
Abstract
Large interindividual variability has been observed in the metabolism of CYP2C19 substrates in vivo. The study aimed to evaluate sources of this variability in CYP2C19 activity, focusing on CYP2C19 diplotypes and the cytochrome P450 oxidoreductase (POR). CYP2C19 gene analysis was carried out on 347 human liver samples. CYP2C19 activity assayed using human liver microsomes confirmed a significant a priori predicted rank order for (S)-mephenytoin hydroxylase activity of CYP2C19*17/*17 > *1B/*17 > *1B/*1B > *2A/*17 > *1B/*2A > *2A/*2A diplotypes. In a multivariate analysis, the CYP2C19*2A allele and POR protein content were associated with CYP2C19 activity. Further analysis indicated a strong effect of the CYP2C19*2A, but not the *17, allele on both metabolic steps in the conversion of clopidogrel to its active metabolite. The present study demonstrates that interindividual variability in CYP2C19 activity is due to differences in both CYP2C19 protein content associated with gene diplotypes and the POR concentration.
Similar content being viewed by others


Introduction
CYP2C19 is a member of the human drug-metabolizing family of cytochrome P450 enzymes that is encoded by the CYP2C19 gene. It is a clinically important enzyme that plays a critical role in the metabolism and related drug–drug interactions of a variety of therapeutic agents, including proton pump inhibitors, antiepileptic agents, antiplatelet drugs and antidepressants.1, 2, 3 Large interindividual variability has been observed in the metabolism of these drugs in vivo, with differences presumed to be due in part to inherited variation in the CYP2C19 gene. Similar to several other human P450 family members, CYP2C19 activity is polymorphic, with sub-populations of poor metabolizers, intermediate metabolizers, extensive metabolizers and ultrarapid metabolizers.4, 5, 6
Multiple allelic variants in the CYP2C19 gene have been described, with over 69 reported to date (http://www.cypalleles.ki.se/cyp2c19.htm, the last update was 8 September 2014). Many of these are derived from nonsynonymous single-nucleotide polymorphisms (nsSNPs) in the coding regions of the CYP2C19 gene. Some of these genetic changes profoundly affect hepatic CYP2C19 expression, with metabolic insufficiency arising from the absence of CYP2C19 protein accumulation. For example, homozygosity for the CYP2C19*2A (19154G>A; rs4244285) allele causes aberrant mRNA splicing, resulting in no detectable hepatic protein and a poor metabolizer phenotype.7, 8 In contrast, the CYP2C19*17 (−806C>T; rs12248560) allele is associated with an increase in transcriptional activity that has been described as causing ultra-rapid metabolism of CYP2C19 substrates.9, 10
In a clinical study involving the proton pump inhibitors omeprazole and lansoprazole, a genotype-dependent increase in drug area under the concentration-time curve and intragastric pH was observed, resulting in a change in the healing rate of peptic ulcer, gastroesophageal reflux disease and/or the Helicobacter pylori eradication rate.11, 12 Another prominent example involves bioactivation of the antiplatelet drug clopidogrel by CYP2C19. CYP2C19*2 carriers receiving the drug are at increased risk for recurring cardiovascular events such as myocardial infarction, cerebral stroke and stent thrombosis than are patients homozygous for the wild-type allele.13, 14, 15 This difference in clinical outcome has been attributed to significantly lower blood levels of the active thiol metabolite of clopidogrel in patients carrying the CYP2C19*2 allele.13 In contrast, the CYP2C19*17 allele has been associated with an enhanced efficacy of clopidogrel in reducing reoccurring cardiovascular events, risk of stent thrombosis and residual platelet aggregation, while increasing the risk of bleeding events.16, 17, 18 Although not all clopidogrel study results are in agreement, the CYP2C19*17 allele has also been associated with enhanced metabolism of several other drugs, including omeprazole, amitriptyline, voriconazole, mephenytoin, pantoprazole and escitalopam.9, 19, 20, 21, 22, 23, 24, 25, 26
There is only a limited understanding of the mechanistic basis for the CYP2C19*17-associated phenotype. The putative causal SNP is found in the promoter of the CYP2C19 gene and ostensibly affects the binding of transcription factors that enhance gene transcription.9 However, a recent paper reported no statistically significant difference in CYP2C19 enzyme activity ((S)-mephenytoin hydroxylation) in microsomes from livers with a heterozygous or homozygous CYP2C19*17 genotype, compared with liver microsomes from homozygous wild-type livers, although directionality of the observed trend supported the hypothesis.10 The CYP2C19*2 and CYP2C19*17 loci appear to be in linkage disequilibrium (LD), and thus one finds a mixed allelic diplotype in the population (that is, CYP2C19*2/CYP2C19*17) that has an uncertain phenotype.26, 27, 28 With this in mind, and the lack of clarity regarding the exact functional consequences of the CYP2C19*17 allele, we sought to characterize interindividual variability of CYP2C19-catalyzed drug ((S)-mephenytoin and clopidogrel) metabolism in a large set of human livers and determine its dependency on the CYP2C19*2 and CYP2C19*17 allelic variants, with special consideration of predicted diplotypes, as well as the specific content of the obligatory coenzyme, cytochrome P450 oxidoreductase (POR). We also assessed the contribution of rare CYP2C19 variation together with clinical and other demographic variables to interindividual differences in hepatic microsomal enzyme activity.
Materials and methods
Chemicals and reagents
(S)-mephenytoin was purchased from Sigma-Aldrich (St Louis, MO, USA). 4-Hydroxymephenytoin-d3 was purchased from Toronto Research Chemicals (Toronto, ON, Canada). Clopidogrel, 2-oxo-clopidogrel, clopidogrel active thiol metabolite and prasugrel were all kindly provided by Eli Lilly (Indianapolis, IN, USA) and the Daiichi Sankyo (Tokyo, Japan). Stable isotope labeled amino acids [13C615N2]-lysine and [13C615N4]-arginine were purchased from Pierce Biotechnology (Rockford, IL, USA). All other chemicals and general reagents were of analytical grade or better and were obtained from various commercial sources such as Cerilliant (Round Rock, TX, USA), Invitrogen (Carlsbad, CA, USA) or Applied Biosystems (Foster City, CA, USA).
Tissue samples
A total of 347 different human livers used in the present study consisted of 65 samples obtained from the University of Washington Human Liver Bank (Seattle, WA, USA), a resource established by a Program Project grant on Drug Interactions from the National Institutes of Health (P01-GM32165) and 282 liver samples from the St Jude Liver Resource at St Jude Children’s Research Hospital (Memphis, TN, USA), a resource established by the Liver Tissue Procurement and Distribution System (National Institutes of Health National Institute of Diabetes and Digestive and Kidney Diseases Contract N01-DK92310) and by the Cooperative Human Tissue Network. The collection and use of these tissues for research purposes was approved by the Human Subjects Institutional Review Boards at the University of Washington and St Jude Children’s Research Hospital. In both cases, all links between archived tissues and the original donors were destroyed to preserve anonymity and facilitate study execution. Additional details about the selection of livers and sample size and investigator blinding for sample analysis can be found in Supplementary Methods.
Genotyping and CYP2C19 exon sequencing
Genomic DNA from human livers was isolated by use of a DNeasy tissue kit. Genotyping of the CYP2C19*2 (rs4244285) and CYP2C19*17 (rs12248560) variants was performed according to the manufacturer’s instructions using TaqMan SNP genotyping assays (Applied Biosystems). The detailed methods used for genotyping and exon sequencing are described in the Supplementary Methods.29
LD analysis
Pairwise LD was calculated and displayed using Haploview 4.2 software.30 The D’, along with its corresponding logarithm of odds (LOD) score, and r2 LD values were determined between rs3758581 (CYP2C19*1A or *1B), rs4244285 (CYP2C19*2) and rs12248560 (CYP2C19*17) SNPs. The frequencies of other alleles were too low for meaningful LD analysis.
