dispersers are tropical regions where the communities represent extremely diversified assemblages with a long evolutionary history13. Here, we study the fauna of the Malay Peninsula, one of the most stable tropical regions with regard to tectonic history and position close to the equator14,15. The tropical rainforests depend on climatic conditions which are generally unstable16, but the monsoon dependant rainforests in South East Asia have long history starting with the origin of the monsoon system after the collision of India and Asia17,18. Although the extent of tropical rainforests dynamically changed due to climate fluctuations since their origin19, the high mountains along the western margin of the Sundaland obtained higher rainfall than central lowlands of the subaerial Sunda Shelf even during periods of extreme aridification20.
We need to expand the spectrum of model groups to investigate the power of individual factors in the speciation process, but research using non-model organisms has suffered from a restricted development of appropriate molecular marker systems with the resolution to study the origins of reproductive isolation. Therefore, the net-winged beetle neotenics have been omitted, although they are a promising model for differentiation and speciation studies. All of them have uniform ecology and occur in ecologically stable habitats which enable long-term survival of animals with low dispersal propensity21,22 and they are known for their very low dispersal propensity and low flying activity of males due to presence of larviform females23. Here, we present a clade of four closely related species of Scarelus net-winged beetles (Fig. 1) as a model system for diversification at an extremely small geographic scale. A series of studies has been recently published on neotenic beetles including Scarelus and most of them focused on diversification in large areas and over long time spans23,24. The phylogenetic analyses identified the deeply split clades of species occurring in each region, the geographically structured species distribution even within individual areas23,24 and in all cases the ranges of neotenics are limited to long-term stable ecosystems22. Although not as brightly coloured as other unpalatable25 net-winged beetles, Scarelus colouration regularly fits in the local mimetic complexes of under-canopy dwelling net-winged beetles and their mimics. Colour patterns adapted to syntopically occurring mimetic complexes have been hypothesised as factors starting the speciation process in net-winged beetles previously26, with the aposematic signal also shown to constitute a barrier to gene-flow in other animal groups27. Conversely, the adult morphology of Scarelus is uniform and differences are limited to the shape of male genitalia and length of antennae28.

Scarelus anthracinus (Coleoptera: Lycidae) in nature.
Here, we describe an extremely spatially limited diversification process within the Main Range of the Malay Peninsula, where no clear barriers are present on a continuous slope of the Cameron Highlands massif. We re-visit a previously identified Scarelus system characterised by female neoteny23 and study in detail a clade of closely related species: a single species is of black colouration with an ‘island’ distribution on a highland plateau surrounded by varying degrees of light coloured species at lower elevations. We use large numbers of anonymous genetic markers (RAD) as well as mitochondrial and nuclear markers to ask whether it is possible to identify genetic structure within this system, including the origin of the highland species and to see if adaptation to local mimicry patterns and morphology fits with genetic divergence and plays a role in the diversification process.
Results
Sanger and NextRAD sequencing, filtering and SNP calling
Three mtDNA fragments, rrnL–tRNA-Leu–nad1, cox1–tRNA-Leu–cox2 and nad5–tRNA-Phe–tRNA-Glu–tRNA-Ser, were obtained for 42 specimens from the Main Range and combined with earlier available nuclear rRNA and mitochondrial DNA data22,23. The aligned dataset consisted of 18S rRNA (1865 homologous positions), 28S rRNA (643 positions), rrnL mtDNA (793 positions), cox1 mtDNA (1109 positions) and nad5 mtDNA (1187 positions). We identified up to 17 unique haplotypes in individual mtDNA fragments for a single species (Supplementary Table S4) and the uncorrected mtDNA pairwise distances among Scarelus species reached up to 21.55% (Supplementary Tables S3, S5–S7).
The nuclear NextRAD data comprised 88–147,000 clusters of potential loci across individuals with a mean depth 9–22 reads per cluster. Raising the clustering threshold increased the number of clusters recovered and causing within-individual heterozygosity to drop sharply above the value 0.8 (Supplementary Figure S5). This trade-off between heterozygosity and cluster number was used to choose a threshold of 0.8. This value is slightly higher than the heterozygosity peak as the introduced bias from erroneous cluster-splitting has been deemed relatively unimportant compared with that from over-clustering and potentially allows the recovery of more loci. A similar trade-off between numbers of accepted clusters and the stabilisation of heterozygosity was used to infer a clustering depth of 8 for the population genetic analyses (Supplementary Figure S4). Final data analysis was performed using the parameters mindepth = 8 clustering threshold = 0.8, minimum 21 individuals to accept locus. For the final dataset of 25 individuals the read numbers and mean coverage per individual were calculated (Supplementary Table S8). Raw reads for all individuals were deposited in GenBank under SRA accession number SRR3679806. We allowed up to 25% missing data to obtain the higher numbers of loci and SNPs in the dataset. The resultant nuclear datasets for the final analyses gave between 92 and 1853 loci comprising 593–12737 SNPs. The SNP data showed 72.90–87.75% identical bases in the 3-species dataset (Supplementary Table S9). From 1354 independent rad tags (loci) tested for signal of selection across all individuals, BAYESCAN determined that no loci displayed signal of selection. The LOSITAN approach suggested that across all individuals 11 loci were under positive selection with 27 loci showing signal of balancing selection. The samples of distantly related S. umbrosus were removed and the tests were repeated across 1777 loci for the remaining three species. The signal of positive selection was seen across 24 loci and balancing selection in five and all of these loci were excluded from subsequent analyses.
Species delimitation
Four closely related species were identified in the Cameron Highland massif and their limits are supported by nuclear and mtDNA datasets. High intraspecific genetic variability was identified in all species and reached up to 0.76, 3.17 and 2.61% for rrnL, cox1 and nad5, respectively (Supplementary Table S4). The interspecific pairwise cox1 distances between S. anthracinus and S. pahangensis/S. pseudoumbrosus, reached 5.74–6.53% and 8.93–10.03%, respectively (Supplementary Table S6, Figure S3). Additionally, the species limits are supported by differences in male genitalia (Fig. 2, Supplementary Figure S9) and colouration of elytra and pronotum which subtly varies within the lowland brown pattern (Fig. 2); only S. anthracinus (Fig. 2i) belongs to a black colour pattern which is endemic to high elevations in the Cameron Highlands. The identified hybrid specimen NG0038 shared mtDNA sequences with S. pseudoumbrosus (Fig. 3b), but its nuclear DNA pointed to much closer relationships with S. anthracinus (Supplementary Figure S7a). The specimen has intermediate morphology and cannot be formally classified with either of the parental species (Fig. 3a, Supplementary Figure S7a,b, Supplementary Table S9).

General appearance and male genitalia of Scarelus.
(a,b) S. umbrosus, (c,d) S. pahangensis, (e,f) S. pseudoumbrosus, (g,h) Scarelus sp., individual NG0038, (i,j) S. anthracinus. (see Supplementary Figure S9 for further information).

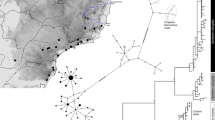
(a) Map of the Cameron Highlands Plateau and adjacent peaks, the distances given for localities sharing related populations of Scarelus anthracinus. The map was produced using the QGIS software package (www.qgis.org/en/site/forusers/download.html) from ASTER GDEM V2 data publicly available from the Land Processes Distributed Active Archive Center (LP DAAC) and J-spacesystems. (b) Phylogenetic hypothesis for the Malay clade of Scarelus based on a maximum likelihood (ML) analysis of five fragments produced by Sanger sequencing. (c) The Bayesian reconstruction of ancestral areas for populations of three species from the Cameron Highlands massif inferred from the RAD dataset. (d) Phylogenetic relationships for populations of three Scarelus species from the Cameron Highlands massif based on the maximum likelihood analysis of the 24-sample RAD dataset. The sample NG0038 omitted, see Supplementary Figure S7 for comparison.
The STRUCTURE analysis of the dataset of 24 individuals after removing S. umbrosus cluster resulted in 1746 SNPs. At K = 3 the three groups separated with high memberships to their own clusters (Qmean > 0.80), i.e. S. pseudoumbrosus, S. pahangenis and S. anthracinus. This mirrors the species partitioning seen in the PCA (Supplementary Figure S6B). Two species, S. pseudoumbrosus and S. pahangensis were seen to be less differentiated relative to each other (FST(W&C 1984) = 0.17) than either compared with S. anthracinus (FST(W&C 1984) = 0.28 for both). Using the POPFLAG admixture determination for the putative hybrid individual NG0038 gave a majority contribution from S. anthracinus with smaller contributions from the other two species (~0.78, 0.14, 0.08, respectively). The RAD genetic distances among of 19 individuals of S. anthracinus of were moderately positively correlated with geographical distances (correlation coefficient r = 0.528, P ≤ 0.001, Fig. 4).

Intraspecific relationships between uncorrected pairwise genetic and Euclidean distances of Scarelus anthracinus.
Sanger and SNP phylogeny, dating and morphology
The maximum likelihood (ML) analysis of the dataset mitochondrial DNA and nuclear rRNA fragments produced the tree with S. umbrosus as a sister to the clade (S. pseudoumbrosus (S. pahangensis, S. anthracinus; Fig. 3b). The partial Bayesian analyses of cox1 and rrnL fragments recovered a similar topology and inferred the split of S. pseudoumbrosus 5.39 and 3.47 mya, P. pahangensis versus S. anthracinus 2.17 and 1.92 mya and the most basal split among S. anthracinus populations 0.409 and 0.681 mya, respectively; Figure S8a,b).
The nuclear ML phylogeny inferred from the SNP dataset was similar to mtDNA/rRNA phylogeny (Fig. 3c,d, Supplementary Figure S7a,b), except the position of the individual NG0038 within S. anthracinus clade in contrast with its position within S. pseudoumbrosus clade in mtDNA phylogeny (Fig. 3b). The BS support for deep splits in the SNP phylogeny was slightly lower when the putative hybrid NG0038 was included in the analysis (Supplementary Figure S7a,b). The same topology was recovered in the Bayesian analyses of the same dataset with high posterior probabilities for all recovered clades (Fig. 3d).