Liver microsomal preparations
Human liver microsomes (HLMs) were prepared using a modified version of previously described methods.31, 32 The detailed method used for HLM preparations is described in the Supplementary Methods. To generate pooled HLMs from the following homozygous diplotypes (CYP2C19*1B/*1B, CYP2C19*2A/*2A and CYP2C19*17/*17), an equal amount of microsomal protein from 115, 5 and 23 individual preparations, respectively, was combined. Total protein concentrations were determined by the BCA Protein Assay (Pierce, Rockford, IL, USA). Total microsomal P450 concentrations for each of the HLM pools were determined by difference spectroscopy of the carbon monoxide-complexed hemoprotein.33
CYP2C19 activity assay in HLMs
(S)-Mephenytoin hydroxylation activity phenotyping and kinetic analysis
Liver microsomal CYP2C19 metabolic activity profiling was conducted at (S)-mephenytoin concentrations of 20 μM (fivefold below reported Km values). For each liver microsomal preparation, incubations were performed in triplicate in solutions containing 100 mM potassium phosphate buffer (KPi, pH 7.4) and final incubation volumes were 100 μl. (S)-mephenytoin and HLM (0.1 mg ml−1) were preincubated for 10 min at 37 °C before reactions were initiated by adding NADPH (1 mM, final concentration). Reactions were terminated after 20 min by adding an equal volume of ice-cold acetonitrile containing 100 nM internal standard (d3-4’-hydroxymephenytoin).
For the determination of kinetic parameters, CYP2C19-catalyzed (S)-mephenytoin 4’-hydroxylation was measured in triplicate over a range of substrate concentrations (1–400 μM), using pooled HLMs with the following homozygous diplotypes: CYP2C19*1B/*1B, *2A/*2A and *17/*17. All incubation conditions and sample workup were the same as that described above for HLM phenotyping. The concentrations of 4’-hydroxy (S)-mephenytoin in the incubations were quantified using a liquid chromatography–tandem mass spectrometry (LC-MS/MS) system (Supplementary Methods).
2-Oxo-clopidogrel formation kinetics
Incubation mixtures contained 0.25 mg ml−1 of diplotype-defined pooled HLM protein, 1% of a 100 × concentrated dimethyl sulfoxide stock solution of clopidogrel (for final substrate concentrations of between 100 nM and 100 μM) and 20 mM ascorbic acid in 100 mM KPi buffer, pH 7.4, containing 100 mM of the hepatic carboxylesterase inhibitor, NaF (to 200 μl final incubation volume). The reaction rate at each substrate concentration was determined in triplicate. After 3 min of preincubation at 37 °C/70 r.p.m. in a water bath, the reactions were initiated by the introduction of an aqueous stock solution of NADPH (1 mM final concentration). Reactions were quenched after 5 min of incubation at 37 °C/70 r.p.m. by the addition of a 50% volume of ice-cold acetonitrile, containing 80 nM prasugrel as internal standard, and were then centrifuged to remove protein. Supernatants were analyzed by LC-MS/MS (Supplementary Methods).
Clopidogrel active metabolite (CAM) formation kinetics
Incubation mixtures again contained 0.25 mg ml−1 microsomal protein, from one of the three CYP2C19 diplotyped HLM pools, 1% of a freshly prepared 100 × concentrated dimethyl sulfoxide stock solution of 2-oxo-clopidogrel (100 nM to 100 μM final concentration) and 20 mM ascorbic acid (included as a glutathione surrogate in order to induce reduction of a presumed sulfenic acid intermediate to the final thiol active metabolite) in 100 mM KPi buffer containing 100 mM NaF (200 μl final volume). The reaction rate at each substrate concentration was determined in triplicate. Reactions were preincubated at 37 °C/70 r.p.m. for 3 min before initiation with NADPH (1 mM final concentration) and were then incubated for 15 min before quenching with a 50% volume of an ice-cold solution of 80 nM prasugrel in acetonitrile. 2-Bromo-3’-methoxyacetophenone (30 μl from a 100 mM dimethyl sulfoxide stock solution) was added as a thiol derivatizing agent and the reactions were incubated for an additional 60 min at 37 °C/70 r.p.m. Protein was then removed by centrifugation and supernatants were analyzed by LC-MS/MS (Supplementary Methods).
Determination of CYP2C19 kinetic parameters
Kinetic parameters for (S)-mephenytoin 4’-hydroxylation by genotype-defined pooled HLMs were estimated by means of nonlinear least-squares analysis, using the MULTI program.34 The affinity of (S)-mephenytoin for CYP2C19 (Km) and the maximal velocity of CYP2C19-catalyzed (S)-mephenytoin 4’-hydroxylation (Vmax) were obtained by fitting the data to the Michaelis–Menten equation (Equation (1)):

where V is the initial metabolism rate of (S)-mephenytoin (pmol min−1 per mg protein), and [S] is the concentration of substrate, (S)-mephenytoin, in the incubation (μM).
Kinetic analyses of 2-oxo-clopidogrel and CAM formation data were performed on GraphPad Prism software, v. 5.03 (San Diego, CA, USA). The following two-enzyme kinetic model was fit to the biphasic data:35

Eadie–Hofstee graphs for both steps of clopidogrel bioactivation were clearly nonlinear across all genotypes, and data comparisons performed in GraphPad Prism using the above two-enzyme model against standard Michaelis–Menten kinetics generated F-values well above 1 for all data sets (9.5<F<120), with P-values ≤0.0012, indicating the relative accuracy of the biphasic model.
Graphical presentation of the data included the best fitted regression line and data points denoting mean values and s.d. error bars from triplicate incubations at each substrate concentration. Tabular presentation of the data represents the mean values and s.d. error bars for parameter estimates determined from three replicate experiments.
Quantitation of total P450 protein content in HLMs
The contents of total P450 protein for pooled HLMs with defined homozygous diplotypes (CYP2C19*1B/*1B, *2A/*2A and *17/*17) were examined by reduced carbon monoxide difference spectral analysis measured on an Olis-modernized Aminco DW-2 spectrophotometer (Olis, Bogart, GA, USA). The detailed methods used are described in the Supplementary Methods.36 All values were determined as means from duplicate experiments.
Quantitation of CYP2C19 and cytochrome P450 oxidoreductase protein content in HLMs
CYP2C19 and POR protein expression in individual or genotype-defined pooled HLM samples were determined by a surrogate peptide-based LC-MS/MS method. Synthetic surrogate peptides of CYP2C19 and POR for LC-MS/MS quantification were selected based on previously reported criteria (Supplementary Table 3).37, 38 The detailed methods used for protein quantitation are described in the Supplementary Methods.39, 40, 41 The CYP2C19 and POR content of each individual microsomal preparation and pooled (based on diplotype) preparations were determined in triplicate. The protein expression data for pooled samples are reported as the mean and s.d. of values obtained from the three experimental determinations. When the measured result was below lower limit of quantification (LLOQ; or not detected), the LLOQ value was introduced as a substitute, although this has a slightly different meaning than <LLOQ.
Statistical analysis
Multivariate linear regression analysis
A multivariate linear regression analysis of demographic and medical variables for the diplotyped liver donors was performed to elucidate major determinants of interindividual variability in CYP2C19 activity (Microsoft Office Excel 2010, Microsoft, Redmond, WA, USA). Full details are given in the Supplementary Methods section. A P-value of <0.05 was considered to be statistically significant for all tests.
Other statistical analyses
The mephenytoin hydroxylation activity data for the full 347 liver data set exhibited a skewed distribution. Accordingly, the nonparametric Mann–Whitney test was performed for between-diplotype group comparisons in the study using individual HLMs. The two-sided unpaired Student’s t-test, with unequal variances, was performed for post hoc between-diplotype group comparisons in the study using pooled HLMs. A correction for multiple comparisons was not applied. A linear trend test (one-way analysis of variance) was performed for the rank order analysis, and a P-value of <0.05 was considered to be statistically significant.