The area of supposed split between S. pahangensis and S. anthracinus is located on the southern slope of Gunung Beremban within an area of a few square kilometres between Robinson Falls and Kg. Kuala Boh (Supplementary Figure S1a,b). The genetic diversity in mtDNA markers suggests intraspecific differentiation during a few hundred years (0.4–0.7 mya, Supplementary Figure S8). The “footprint” of the dispersal directions within the S. anthracinus clade was identified only in the nuclear data when the first splits among S. anthracinus populations were identified along on the southern slope of Mt. Beremban in very close proximity to the range of its sister-species S. pahangensis (8.68 km away, Fig. 3a,c) and shallower splits were inferred gradually on the western and northern slope of Mt Beremban and further populations reached Mt. Brinchang in the north and Mt. Jasar in the west. The most distant S. anthracinus populations were collected 5.6 km apart (Fig. 3a, Supplementary Table S3).
Discussion
The lycid neotenics are known for extremely low dispersal propensity and complete species turnover between individual mountain ranges23,24. Here, for the first time we study their diversification process at an extremely fine geographic scale of a few kilometres in the Cameron Highlands Plateau and adjacent peaks. We assume that mountain populations of S. anthracinus became genetically separated from the nearby occurring lowland species by both distance and environmental differences. As these beetles develop in soil and females remain larviform, the barriers ineffective in restricting other flying insects such as small mountain streams, small breaks in forest cover, rock massifs or forest heterogeneity due to microclimatic conditions can substantially limit their dispersal. Given the distances <15 km between localities of all four allopatrically distributed species in the Cameroon Highlands massif (Fig. 3, Supplementary Table S3, Supplementary Figure S1), we consider it to be very unlikely that they have ever been completely geographically isolated by anything other than such non-permanent and small-scale kinds of barriers. Therefore we conclude that the speciation process proceeded without geographic isolation. The predicted ability to form genetically divergent subpopulations at small geographic scales is supported by the observed high genetic intraspecific divergence in S. anthracinus (Supplementary Table S4–S7, Figure S3). The role of distance in diversification is well known29, but the Scarelus system substantially differs in the extremely small scale at which this differentiation proceeds (Supplementary Table S3).
Our data are consistent with the single dispersal event leading to separation of S. anthracinus from S. pahangensis occurring in the lower elevations on the eastern slope of the Main Range (Fig. 3). We expect the primary hybrid zone between the middle part of the valley north of Sungai Bertam (locality of S. pahangensis) and the south-eastern margin of the Cameron Highlands plateau (Robinson Falls, 8.7 km away, Supplementary Figure S1). The SNP phylogeny identifies the individuals from the Robinson Falls among the deepest splits within the S. anthracinus clade and the dispersal directions can be identified to the north and east with distance steps <3 km (Fig. 3c). Our identification of the place, where the first splits among the populations of S. anthracinus are inferred, is supported by the adjacent range of the putative sister species, S. pahangensis. The dated phylogeny (Supplementary Figure S8) suggests very slow dispersal across a few kilometres of the plateau (0.41 mya since the basal split in the S. anthracinus clade; Fig. 3a–d, Supplementary Figure S8). Even if a substantially higher cox1 rate might be considered for poor dispersers21,30, the time needed for the colonisation of the Cameron Highlands plateau would be estimated in tens of thousand years. The 5.6 km distance between the localities of the inferred deepest split of S. anthracinus populations and the terminal split in the Gunung Brinchang area (Fig. 3c) means that the average dispersal rate is extremely low and can be estimated in centimetres per year. Such dispersal rate is at least ten times lower than previously reported rate for such poor dispersers as minute land snails31.
The split between S. anthracinus and two closely related species is recent (Supplementary Figure S8) and not surprisingly, ongoing gene flow was identified among S. anthracinus and S. pseudoumbrosus when the putative hybrid specimen with intermediate morphology was collected in the contact zone between respective ranges (Fig. 3a, Supplementary Figure S1). Nevertheless, we consider such cases of introgression as rare and not threatening the morphological and genetical coherence of the S. anthracinus as we collected further 78 individuals and using morphology, all these samples were easily categorised as S. anthracinus using colouration and male genitalia (Fig. 2i,j).
Besides the process of neutral divergence3,9, two further factors could start genetic differentiation and subsequently keep coherence of the incipient species: (1) almost all highland net-winged beetles and their mimics are black and this colour pattern was adopted by S. anthracinus (Fig. 2i). The net-winged beetles are protected by the presence of pungent and slightly poisonous substances32 and they form local mimetic complexes. The endemic colour pattern can limit survival outside the range33 and act as an isolating mechanism26. Low intraspecific variability of colouration suggests the presence of selection for similarity within mimetic complexes. (2) The seasonal activity of the adult net-winged beetles strongly depends on moisture due to limited sclerotisation. The mountain plateau has similar rainfall to the lower elevations (Supplementary Figure S2c) but the high level of humidity is also maintained by water trapped by condensation in the cloud covered plateau and due to the lower temperatures (Supplementary Figure S2b). Consequently, organic material, in which all Scarelus develop, heavily accumulates on the soil surface and Scarelus is much more common in higher elevations and active throughout the year. Unlike this favourable habitat, Scarelus are rare and seasonal in the lower elevations (<1000 m, field observations, Supplementary Table S1). S. pahangensis occur on the eastern slope of the Main Range, where the autumn monsoon is stronger and the rest of the year has long dry periods during which their activity is limited or absent (Supplementary Figure S2d). This additional factor might limit opportunity of the highland species to encounter a mate from the parental population despite their geographical proximity.
We observe an extremely narrow zone where a subpopulation splits off from the parental species and established a biological species when ecological factors were the only variable parameters and they changed gradually with elevation. The low dispersal propensity and resulting low gene flow are characteristic for all four species. We assume that low dispersal power alone lead to genetic differentiation without morphological and ecological divergence as seen e.g. in high genetic divergence within S. umbrosus and the lower genetic differentiation between P. pseudoumbrosus and P. pahangensis than between either of them and S. anthracinus. Similar geographically structured differentiation and morphological uniformity as among lowland Scarelus was identified in other net-winged beetles3. Therefore, we conclude that ecology is the necessary component supporting morphological differentiation and origin of the new species in the Scarelus system. Low gene flow in the neotenic lineages defines the extremely small geographic scale, but alone it leads to gradual, neutral evolution and such process does not necessarily lead to the clear morphological differentiation and the origin of a biological species3.
The net-winged beetle neotenics occur exclusively in the stable habitats22 and have regularly small ranges23,24. The observed pattern can be a result of frequent loss of their diversity in unstable habitats due to deep disturbances such as extensive desertification. The elateroid lineages contain numerous neotenic lineages and their independent, relatively frequent origin is well supported34,35,36. Nevertheless, their current diversity is limited to the humid tropics22 or they become soil-dependent if they survive in the less humid but otherwise stable ecosystems such as the Mediterranean (e.g. Omalisidae and Iberobaeniidae34,37). As an alternative to the selection by habitat conditions, we propose that they evolve in any temporarily favourable ecosystems, but survive for long only in those with long stability. Only then we can observe the deep diversification within the clades of the non-flying beetles23,24. Therefore, the stable habitats can be museums of diversity predominantly due to habitat stability leading to the lower extinction rate38.
The winged insects can colonise distant regions39, they easily diversify by multiple rapid altitudinal shifts5,6 and simultaneously they can effectively react if new dispersal opportunities are available40. The Scarelus lineage with their forest dependent immobile neotenic females represents a different and very unusual system. Their ecological characteristics permits only very slow dispersal accompanied by continuous microallopatric differentiation within very small regions (Figs 2 and 4). As a result the ranges of newly split species can be as small as a few dozens of km2 and the likelihood of extinction in the changing environment is much higher than in mobile animals keeping dispersal power. Therefore, the occurrence of diversified neotenics in an area can serve as an indicator of uninterrupted evolutionary history of highly productive tropical ecosystems. The processes of the space-limited differentiation lead to the observed high level of endemism in the Cameron Highlands mountain “island” which can be easily destroyed either by human activity or climate fluctuation.
Methods
Sampling and Sanger sequencing
Four species of Scarelus net-winged beetles (Coleoptera: Lycidae: Ateliinae) from the Cameron highlands in the Malay Peninsula were included in the combined mtDNA and rRNA phylogeny of the whole genus: Scarelus anthracinus (31 spec.), S. pahangensis (3 spec.), S. pseudoumbrosus (4 spec.) and S. umbrosus (3 spec.). All four species were collected in an area of ~400 km2 in elevations 500–1900 m above sea level and only a single species was recorded in each locality (Table 1, Fig. 3, Supplementary Table S1, Supplementary Figure S1). DNA was extracted from the metathoracic muscles of each specimen using a DNeasy tissue kit (Qiagen). Three mitochondrial fragments were amplified: rrnL–tRNA-Leu–nad1 (~760 bp), the 3′end of cox1–tRNA-Leu–cox2 (1090 bp) and nad5–tRNA-Phe–tRNA-Glu–tRNA-Ser (~1150 bp; for primers and conditions see Supplementary Table S2). The fragments are further referred as rrnL, cox1 and nad5, only. The PCR products were purified using PCRu96 Plates (Millipore) and sequenced by an ABI 3130 automated sequencer using the Big Dye Sequencing Kit 1.1 (Life Technologies). The chromatograms produced by Sanger sequencing were edited using Sequencher 4.8 (Genecodes Inc.) and the new data (GenBank accession codes KU319589–KU319671) were merged with the previously published dataset containing additionally full-length18S rRNA (~1855 bp) and the D2 region of 28 S rRNA (~630 bp) for all species included in the analysis23,24.