Results
Liver demographics
Characteristics of liver donors and the tissue itself are summarized in Table 1. A total of 347 liver samples from 200 male donors (mean age 37.7 years, range 0–81 years), 142 female donors (mean age 42.8 years, range 0–87 years) and 5 unknown were tested. The majority (96.3 %) of samples were of Caucasoid (White) origin. Of the 347 donors, 10 (2.88%) were not receiving medications at the time of death or tissue procurement, 154 (44.4%) were receiving one or more medications and 183 (52.7%) had no medication history reported. In addition, 119 donors (34.3%) had no disease at the time of death; 52 (15.0%) had liver disease including fatty liver, chronic hepatitis, fibrotic liver, hepatoma, acute injury and biliary atresia; 74 (21.3%) had any other diseases including cardiovascular disturbance, diabetes, cystic fibrosis and colorectal cancer; 102 (29.4%) were unknown regarding disease information. The cause of death was recorded by a forensic pathologist as: 99 donors (28.5%) had brain damage including cerebral vascular accident, head trauma, subarachnoid hemorrhage and intracerebral hemorrhage; 14 (4.03%) had suffered another accident including auto accident and gunshot; 226 (65.1%) were unknown regarding the cause of death.
CYP2C19 allele and diplotype frequencies
In the present study, 18 different variant alleles of CYP2C19 were identified and they are listed in Table 2. Of these, CYP2C19*1C (80161A>G), *2A (99C>T, 19154G>A, 80160C>T, 80161A>G) and *17 (−3402C>T, −806C>T, 99C>T, 80161A>G) consisted of synonymous or noncoding SNPs. Alleles containing nonsynonymous coding SNPs were CYP2C19*1A, *4B (−3402C>T, −806C>T, 1A>G, 99C>T, 80161A>G), *6 (99C>T, 12748G>A, 80161A>G), *8 (12711T>C), *11 (99C>T, 12802G>A, 80161A>G), *13 (80161A>G, 87290C>T), L14S (37T>C), Q22R (65A>G), G62R (12368G>C), R125H (12727G>A), R150C (12801C>T), G228E (19155G>A), D341H (80191G>C) and D360N (80248G>A). These nsSNPs were classified into deleterious nsSNPs (CYP2C19*4B, *6, *8, L14S, G62R, R125H, G228E, D341H and D360N variant alleles) or benign nsSNPs (CYP2C19*1A, *11, *13, Q22R and R150C variant alleles) for the functional consequence (Supplementary Table 2). The other CYP2C19*2 variant, *2B (99C>T, 12460G>C, 19154G>A, 80160C>T, 80161A>G), was also detected in the set of human livers studied, but it was found at a low frequency (<5%), compared with CYP2C19*2A. The coding change (Glu92Asp) is predicted to be benign, but the phenotype is probably null because of the presence of the splice site variant (19154G>A) in the haplotype.
To characterize the association between gene diplotype and enzyme activity, we focused on the following alleles: CYP2C19*1B, *1C, *2A and *17. From the panel of 347 human liver samples, the frequencies of the respective CYP2C19*1B/*1B, *1B/*1C, *1C/*1C, *1B/*2A, *2A/*2A, *1B/*17, *17/*17, *1C/*2A, *1C/*17 and *2A/*17 diplotypes were 32.6% (n=113), 0.576% (n=2), 0% (n=0), 12.1% (n=42), 1.44% (n=5), 19.0% (n=66), 6.63% (n=23), 0.288% (n = 1), 0% (n=0) and 7.49% (n=26), respectively (Table 1). The reference allele, CYP2C19*1A, was detected in only 24 livers. The frequencies of the liver groups with deleterious and benign nsSNPs were 2.59% (n=9) and 17.3% (n=60), respectively.
Impact of gene diplotype on CYP2C19-catalyzed metabolism of (S)-mephenytoin
CYP2C19 activity was measured using HLMs isolated from 347 genotyped liver samples and (S)-mephenytoin as a prototypical substrate. We saw no difference between the (S)-mephenytoin 4’-hydroxylase activity of HLMs from livers with the *1A/*1B and *1B/*1B diplotypes (data not shown). Because the frequency of CYP2C19*1A and *1C alleles was remarkably low compared with that of other alleles, only CYP2C19*1B, *2A and *17 alleles were used to test diplotype–activity association in subsequent analyses.
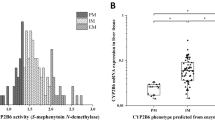
The Mann–Whitney test was performed for between-diplotype group comparisons (Table 3 and Supplementary Figure 1). In the group of HLMs with a CYP2C19*2A/*2A diplotype, the mean activity was only 5.14% of that found for the wild-type HLMs with a CYP2C19*1B/*1B diplotype. The mean activity for the CYP2C19*1B/*2A diplotype was 35.4% lower than that of the CYP2C19*1B/*1B diplotype, although the difference did not reach statistical significance. There was a trend for higher CYP2C19 activity in livers with a CYP2C19*1B/*17 (1.05-fold) and *17/*17 (1.28-fold) diplotype, compared with the CYP2C19*1B/*1B diplotype, but again the head-to-head comparisons were not significantly different. In addition, the metabolic activity of the CYP2C19*2A/*17 diplotype livers was on average 39.6% lower than that of the CYP2C19*1B/*1B diplotype livers, suggesting that there was not an equal offset of effects from the *2 and *17 alleles, but once again the difference was not significant. For the livers from donors carrying a nonsynonymous coding variant, the mean activity in the livers with deleterious nsSNPs was 14.9% lower than that in the livers with a CYP2C19*1B/*1B diplotype, although the difference did not reach statistical significance (Table 3). The metabolic activity of livers from donors carrying a benign nsSNP was not different from that of the CYP2C19*1B/*1B diplotype. Using a linear trend analysis, the a priori assigned rank order for enzyme activity of the common diplotypes (CYP2C19*17/*17 > *1B/*17 > *1B/*1B > *2A/*17 > *1B/*2A > *2A/*2A) was statistically significant (P<0.05) (Figure 1).

Association between CYP2C19 diplotype and CYP2C19 metabolic activity. CYP2C19-catalyzed (S)-mephenytoin 4’-hydroxylation activity of 347 different human liver microsome (HLM) preparations was measured and segregated according to the observed CYP2C19 diplotypes. Data (closed bars) are presented as mean±s.d. The insert is a box-and-whisker plot (Tukey’s box plot). The box represents the 25th and 75th percentiles enclosing a median bar, and the whiskers represent the highest and lowest values that are not outliers or extreme values. Outliers (values that are more than 1.5 times the interquartile range) are individually represented by open circles beyond the whiskers. A linear trend test (one-way analysis of variance (ANOVA)) was performed for the rank order and was found to be significant (P<0.05). A two-sided, unpaired Student’s t-test was employed for post hoc, pairwise comparisons; †P<0.05, significantly different from metabolic activity in HLM with CYP2C19*1B/*1B diplotype.
LD between CYP2C19*1B, *2A and *17 alleles
The LD between the common CYP2C19 SNPs is visualized in Figure 2. The calculations show complete LD (|D’|=1, LOD > 2) between CYP2C19*2A and *17. Complete LD was also observed between CYP2C19*1B and *17. However, although the analysis revealed |D’|=1 between CYP2C19*1B and *2A alleles, the LOD score <2 indicates that the loci are not significantly linked. Even though CYP2C19*2A and *17 alleles are in complete LD, the correlation coefficient (r2) between them was only 0.057. Low r2 values were also observed between CYP2C19*1B and CYP2C19*2A and between CYP2C19*1B and *17 (r2=0.003 and 0.017, respectively).

Linkage disequilibrium (LD) map for the common CYP2C19 allelic variants. An LD map of the CYP2C19*1B, *2A and *17 alleles was created using Haploview 4.2. The dark gray (the red in a full colour version) squares show complete LD with statistical significance (|D’|=1, LOD > 2), and the light gray (the blue in a full colour version) square shows complete LD, but lacking statistical significance (|D’|=1, LOD<2). LOD, logarithm of odds. A full colour version of this figure is available at the The Pharmacogenomics Journal journal online.
Correlation between CYP2C19 metabolic activity and protein expression
The CYP2C19 protein content for pooled HLMs from donors with a defined CYP2C19 diplotype is presented in Figure 3. As expected, in the group of HLMs with a CYP2C19*2A/*2A diplotype, the mean CYP2C19 protein level was negligible compared with that found for the wild-type HLMs with a CYP2C19*1B/*1B diplotype. The mean protein level for the CYP2C19*1B/*2A diplotype was 46.9% lower than that of the CYP2C19*1B/*1B diplotype (P<0.05). There was a trend for a higher CYP2C19 protein level in livers with a CYP2C19*17/*17 (1.22-fold) diplotype, compared with the CYP2C19*1B/*1B diplotype, although the differences did not reach statistical significance. The protein level of the CYP2C19*2A/*17 diplotype livers was significantly lower than that of the CYP2C19*1B/*1B diplotype livers. Using a linear trend analysis, the rank order of enzyme protein level for the common diplotypes (CYP2C19*17/*17 > *1B/*17 > *1B/*1B > *2A/*17 > *1B/*2A > *2A/*2A) was significant (P<0.0001). A strong correlation was observed between CYP2C19 activity and protein content in these six genotype-defined HLMs groups (r2=0.984; Figure 4).

Association between CYP2C19 diplotype and CYP2C19 protein content in genotype-defined pooled human liver microsomes (HLMs). The absolute abundance of CYP2C19 protein was quantified by surrogate peptide-based liquid chromatography–tandem mass spectrometry (LC-MS/MS) analysis. Data (closed bars) are presented as means±s.d. (n=3 replicate determinations). A linear trend test (one-way analysis of variance (ANOVA)) was performed for the rank order and was found to be statistically significant (P<0.0001). A two-sided, unpaired t-test was employed for post hoc, pairwise comparisons; †P<0.05, significantly different from CYP2C19 protein content in HLMs with CYP2C19*1B/*1B diplotype. ‡P<0.01, significantly different from CYP2C19 protein content in HLMs with CYP2C19*1B/*1B diplotype.

Correlation between CYP2C19 metabolic activity and protein content in genotype-defined pooled human liver microsomes (HLMs). CYP2C19-catalyzed (S)-mephenytoin 4’-hydroxylation activity and CYP2C19 protein content for genotype-defined pooled HLMs (CYP2C19*1B/*1B (filled circles), CYP2C19*2A/*2A (filled squares), CYP2C19*17/*17 (filled triangles), CYP2C19*1B/*2A (open circles), CYP2C19*1B/*17 (open squares) and CYP2C19*2A/*17 (open triangles) diplotypes) were measured. Individual data points represent the mean of triplicate protein and activity determinations. Approximately 98% of the variability in CYP2C19 activity in the different diplotype pools could be explained by CYP2C19 protein variability (r2=0.984).
POR content in HLMs
The mean POR protein contents for HLMs from donors with CYP2C19*1B/*1B, *1B/*2A, *2A/*2A, *1B/*17, *17/*17 and *2A/*17 diplotypes were 22.5±12.6, 31.6±16.4, 24.9±17.2, 21.8±11.4, 27.7±11.9 and 22.0±9.5 pmol per mg protein, respectively. There was no significant difference in the mean POR protein content of the different diplotype groups. A linear trend analysis also showed no significant rank order correlation between CYP2C19 diplotype and POR level, indicating that POR variability could be an independent determinant of CYP2C19-catalyzed drug metabolism. Similar results for the POR protein contents were obtained from pooled HLMs (Supplementary Figure 2).
Multivariate linear regression analysis for factors affecting interindividual variability of CYP2C19-catalyzed (S)-mephenytoin metabolism
The results of the multivariate linear regression analysis, with missing values imputed, for the identification of predictors that affect interindividual variability in CYP2C19-catalyzed (S)-mephenytoin 4’-hydroxylation can be found in Table 4. The coefficient of determination (R2 value) for the multivariate linear regression was 0.129, indicating that ∼13% of interindividual variability of CYP2C19 activity is explained by the predictors in the model. The CYP2C19*2A and POR are significant predictors of CYP2C19 activity, with P-values of 0.000719 and 8.00 × 10−6, respectively. The effect size (β-value) of the CYP2C19*2A allele was −10.5, indicating that each copy of the *2A allele inherited by an individual results in an average decrease of −10.5 pmol min−1 per mg protein in CYP2C19 activity. All other covariates were not significant, including CYP2C19*17, that had a P-value of 0.585. We also conducted a multivariate regression analysis without imputing missing data and including only CYP2C19*2A, CYP2C19*17 and POR as covariates (Supplementary Table 4). Here the results were similar, where CYP2C19*2A and POR are significant predictors of CYP2C19 activity in the reduced model, with P-values of 0.00106 and 3.61 × 10−6, respectively. Again, CYP2C19*17 was not a significant factor in the reduced model. The R2 value for the multivariate linear regression was 0.103.
Kinetic analysis of CYP2C19-catalyzed (S)-mephenytoin metabolism by pooled HLMs from donors with homozygous CYP2C19 diplotypes
The mechanism for the effect of CYP2C19 diplotype on enzyme activity was examined, in part, by determining the enzyme concentration dependence of the CYP2C19-catalyzed 4’-hydroxylation of (S)-mephenytoin in pooled HLMs with defined homozygous diplotypes (CYP2C19*1B/*1B, *2A/*2A and *17/*17) (Figure 5). The average expression levels of P450, CYP2C19 and POR proteins in these pooled HLMs are shown in Figure 6. The CYP2C19 activities were normalized to HLM protein (Figure 5a), total P450 (Figure 5b) and CYP2C19 protein levels (Figure 5c).

Kinetic analysis of CYP2C19-catalyzed (S)-mephenytoin metabolism by pooled human liver microsomes (HLMs) from donors with homozygous CYP2C19 diplotypes. (S)-Mephenytoin 4’-hydroxylation by HLMs with homozymous diplotype (CYP2C19*1B/*1B (filled circles), CYP2C19*2A/*2A (filled squares) and CYP2C19*17/*17 (filled triangles)) was measured at substrate concentrations between 1 and 400 μM. (a) Results normalized for HLM protein content. (b) Results normalized for functional P450 content. (c) Results normalized for CYP2C19 protein content. Each data point is the mean velocity of the reaction±s.d. (from three replicate determinations).

Total P450 protein, CYP2C19 protein and cytochrome P450 oxidoreductase (POR) protein expression levels in pooled human liver microsomes (HLMs) from donors with homozygous CYP2C19 diplotypes. Total P450 protein expression level (open bars) in HLMs with homozygous diplotype (CYP2C19*1B/*1B, CYP2C19*2A/*2A and CYP2C19*17/*17) was measured by reduced 2-oxo-clopidogrel (CO) difference spectral analysis. CYP2C19 protein (closed bars) and POR protein (hatched bars) expression levels in HLMs with homozygous diplotype were measured by liquid chromatography–tandem mass spectrometry (LC-MS/MS) analysis. Data are shown as means±s.d. from three replicate determinations. A two-sided, unpaired t-test was employed for post hoc, pairwise comparisons; †P<0.05, significantly different from total P450 protein expression in wild-type HLMs with CYP2C19*1B/*1B diplotype. ††P<0.05, significantly different from CYP2C19 protein expression in wild-type HLMs with CYP2C19*1B/*1B diplotype.