NextRAD sequencing, filtering and SNP calling
High numbers of single nucleotide polymorphisms (SNPs) were used to investigate relationships among closely related species of Scarelus and their populations. Such an approach has the power to detect even low level consequences of demography and introgression41, enables a study to use a non-model group42 and provides sufficient data to make such analyses feasible43,44. The set of 28 samples representing individuals of four species (S. umbrosus, S. pseudoumbrosus, S. pahangensis and S. anthracinus) and their populations from 11 localities in the Cameron Highlands was prepared for RAD sequencing (Table 1, Fig. 3a, Supplementary Figure S1). The samples were processed for nuclear markers with the NextRAD genomic sequencing approach (SNPsaurus) using the Illumina HiSeq 2500 system. NextRAD sequencing produces primer-based individually barcoded single-end read amplifications with average length ~75 bp representing loci scattered across the genome. These data were processed for all individuals with separate identifiers. The pyRAD 3.0.445 workflow was employed to perform the filtering and SNP recovery process. Data were evaluated in order to maximise the numbers of SNPs recovered and in so doing minimise any bias introduced by the filtering process46,47. Initial full runs (all individuals) were used to select a reduced representation of six individuals representing the spatial extent of the sampling which were then run at a range of clustering thresholds between 65 and 90%. Within individual heterozygosity scores were averaged and these were used to evaluate clustering success48. A sensitivity analysis was subsequently conducted to ascertain the influence of the minimum coverage depth required for locus inclusion (values of 4–9). Because of the large divergence of S. umbrosus individuals, a 25-sample dataset was created excluding three S. umbrosus samples. Analysis of this subset of data allowed a higher proportion of accepted marker loci for further analyses. Some processing and calculations for the methods were run through R49. Required file formats not generated in pyRAD were converted by PGDSpider50.
We tested all loci for signal of selection across populations in order to remove those potentially violating the assumption of neutrality. We applied two FST outlier approaches performed in BAYESCAN51 and LOSITAN52. The methods use the divergence in allele frequencies between populations and a common gene pool to generate a subpopulation specific FST coefficient. These analyses were performed on four- and three-species datasets, with and without S. umbrosus.
We further employed a population genetic approach to investigate introgression using the STRUCTURE53 application and the Principal Component Analysis (PCA)54. Genetic cluster proportions over populations were averaged across 20 runs at 105 steps of burn-in followed by 105 steps for data collection. The individual NG0038 was identified by PCA centrally positioned with no clear group membership in the 25-sample dataset and an estimate of admixture proportions was calculated for this individual against the three genetic clusters representing S. pseudoumbrosus, S. pahangensis and S. anthracinus. Population genetic summary statistics were calculated in each species for the NextRAD data in R and population differentiation information measured in 4P55. Two datasets were considered; all individuals of four species and a dataset excluding S. umbrosus. Further, data matrices were assembled to compare geographical distances and RAD genetic distances among 19 individuals of S. anthracinus. The correlation coefficients for geographic and RAD genetic distances and their corresponding P-values (Mantel tests, number of repetitions nrepet = 10000) were analysed in R 3.1.149.
Phylogenetic analyses of Sanger data and mtDNA based dating
Nuclear rRNA and mitochondrial DNA fragments were aligned using default parameters of Mafft 7.256 and the alignments of the protein coding genes were checked for amino acid reading frames. We conducted Maximum Likelihood (ML) phylogenetic analyses using RAxML-HPC2 8.1.24 as implemented in the CIPRES web server57,58 with data partitioned by genes and by codons, respectively (18 partitions). The analysis was performed using 100 searches for the best tree under a GTR + G + I model identified using jModelTest 259, with bootstrap (BS) values calculated using the rapid algorithm60 with 1,000 bootstrap iterations under the GTRCAT model. We dated the splits between species using single-fragment datasets for the Cameron Highlands Scarelus clade in Beast 1.8.161 using the GTR + G + I model identified as above, Yule Process and Lognormal Uncorrelated Relaxed Clock as proposed in the Beast manual62. The molecular clock was tested for cox1 and rrnL mtDNA datasets using a likelihood ratio test. The null hypothesis L0 is that molecular clock holds and L1 hypothesis relaxes the clock constraint. The chi-square value is given by 2logL = 2(logL0 − logL1) where L0 and L1 are likelihoods of the tree under given constraints. The p-value is counted for s-2 degrees of freedom where s is the number of terminal branches on the tree63. The null model was rejected and an Uncorrelated Relaxed Clock was used in the analyses. As the fossils are unavailable, the 0.0177 ± 0.0019 substitutions/site/my/lineage for cox1 fragment and the 0.0054 ± 0.0009 subs/s/my/l for rrnL fragment21 were used for calibration. The MCMC parameters were fixed to 30 million generations with sampling every 10,000 generations and Effective Sample Size values (ESS) and the pre-stationary phase were identified in Tracer 1.664. The initial generations were discarded as burn-in.