The formation of 4’-hydroxy (S)-mephenytoin by pooled HLMs from donors with CYP2C19*1B/*1B and *17/*17 diplotypes was saturable, but not so for the CYP2C19*2A/*2A diplotype. Parameters obtained from the kinetic analysis are summarized in Table 5. Compared with HLMs from donors with a CYP2C19*1B/*1B diplotype, HLMs from donors with a CYP2C19*17/*17 diplotype exhibited a significantly increased Vmax value for the reaction, when normalized for total HLM protein content, and no significant change in Km value. Overall, the metabolic efficiency, evaluated as Vmax/Km value, was significantly increased by 20.1%. When the activities were normalized to either the total P450 or CYP2C19 protein levels, to assess the intrinsic function of CYP2C19.1B and other variant CYP2C19 enzymes, the Vmax/Km values were found to be comparable for CYP2C19*1B/*1B and *17/*17 diplotypes. The Vmax value on the basis of CYP2C19 protein level was also comparable for CYP2C19*1B/*1B and *17/*17 diplotypes, whereas the Vmax values on the basis of P450 protein level was significantly lower in CYP2C19*17/*17 diplotype than in the CYP2C19*1B/*1B diplotype. In the case of HLMs with a CYP2C19*2A/*2A diplotype, nonsaturable product formation kinetics was observed. Vmax/Km values derived from the linear curves revealed a significant decrease compared with HLMs with a CYP2C19*1B/*1B diplotype, indicative of complete loss of CYP2C19 activity, with low residual activity represented by another nonsaturable enzyme (Table 5). Similarly, a significant decrease in the Vmax/Km values were observed even though the activities were normalized to P450 protein levels. However, as shown in Figure 6, CYP2C19 protein level was negligible in HLMs with CYP2C19*2A/*2A diplotype. Therefore, normalization of the activity to the CYP2C19 protein gives an exaggerated rate of metabolism, when compared with the other diplotypes, where CYP2C19 is expressed.
Kinetic analysis of CYP2C19-catalyzed clopidogrel and 2-Oxo-clopidogrel metabolism by pooled HLMs from donors with homozygous CYP2C19 diplotypes
To explore the impact of the CYP2C19 diplotype on the metabolism of a clinically relevant substrate for the encoded enzyme, kinetic analysis of CYP2C19-catalyzed clopidogrel and 2-oxo-clopidogrel metabolism was undertaken in pooled HLMs with defined homozygous diplotypes (Figure 7). Formation of the active thiol metabolite of clopidogrel (CAM) is thought to proceed through two consecutive P450-dependent oxidation steps: clopidogrel to 2-oxo-clopidogrel and then 2-oxo-clopidogrel to a sulfenic acid intermediate that undergoes subsequent glutathione-mediated reduction to give CAM. Therefore, the kinetic characteristics of each step were studied separately with pooled HLMs with defined homozygous diplotypes, as described above for the marker substrate, (S)-mephenytoin. In contrast to (S)-mephenytoin, both clopidogrel bioactivation steps exhibited multiphasic kinetics that could be described adequately by a two-enzyme model. Because of the significant difference in total P450 content between the different CYP2C19 diplotype pools and the multienzyme nature of the clopidogrel bioactivation steps, for our primary analysis, we elected to compare catalytic activities that were normalized for total P450 (Figures 7c and d) rather than total HLM protein (Figures 7a and b), although data normalized for total HLM protein and CYP2C19 protein levels (Figures 7e and f) are presented for completeness. Regardless of the pathway studied, all extracted kinetic parameters for the pooled HLMs with CYP2C19*1B/*1B and CYP2C19*17/*17 diplotypes were of similar magnitude, when normalized for total P450 content (Table 6). In contrast, the HLMs from donors with a CYP2C19*2A/*2A diplotype exhibited lower intrinsic clearances (Vmax/Km) to both 2-oxo-clopidogrel and CAM for the high- and low-affinity enzymatic components. For either reaction, it is likely that only the high-affinity enzymes are relevant to the in vivo metabolism of the drug, and hence relative enzymatic efficiencies are discussed below only in terms of this component.

Kinetic analysis of CYP2C19-catalyzed clopidogrel and 2-oxo-clopidogrel (OC) metabolism by pooled human liver microsomes (HLMs) from donors with homozygous CYP2C19 diplotypes. Kinetics of formation of (a, c and e) OC from clopidogrel and (b, d and f) clopidogrel active metabolite (CAM) from OC was studied separately by HLMs with a homozygous diplotype (CYP2C19*1B/*1B (filled circles), CYP2C19*2A/*2A (filled squares) and CYP2C19*17/*17 (filled triangles)). (a and b) Results normalized for HLM protein content. (c and d) Results normalized for functional P450 content. (e and f) Results normalized for CYP2C19 protein content. Each data point is the mean velocity of the reaction±s.d. from three replicate determinations.
The high-affinity intrinsic clearance (Vmax/Km) to 2-oxo-clopidogrel was reduced by 52% in the pooled HLMs with the CYP2C19*2A/*2A diplotype relative to CYP2C19*1B/*1B diplotype, and by 84% for the second bioactivation step to CAM. For CAM formation, the Km value increased fivefold (to ~5 μM) in the pooled HLMs with the CYP2C19*2A/*2A diplotype, reflective of the absence of CYP2C19 enzyme, and likely represents catalysis by other slightly lower affinity P450s such as CYP3A4 and CYP2C9.42 Interestingly, no significant change in Km value was observed for the first bioactivation step in the pooled HLMs with the CYP2C19*2A/*2A diplotype, but again this is in agreement with the microsomal kinetic studies of Kazui et al.42 who observed that CYP1A2 and CYP2B6 possessed similarly low Km values to CYP2C19 for this reaction. Normalization of the activity to the CYP2C19 protein level exhibited more variable Vmax and Vmax/Km values compared with those on the basis of P450 protein level (Table 6). These findings support the consideration described above that 2-oxo-clopidogrel and CAM formations are catalyzed by multiple P450 enzymes with different affinity for each reaction.
Discussion
Of the 19 CYP2C19 alleles observed, 3 are thought to confer wild-type activity (CYP2C19*1A, CYP2C19*1B and CYP2C19*1C). The 80161G (331V) variant, representing the CYP2C19*1B allele, was more frequently observed in our study population when compared with the 80161A (331I) allele, designated CYP2C19*1A.43 Accordingly, the CYP2C19*1A/*1B and *1B/*1B diplotypes were also more frequently encountered in our sample set (Supplementary Table 5). Based on prevalence and catalytic activity, we used CYP2C19*1B/*1B microsomes as the reference for all subsequent diplotype comparisons.
The CYP2C19*17 and *2A variants accounted for the remainder of the common diplotypes, with frequencies ranging from 7.5 to 33%. Given that 96.3% of the liver donors in this study were identified racially as White, the diplotype frequencies were as expected and consistent with a previous report showing that CYP2C19*17 allele is more frequently observed compared with the CYP2C19*2 allele in Whites.10
Three of the nine possible diplotype combinations from the *1B, *17 and *2A alleles, *1B*2A/*17*17, *2A*2A/*1B*17 and *2A*2A/*17*17, were not observed in our samples, implying the absence of the *2A/*17 haplotype (Table 1). Further analysis revealed that CYP2C19*2A and *17 alleles were in complete LD (|D’|=1, LOD >2) (Figure 2). Complete LD was also observed between CYP2C19*1B and *17 alleles. However, despite the fact that the CYP2C19*17 (−806T) variant appeared to be always found on the haplotype lacking the CYP2C19*2A (19154A) variant, the r2 value between the two loci was low (r2=0.057). This is likely because the reference allele at the CYP2C19*2A locus is much more common than CYP2C19*17 and hence is also seen with the reference allele at the CYP2C19*17 locus. This finding is similar to that reported in recent studies, where it was also concluded that individuals heterozygous or homozygous for the CYP2C19*17 allele are less likely to carry the CYP2C19*2 allele, whereas those who are wild type at the CYP2C19*17 locus are more likely to carry the CYP2C19*2 allele.26, 44
A linear trend analysis revealed that the rank order for CYP2C19 activity (*17/*17 > *1B/*17 > *1B/*1B > *2A/*17 > *1B/*2A > *2A/*2A) was statistically significant. This is in good agreement with a clinical report describing an association between CYP2C19 polymorphisms and single-dose pharmacokinetics of oral pantoprazole in healthy volunteers. The under the concentration-time curve of pantoprazole was lowest in subjects with a reported CYP2C19*17/*17 diplotype, followed by CYP2C19*1/*17, *1/*1, *2/*17 and *1/*2 diplotypes, and highest in subjects with the *2/*2 diplotype.25 Individuals with the CYP2C19*17 variant are also reported to exhibit enhanced metabolic clearance of several other drugs, including omeprazole, amitriptyline, voriconazole and escitalopam.9, 20, 21, 22, 23, 24, 25 Of the diplotypes that were observed in our study, the mean activity was greatly decreased in HLMs with the CYP2C19*2A/*2A diplotype compared with HLMs with a CYP2C19*1B/*1B diplotype. Moreover, HLMs from CYP2C19*2A/*2A diplotype livers exhibited nonsaturable metabolic kinetics, indicative of a complete loss of CYP2C19 expression. Indeed, CYP2C19 protein was negligible in all livers with a homozygous CYP2C19*2A/*2A diplotype.