SNP based phylogeny and reconstruction of ancestral distribution
The datasets of SNP sequences representing three Cameron Highlands species and 25 or 24 individuals were produced for phylogenetic analyses (Table 1). S. umbrosus was excluded from both datasets due to the high level of missing data and alternatively the sample NG0038 due to the putative hybrid origin. The NextRAD data were used for all individuals, accepting up to 25% missing data. Datasets were analysed under the ML criterion using RAxML-HPC2 8.1.24 as implemented in the CIPRES web server57,58 and data were unpartitioned. The analyses were performed using 100 searches for the best tree under a GTR + G model identified using jModelTest 259, with bootstrap (BS) values calculated using the rapid algorithm60 with 1,000 bootstrap iterations under the GTRCAT model. Additionally, we used the Bayesian inference implemented in the Beast 1.8.161 to construct the phylogenetic tree from the 24-sample dataset using the HKY model62, Birth-Death process, Strict and Lognormal relaxed clock50, respectively and the clock rate fixed to 1.0. The analyses using the GTR + G model proposed by jModelTest 259 did not reach a stationary phase, therefore the second best identified model was applied in the Bayesian analyses. The stationary phase and ESS values were evaluated in Tracer 1.664. Additionally, the ancestral areas were inferred in Beast 1.8.161 using the HKY model as above, Coalescent Model for the speciation process, Constant Population Size, Strict Clock and constrained topology gained from the ML analysis of the same dataset. The parameters of the analysis follow the recommendation in the Beast manual62. The localities were coded as given at terminals in Fig. 3c.
Morphology
The morphological characteristics of each sampled individual were investigated using external morphology and male genitalia. The dry preserved collection and sequenced individuals contained S. anthracinus (78 spec., all from the Cameron Highlands), S. umbrosus (6 spec. from the Fraser Hills, Gn. Benom and Tapah–Ringlet Rd), S. pseudoumbrosus (6 spec. from Korbu Mts and Ipoh–Kg. Raja Rd) and S. pahangensis (5 spec. Kg. Kuala Boh and Mt. Benom). Genitalia were dissected, cleaned from muscles and fat bodies and photographed using a camera attached to a binocular microscope.
Additional Information
How to cite this article: Bray, T. C. and Bocak, L. Slowly dispersing neotenic beetles can speciate on a penny coin and generate space-limited diversity in the tropical mountains. Sci. Rep. 6, 33579; doi: 10.1038/srep33579 (2016).
References
Barley, A. J., White, J., Diesmos, A. C. & Brown, R. M. The challenge of species delimitation at the extremes: diversification without morphological change in Philippine sun skinks. Evolution, 67, 3556–3572 (2013).
Pinzon, J. H. & LaJeunesse, T. Species delimitation of common reef corals in the genus Pocillopora using nucleotide sequence phylogenies, population genetics and symbiosis ecology. Mol Ecol 20, 311–325 (2011).
Li, Y., Gunter, N. L., Pang, H. & Bocak, L. DNA-based species delimitation separates highly divergent populations within morphologically coherent clades of poorly dispersing beetles. Zool J Linn Soc 175, 59–72 (2015).
Tänzler, R. et al. Macroevolution of hyperdiverse flightless beetles reflects the complex geological history of the Sunda Arc. Sci Rep 6, 18793 (2016).
Toussaint, E. F. A. et al. The towering orogeny of New Guinea as a trigger for arthropod megadiversity. Nat Comm 5, 4001 (2014).
Merckx, S. F. T. et al. Evolution of endemism on a young tropical mountain. Nature, 524, 347–350 (2015).
Coyne, J. A. & Orr, H. A. Speciation. Sinauer Associates, Sunderland, Massachusetts (2004).
Ikeda, H., Nishikawa, M. & Sota, T. Loss of flight promotes beetle diversification. Nat Comm 3, 648 (2011).
Baselga, A. et al. Whole-community DNA barcoding reveals a spatio-temporal continuum of biodiversity at species and genetic levels. Nat Comm 4, 1892 (2013).
DeVries, P. J., Murray, D. & Lande, R. Species diversity in vertical, horizontal and temporal dimensions of a fruit-feeding butterfly community in an Ecuadorian rainforest. Biol J Linn Soc 62, 343–364 (1997).
Savolainen, V. et al. Sympatric speciation in palms on an oceanic island. Nature, 441, 210–213 (2006).
Marten, A., Braendle, M. & Brandl, R. Habitat type predicts genetic population differentiation in freshwater invertebrates. Mol Ecol 15, 2643–2651 (2006).
Park, C. C. Tropical rainforests. Routledge, London and New York (2002).
Hall, R. Cenozoic geological and plate tectonic evolution of SE Asia and the SW Pacific: computer-based reconstructions, model and animations. J Asian Earth Sci 20, 353–431 (2002).
Cottam, M. A., Hall, R. & Ghani, A. A. Late Cretaceous and Cenozoic tectonics of the Malay Peninsula constrained by thermochronology J Asian Earth Sci 76, 241–257 (2013).
DiNezio, P. N. & Tierney, J. E. The effect of sea level on glacial Indo-Pacific climate. Nat Geosci 6, 485–491 (2013).
Najman, Y., Pringle, M., Godlin, L. & Oliver, G. Dating of the oldest continental sediments from the Himalayan foreland basin. Nature 410, 194–197 (2001).
Wu, G. X. et al. Thermal Controls on the Asian Summer Monsoon. Sci. Rep. 2, 404 (2012).