In contrast, our findings demonstrated that inheritance of the CYP2C19*17 allele is associated with increased hepatic CYP2C19 protein content, as indicated by an increased Vmax value for (S)-mephenytoin 4’-hydroxylation and CYP2C19 protein content for pooled livers homozygous for the *17 allele and lacking the *2A allele in comparison with those homozygous for CYP2C19*1B. This same trend was observed when liver activity was examined individually, although the considerable within-diplotype group variability reduced the power of the analysis; only a trend test for the a priori rank order assignment was significant. These results support the hypothesis that the SNP described by the *17 locus enhances CYP2C19 transcription and protein synthesis, although the effect size is modest. This conclusion is also bolstered by the fact that the Km value for (S)-mephenytoin 4’-hydroxylation in the CYP2C19*17/*17 livers was not altered in comparison with that for the CYP2C19*1B/*1B diplotype; the structure of the protein generated from each allele would be unchanged. Of note, the Km value for (S)-mephenytoin 4’-hydroxylation associated with the CYP2C19*1B/*1B diplotype was comparable to a previously reported value reported for microsomes isolated from yeast cells expressing CYP2C19 alleles.45
The Clinical Pharmacogenetics Implementation Consortium has classified CYP2C19*17 carriers as ultrarapid metabolizers in the context of clopidogrel metabolism to an active thiol metabolite (CAM).46 However, we were unable to demonstrate a significant difference in the formation of 2-oxo-clopidogrel from clopidogrel or CAM from 2-oxo-clopidogrel by HLMs from donors with a CYP2C19*17/*17 diplotype compared with the CYP2C19*1B/*1B diplotype. This appears to be a consequence of the fact that CYP2C19 is only one of multiple enzymes catalyzing each reaction and, thus, there is reduced penetrance from a relatively weak enzyme activity modifier (*17). Our finding is consistent with the recent observations of Shuldiner and colleagues,26 who saw no significant change in CAM or adenosine diphosphate-stimulated platelet aggregation in individuals with CYP2C19*1/*1, *1/*17 and *17/*17 diplotypes. Furthermore, CYP2C19*1/*2 and *2/*17 diplotypes also yielded similar results. In contrast, HLMs from donors with a CYP2C19*2A/*2A diplotype exhibited lower activity for the conversion of both clopidogrel to 2-oxo-clopidogrel and 2-oxo-clopidogrel to CAM, although the relative effect size of the variant SNP at the *2 locus on the high-affinity (low Km) component of the clopidogrel and 2-oxo-clopidogrel oxidation reaction was smaller than that observed for (S)-mephenytoin 4’-hydroxylation. This matches with what has been observed in vivo with regard to clopidogrel metabolism and that of more selective CYP2C19 substrates.11, 26, 47, 48
There were a number of CYP2C19 nsSNPs detected in our panel of livers (∼20% of the total), and hence it is possible that they contributed to interindividual variability of CYP2C19-catalyzed drug metabolism (Table 1). Although there was a trend for lower CYP2C19 activity in livers with a deleterious nsSNPs as compared with the CYP2C19*1B/*1B diplotype, the head-to-head differences were not significant. Individually, the benign nsSNPs were too infrequent for comparison with the observed metabolic activities, particularly given the considerable within-group variability of the CYP2C19*1B/*1B diplotype. Further studies with a larger sample size would be needed to dissect out any impact of nsSNPs on CYP2C19 activity.
It is also possible that extreme variability in CYP2C19 activity and protein content detected in vitro is an artifact of conditions present following brain death and organ procurement. A number of the liver donors were receiving medications with the potential to modulate metabolizing enzyme activity up and down, such as dexamethasone, ketoprofen, prednisone, phenytoin, cimetidine, amiodarone and phenobarbital.49, 50, 51, 52, 53 Moreover, 15.0% of liver donors were patients with evidence of liver disease and/or had liver surgery, including fatty liver, chronic hepatitis, fibrotic liver, cirrhosis, hepatoma, acute injury, biliary atresia and cholangiocarcinoma. It was reported that CYP2C19-mediated metabolism of (S)-mephenytoin is quite sensitive to the presence of liver disease (CYP2C19 activity is reduced at an early stage of disease).54 Therefore, the extreme distribution of our measured activity data may be explained in part by the presence of liver disease. In addition, 28.5% of liver donors had brain damage, including cerebral vascular accident, head trauma, subarachnoid hemorrhage and intracerebral hemorrhage, as the cause of death. Because an increase in inflammatory cytokines have been noted in both the serum and cerebrospinal fluid of patients with traumatic brain injury, it may be important to consider the influence of the endogenous cytokines on expression of CYP enzymes in donor organs.55 Indeed, it was previously shown that various cytokines, such as interleukin-1β, interleukin-4, interleukin-6, tumor necrosis factor and interferon, act directly on human hepatocytes to affect expression of major P450 genes (CYP1A2, CYP2C, CYP2E1 and CYP3A) and associated enzymatic activity.56 Similarly, altered hepatic CYP expression and antioxidant activity have been reported following endotoxin (lipopolysaccharide) or proinflammatory cytokine administration.57 Although pre-existing liver disease and induced inflammation remain possible sources of interindividual variability in hepatic CYP2C19 activity, results from our multivariate analysis indicated no significant effect, possibly because of incomplete medical information in the donor population. Another possible explanation for a substantial fraction of interliver differences in CYP2C19 activity is the effect of gender on CYP2C19 activity and protein content. Indeed, recent report showed sex differences in the expression of hepatic drug-metabolizing enzymes.58, 59 Interestingly, growth hormone exerts sex-dependent effects on the liver in many species, with many hepatic genes, most notably genes coding for P450 enzymes, being transcribed in a sex-dependent manner. In the present study, we also examined CYP2C19 activity and protein content as a function of gender but found no significant difference (Table 1). As described in Table 4, the results of our multivariate linear regression analysis revealed that gender, as well as race, medication, liver disease and brain damage, was not a significant contributor to variability in the CYP2C19 activity in HLMs. This apparent inconsistency between our results and that of other investigators could be the result of inadequate power in our study. Finally, other uncontrolled aspects of the cellular environment, such as the impact of diet and disease, could contribute to the variability observed, possibly by epigenetic mechanisms.
We also explored the possibility that the availability of POR affected CYP2C19 activity, given its essential role in the catalytic process as an electron donor and prior observations that genetic variation in the POR gene can modulate P450 enzyme activity. The results of our multivariate linear regression analysis revealed that POR protein, as well as presence of the CYP2C19*2A variant, was a significant contributor to variability in the CYP2C19 activity in HLMs. POR activity would likely have been a better measure of POR function in supporting CYP2C19 reactions than microsomal POR protein concentration, particularly if there were intrinsic differences in the flavin content of the POR flavoprotein or if there was differential flavin loss during microsomal isolation and storage. However, because of limited availability of microsomes from many of the samples, we were unable to conduct comprehensive POR activity measurements. Working on the assumption that POR activity is dependent on POR protein content, we utilized the latter parameter in our multivariate analysis. If hepatic POR content can be linked to specific POR variants, testing for these variants may have utility for the individualization of clopidogrel therapy.
In conclusion, the present study revealed that both the CYP2C19*2A and CYP2C19*17 alleles contributed to variability in hepatic CYP2C19 protein content and (S)-mephenytoin 4’-hydroxylase activity. However, only the CYP2C19*2A allele had a significant effect on clopidogrel and 2-oxo-clopidogrel metabolism, consistent with the multienzyme nature of these reactions. In addition, HLMs from donors with a CYP2C19*2A/*17 diplotype exhibited (S)-mephenytoin 4’-hydroxylase activity most similar to that of HLMs for the CYP2C19*1B/*2A diplotype, consistent with the much stronger functional impact of the *2A null allele. A substantial fraction of interliver differences in CYP2C19 activity was unexplained, but some of it could be attributed to variation in POR content. Potential sources for future investigation include variation in CYP2C19 regulatory genes and other endogenous and exogenous environmental factors (for example, regulatory hormone levels, diet and stress metrics).
References
Li-Wan-Po A, Girard T, Farndon P, Cooley C, Lithgow J . Pharmacogenetics of CYP2C19: functional and clinical implications of a new variant CYP2C19*17. Br J Clin Pharmacol 2010; 69: 222–230.
Shah BS, Parmar SA, Mahajan S, Mehta AA . An insight into the interaction between clopidogrel and proton pump inhibitors. Curr Drug Metab 2012; 13: 225–235.
Shirasaka Y, Sager JE, Lutz JD, Davis C, Isoherranen N . Inhibition of CYP2C19 and CYP3A4 by omeprazole metabolites and their contribution to drug-drug interactions. Drug Metab Dispos 2013; 41: 1414–1424.
Wedlund PJ . The CYP2C19 enzyme polymorphism. Pharmacology 2000; 61: 174–183.
McGraw J, Waller D . Cytochrome P450 variations in different ethnic populations. Expert Opin Drug Metab Toxicol 2012; 8: 371–382.
Hicks JK, Swen JJ, Thorn CF, Sangkuhl K, Kharasch ED, Ellingrod VL et al. Clinical Pharmacogenetics Implementation Consortium guideline for CYP2D6 and CYP2C19 genotypes and dosing of tricyclic antidepressants. Clin Pharmacol Ther 2013; 93: 402–408.
De Morais SM, Wilkinson GR, Blaisdell J, Nakamura K, Meyer UA, Goldstein JA . The major genetic defect responsible for the polymorphism of S-mephenytoin metabolism in humans. J Biol Chem 1994; 269: 15419–1522.
Ferguson RJ, De Morais SM, Benhamou S, Bouchardy C, Blaisdell J, Ibeanu G et al. A new genetic defect in human CYP2C19: mutation of the initiation codon is responsible for poor metabolism of S-mephenytoin. J Pharmacol Exp Ther 1998; 284: 356–361.
Sim SC, Risinger C, Dahl ML, Aklillu E, Christensen M, Bertilsson L et al. A common novel CYP2C19 gene variant causes ultrarapid drug metabolism relevant for the drug response to proton pump inhibitors and antidepressants. Clin Pharmacol Ther 2006; 79: 103–113.
Sanford JC, Guo Y, Sadee W, Wang D . Regulatory polymorphisms in CYP2C19 affecting hepatic expression. Drug Metabol Drug Interact 2013; 28: 23–30.
Furuta T, Ohashi K, Kosuge K, Zhao XJ, Takashima M, Kimura M et al. CYP2C19 genotype status and effect of omeprazole on intragastric pH in humans. Clin Pharmacol Ther 1999; 65: 552–561.
Klotz U . Clinical impact of CYP2C19 polymorphism on the action of proton pump inhibitors: a review of a special problem. Int J Clin Pharmacol Ther 2006; 44: 297–302.
Mega JL, Close SL, Wiviott SD, Shen L, Hockett RD, Brandt JT et al. Cytochrome p-450 polymorphisms and response to clopidogrel. N Engl J Med 2009; 360: 354–362.
Mega JL, Simon T, Collet JP, Anderson JL, Antman EM, Bliden K et al. Reduced-function CYP2C19 genotype and risk of adverse clinical outcomes among patients treated with clopidogrel predominantly for PCI: a meta-analysis. JAMA 2010; 304: 1821–1830.
Hwang SJ, Jeong YH, Kim IS, Koh JS, Kang MK, Park Y et al. The cytochrome 2C19*2 and *3 alleles attenuate response to clopidogrel similarly in East Asian patients undergoing elective percutaneous coronary intervention. Thromb Res 2011; 127: 23–28.
Sibbing D, Koch W, Gebhard D, Schuster T, Braun S, Stegherr J et al. Cytochrome 2C19*17 allelic variant, platelet aggregation, bleeding events, and stent thrombosis in clopidogrel-treated patients with coronary stent placement. Circulation 2010; 121: 512–518.
Tiroch KA, Sibbing D, Koch W, Roosen-Runge T, Mehilli J, Schömig A et al. Protective effect of the CYP2C19 *17 polymorphism with increased activation of clopidogrel on cardiovascular events. Am Heart J 2010; 160: 506–512.
Paré G, Mehta SR, Yusuf S, Anand SS, Connolly SJ, Hirsh J et al. Effects of CYP2C19 genotype on outcomes of clopidogrel treatment. N Engl J Med 2010; 363: 1704–1714.
Goldstein JA, Faletto MB, Romkes-Sparks M, Sullivan T, Kitareewan S, Raucy JL et al. Evidence that CYP2C19 is the major (S)-mephenytoin 4'-hydroxylase in humans. Biochemistry 1994; 33: 1743–1752.
Baldwin RM, Ohlsson S, Pedersen RS, Mwinyi J, Ingelman-Sundberg M, Eliasson E et al. Increased omeprazole metabolism in carriers of the CYP2C19*17 allele; a pharmacokinetic study in healthy volunteers. Br J Clin Pharmacol 2008; 65: 767–774.
Ohlsson Rosenborg S, Mwinyi J, Andersson M, Baldwin RM, Pedersen RS, Sim SC et al. Kinetics of omeprazole and escitalopram in relation to the CYP2C19*17 allele in healthy subjects. Eur J Clin Pharmacol 2008; 64: 1175–1179.
Wang G, Lei HP, Li Z, Tan ZR, Guo D, Fan L et al. The CYP2C19 ultra-rapid metabolizer genotype influences the pharmacokinetics of voriconazole in healthy male volunteers. Eur J Clin Pharmacol 2009; 65: 281–285.
De Vos A, van der Weide J, Loovers HM . Association between CYP2C19*17 and metabolism of amitriptyline, citalopram and clomipramine in Dutch hospitalized patients. Pharmacogenomics J 2011; 11: 359–367.
Dolton MJ, McLachlan AJ . Clinical importance of the CYP2C19*17 variant allele for voriconazole. Br J Clin Pharmacol 2011; 71: 137–138.
Gawrońska-Szklarz B, Adamiak-Giera U, Wyska E, Kurzawski M, Gornik W, Kaldonska M et al. CYP2C19 polymorphism affects single-dose pharmacokinetics of oral pantoprazole in healthy volunteers. Eur J Clin Pharmacol 2012; 68: 1267–1274.
Lewis JP, Stephens SH, Horenstein RB, O'Connell JR, Ryan K, Peer CJ et al. The CYP2C19*17 variant is not independently associated with clopidogrel response. J Thromb Haemost 2013; 11: 1640–1646.