Cannon, C. H., Morley, R. J. & Bush, A. B. G. The current refugial rainforests of Sundaland are unrepresentative of their biogeographic past and highly vulnerable to disturbance. Proc Nat Acad Sci USA, 106, 11188–11193 (2009).
Morley, R. J. Origin and evolution of tropical rain forests. John Wiley and Sons Ltd., Chichester (2000).
Papadopoulou, A., Anastasiou, I., Keskin, B. & Vogler, A. P. Comparative phylogeography of tenebrionid beetles in the Aegean archipelago: the effect of dispersal ability and habitat preference. Mol Ecol 18, 2503–2517 (2009).
Bocak, L., Bocakova, M., Hunt, T. & Vogler, A. P. (2008) Multiple ancient origins of neoteny in Lycidae (Coleoptera): consequences for ecology and macroevolution. Proc R Soc B-Biol Sci 275, 2015–2023 (2008).
Malohlava, V. & Bocak, L. Evidence of extreme habitat stability in a Southeast Asian biodiversity hotspot based on the evolutionary analysis of neotenic net-winged beetles. Mol Ecol 19, 4800–4811 (2010).
Masek, M., Palata, V., Bray, T. C. & Bocak, L. Molecular phylogeny reveals high diversity, geographic structure and limited ranges in neotenic net-winged beetles Platerodrilus (Coleoptera: Lycidae). PloS One 10, e0123855 (2015).
Eisner, T. et al. Defensive chemistry of lycid beetles and of mimetic cerambycid beetles that feed on them. Chemoecology 18, 109–119 (2008).
Bocak, L. & Yagi, T. Evolution of mimicry patterns in Metriorrhynchus (Coleoptera: Lycidae): the history of dispersal and speciation in Southeast Asia. Evolution 64, 39–52 (2010).
Chouteau, M. & Angers, B. The role of predators in maintaining the geographic organization of aposematic signals. Am Nat 178, 810–817 (2011).
Malohlava, V. & Bocak, L. A revision of the neotenic net-winged beetle Scarelus (Coleoptera: Lycidae). Ann Soc entomol Fr 47, 176–192 (2011).
Grimbacher, P. S. & Catterall, C. P. How much do site age, habitat structure and spatial isolation influence the restoration of rainforest beetle species assemblages? Biol Cons 135, 107–118 (2007).
Fujisawa, T., Vogler, A. P. & Barraclough, T. G. Ecology has contrasting effects on genetic variation within species versus rates of molecular evolution across species in water beetles. Proc R Soc B-Biol Sci 282, 20142476 (2015).
Hausdorf, B. & Hennig, C. Nestedness of north-west European land snail ranges as a consequence of differential immigration from Pleistocene glacial refuges. Oecologia 35, 102–109 (2003).
Moore, B. P. & Brown, W. V. Identification of warning odour components, bitter principles and antifeedants in an aposematic beetle–Metriorrhynchus rhipidium (Coleoptera: Lycidae). Ins Biochem 15, 493–499 (1981).
Martin, S. H. et al. Genome-wide evidence for speciation with gene flow in Heliconius butterflies. Gen Res 23, 1817–1828 (2013).
Bocakova, M., Bocak, L., Hunt, T., Teravainen, M. & Vogler, A. P. Molecular phylogenetics of Elateriformia (Coleoptera): evolution of bioluminescence and neoteny. Cladistics 23, 477–496 (2007).
Kundrata, R., Bocakova, M. & Bocak, L. The comprehensive phylogeny of Elateroidea (Coleoptera): molecular evidence for evolution of soft-bodiedness, neoteny and bioluminiscence. Mol Phyl Evol 76, 162–171 (2014).
Bocak, L., Grebennikov, V. V. & Sklenarova, K. Cautires apterus, a new species and the first record of wingless male Lycidae (Coleoptera) discovered in the North Pare Mountains, Tanzania. Ann Zool 64, 1–7 (2014).
Bocak, L., Kundrata, R., Andújar Fernández, C. & Vogler, A. P. The discovery of Iberobaeniidae (Coleoptera: Elateroidea): a new family of beetles from Spain, with immatures detected by environmental DNA sequencing. Proc R Soc B-Biol Sci 283, 20152350 (2016).
Liebherr, J. K. Gene flow in ground beetles (Coleoptera: Carabidae) of differing habitat preference and hight-wing development. Evolution 42, 129–137 (1988).
Su, Y. C., Wang, J. F., Villanueva, R. J. T., Nuneza, O. M. & Lin, C. P. Hopping out of Mindanao: Miocene-Pliocene geological processes and cross-island dispersal as major drivers of diversity for Philippine treehoppers. J Biogeogr 41, 1277–1290 (2014).
Balke, M. et al. New Guinea highland origin of a widespread arthropod supertramp. Proc R Soc B-Biol Sci 276, 2359–2367 (2009).
Twyford, A. D. & Ennos, R. A. Next-generation hybridization and introgression. Heredity 108, 179–189 (2012).
Willing, E.-M., Dreyer, C. & van Oosterhout, C. Estimates of genetic differentiation measured by FST do not necessarily require large sample sizes when using many SNP markers. PLoS One 7, e42649 (2012).
Eaton, D. A. R. & Ree, R. H. Inferring Pphylogeny and introgression using RADseq data: an example from flowering plants (Pedicularis: Orobanchaceae). Syst Biol 62, 689–706 (2013).
Excoffier, L., Dupanloup, I., Huerta-Sanchez, E., Sousa, V. C. & Foll, M. Robust demographic inference from genomic and SNP data. PLoS Genet 9, e1003905 (2013).
Eaton, D. A. R. PyRAD: assembly of de novo RADseq loci for phylogenetic analyses. Bioinformatics 30, 1844–1849 (2014).
Chattopadhyay, B., Garg, K. M. & Ramakrishnan, U. Effect of diversity and missing data on genetic assignment with RAD-Seq markers. BMC Res Notes 7, 841 (2014).
Ilut, D. C., Nydam, M. L. & Hare, M. P. Defining loci in restriction-based reduced representation genomic data from nonmodel species: sources of bias and diagnostics for optimal clustering. Bio Med Res Int 675158 (2014).
Harvey, M. G. et al. Similarity thresholds used in short read assembly reduce the comparability of population histories across species PeerJ 3, e895 (2015).
R Core Team R: A language and environment for statistical computing. R Foundation for Statistical Computing, Vienna, Austria. http://www.R-project.org/ (2016).
Lischer, H. E. L. & Excoffier, L. PGDSpider: an automated data conversion tool for connecting population genetics and genomics programs. Bioinformatics 28, 298–299 (2012).
Foll, M. & Gaggiotti, O. E. A genome scan method to identify selected loci appropriate for both dominant and codominant markers: A Bayesian perspective. Genetics 180, 977–993 (2008).
Antao, T., Lopes, A., Lopes, R. J., Beja-Pereira, A. & Luikart, G. LOSITAN: a workbench to detect molecular adaptation based on a Fst-outlier method. BMC Bioinform. 9, 323 (2008).
Pritchard, J. K., Stephens, M. & Donnelly, P. Inference of population structure using multilocus genotype data. Genetics 155, 945–959 (2000).
Price, A. L. et al. Principal components analysis corrects for stratification in genome- wide association studies. Nat Genet 38, 904–909 (2006).
Benazzo, A., Panziera, A. & Bertorelle, G. 4P: fast computing of population genetics statistics from large DNA polymorphism panels. Ecol Evol 5, 172–175 (2015).
Katoh, K. & Standley, D. M. MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol Biol Evol 30, 772–780 (2013).
Stamatakis, A. RAxML Version 8: A tool for phylogenetic analysis and post- analysis of large phylogenies. Bioinformatics 30, 1312–1313 (2014).
Miller, M. A., Pfeiffer, W. & Schwartz, T. “Creating the CIPRES Science Gateway for inference of large phylogenetic trees” in Proceedings of the Gateway Computing Environments Workshop, 14 Nov. 2010, New Orleans, LA, pp 1–8 (2010).
Darriba, D., Taboada, G. L., Doallo, R. & Posada, D. jModelTest 2: more models, new heuristics and parallel computing. Nat Meth 9, 772 (2012).
Stamatakis, A., Hoover, P. & Rougemont, J. A rapid bootstrap algorithm for the RAxML web servers. Syst Biol 57, 758–771 (2008).
Drummond, A. J. & Bouckaert, R. R. Bayesian Evolutionary Analysis with Beast. Cambridge University Press, Cambridge (2015).
Drummond, A. J., Suchard, M. A., Xie, D. & Rambaut, A. Bayesian phylogenetics with BEAUti and the BEAST 1.7. Mol Biol Evol 29, 1969–1973 (2012).
Felsenstein, J. Evolutionary trees from DNA sequences: a maximum likelihood approach. J Mol Evol 17, 368–376 (1981).
Rambaut, A., Suchard, M. A., Xie, D. & Drummond, A. J. Tracer 1.6, Available from http://beast.bio.ed.ac.uk/Tracer (2014).
Acknowledgements
This project was supported by the Czech Science Foundation (P506/11/1757), the European Social Fund and the Czech Ministry of Education (CZ.1.07/2.3.00/30.0004). We thank to A. P. Vogler, A. Jansen van Rensburg and the colleagues in the Department of Evolutionary Biology and Environmental Studies, University of Zurich for advice and discussion, M. Bednarik, L. Dembicky and E. Jendek for specimens, M. Masek for preparation of maps, M. Motyka for discussion on Beast analyses and to Ms. Renata Bilkova for the technical assistance.
Author information
Authors and Affiliations
Contributions
L.B. designed the study. T.C.B. analysed RAD data, L.B. conducted phylogenetic analyses, T.C.B. and L.B. wrote the manuscript. Both authors approved the final version of the manuscript.
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Electronic supplementary material
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Bray, T., Bocak, L. Slowly dispersing neotenic beetles can speciate on a penny coin and generate space-limited diversity in the tropical mountains. Sci Rep 6, 33579 (2016). https://doi.org/10.1038/srep33579
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep33579
- Springer Nature Limited