Pedersen RS, Brasch-Andersen C, Sim SC, Bergmann TK, Halling J, Petersen MS et al. Linkage disequilibrium between the CYP2C19*17 allele and wildtype CYP2C8 and CYP2C9 alleles: identification of CYP2C haplotypes in healthy Nordic populations. Eur J Clin Pharmacol 2010; 66: 1199–1205.
Anichavezhi D, Chakradhara Rao US, Shewade DG, Krishnamoorthy R, Adithan C . Distribution of CYP2C19*17 allele and genotypes in an Indian population. J Clin Pharm Ther 2012; 37: 313–318.
Blaisdell J, Mohrenweiser H, Jackson J, Ferguson S, Coulter S, Chanas B et al. Identification and functional characterization of new potentially defective alleles of human CYP2C19. Pharmacogenetics 2002; 12: 703–711.
Barrett JC, Fry B, Maller J, Daly MJ . Haploview: analysis and visualization of LD and haplotype maps. Bioinformatics 2005; 21: 263–265.
Thummel KE, Kharasch ED, Podoll T, Kunze K . Human liver microsomal enflurane defluorination catalyzed by cytochrome P-450 2E1. Drug Metab Dispos 1993; 21: 350–357.
Paine MF, Khalighi M, Fisher JM, Shen DD, Kunze KL, Marsh CL et al. Characterization of interintestinal and intraintestinal variations in human CYP3A-dependent metabolism. J Pharmacol Exp Ther 1997; 283: 1552–1562.
Guengerich FP, Martin MV, Sohl CD, Cheng Q . Measurement of cytochrome P450 and NADPH-cytochrome P450 reductase. Nat Protoc 2009; 4: 1245–1251.
Yamaoka K, Tanigawara Y, Nakagawa T, Uno T . A pharmacokinetic analysis program (multi) for microcomputer. J Pharmacobiodyn 1981; 4: 879–885.
Korzekwa KR, Krishnamachary N, Shou M, Ogai A, Parise RA, Rettie AE et al. Evaluation of atypical cytochrome P450 kinetics with two-substrate models: evidence that multiple substrates can simultaneously bind to cytochrome P450 active sites. Biochemistry 1998; 37: 4137–4147.
Omura T, Sato R . The carbon monoxide-binding pigment of liver microsomes. I. evidence for its hemoprotein nature. J Biol Chem 1964; 239: 2370–2378.
Kamiie J, Ohtsuki S, Iwase R, Ohmine K, Katsukura Y, Yanai K et al. Quantitative atlas of membrane transporter proteins: development and application of a highly sensitive simultaneous LC/MS/MS method combined with novel in-silico peptide selection criteria. Pharm Res 2008; 25: 1469–1483.
Prasad B, Unadkat JD . Optimized approaches for quantification of drug transporters in tissues and cells by MRM proteomics. AAPS J 2014; 16: 634–648.
Edson KZ, Prasad B, Unadkat JD, Suhara Y, Okano T, Guengerich FP et al. Cytochrome P450-dependent catabolism of vitamin K: omega-hydroxylation catalyzed by human CYP4F2 and CYP4F11. Biochemistry 2013; 52: 8276–8285.
Prasad B, Evers R, Gupta A, Hop CE, Salphati L, Shukla S et al. Interindividual variability in hepatic organic anion-transporting polypeptides and P-glycoprotein (ABCB1) protein expression: quantification by liquid chromatography tandem mass spectroscopy and influence of genotype, age, and sex. Drug Metab Dispos 2014; 42: 78–88.
Solstad T, Bjørgo E, Koehler CJ, Strozynski M, Torgersen KM, Taskén K et al. Quantitative proteome analysis of detergent-resistant membranes identifies the differential regulation of protein kinase C isoforms in apoptotic T cells. Proteomics 2010; 10: 2758–2768.
Kazui M, Nishiya Y, Ishizuka T, Hagihara K, Farid NA, Okazaki O et al. Identification of the human cytochrome P450 enzymes involved in the two oxidative steps in the bioactivation of clopidogrel to its pharmacologically active metabolite. Drug Metab Dispos 2010; 38: 92–99.
Fukushima-Uesaka H, Saito Y, Maekawa K, Ozawa S, Hasegawa R, Kajio H et al. Genetic variations and haplotypes of CYP2C19 in a Japanese population. Drug Metab Pharmacokinet 2005; 20: 300–307.
Gurbel PA, Tantry US, Shuldiner AR . Letter by Gurbel et al regarding article, "Cytochrome 2C19*17 allelic variant, platelet aggregation, bleeding events, and stent thrombosis in clopidogrel-treated patients with coronary stent placement". Circulation 2010; 122: e478.
Hanioka N, Tsuneto Y, Saito Y, Sumada T, Maekawa K, Saito K et al. Functional characterization of two novel CYP2C19 variants (CYP2C19*18 and CYP2C19*19) found in a Japanese population. Xenobiotica 2007; 37: 342–355.
Scott SA, Sangkuhl K, Gardner EE, Stein CM, Hulot JS, Johnson JA et al. Clinical Pharmacogenetics Implementation Consortium guidelines for cytochrome P450-2C19 (CYP2C19) genotype and clopidogrel therapy. Clin Pharmacol Ther 2011; 90: 328–332.
Sibbing D, Gebhard D, Koch W, Braun S, Stegherr J, Morath T et al. Isolated and interactive impact of common CYP2C19 genetic variants on the antiplatelet effect of chronic clopidogrel therapy. J Thromb Haemost 2010; 8: 1685–1693.
Qin XP, Xie HG, Wang W, He N, Huang SL, Xu ZH et al. Effect of the gene dosage of CgammaP2C19 on diazepam metabolism in Chinese subjects. Clin Pharmacol Ther 1999; 66: 642–646.
Raucy JL, Mueller L, Duan K, Allen SW, Strom S, Lasker JM . Expression and induction of CYP2C P450 enzymes in primary cultures of human hepatocytes. J Pharmacol Exp Ther 2002; 302: 475–482.
Foti RS, Wahlstrom JL . CYP2C19 inhibition: the impact of substrate probe selection on in vitro inhibition profiles. Drug Metab Dispos 2008; 36: 523–528.
Moody GC, Griffin SJ, Mather AN, McGinnity DF, Riley RJ . Fully automated analysis of activities catalysed by the major human liver cytochrome P450 (CYP) enzymes: assessment of human CYP inhibition potential. Xenobiotica 1999; 29: 53–75.
Furuta S, Kamada E, Suzuki T, Sugimoto T, Kawabata Y, Shinozaki Y et al. Inhibition of drug metabolism in human liver microsomes by nizatidine, cimetidine and omeprazole. Xenobiotica 2001; 31: 1–10.
Ohyama K, Nakajima M, Suzuki M, Shimada N, Yamazaki H, Yokoi T . Inhibitory effects of amiodarone and its N-deethylated metabolite on human cytochrome P450 activities: prediction of in vivo drug interactions. Br J Clin Pharmacol 2000; 49: 244–253.
Frye RF, Zgheib NK, Matzke GR, Chaves-Gnecco D, Rabinovitz M, Shaikh OS et al. Liver disease selectively modulates cytochrome P450—mediated metabolism. Clin Pharmacol Ther 2006; 80: 235–245.
Stein DM, Lindell A, Murdock KR, Kufera JA, Menaker J, Keledjian K et al. Relationship of serum and cerebrospinal fluid biomarkers with intracranial hypertension and cerebral hypoperfusion after severe traumatic brain injury. J Trauma 2011; 70: 1096–1103.
Abdel-Razzak Z, Loyer P, Fautrel A, Gautier JC, Corcos L, Turlin B et al. Cytokines down-regulate expression of major cytochrome P-450 enzymes in adult human hepatocytes in primary culture. Mol Pharmacol 1993; 44: 707–715.
Warren GW, van Ess PJ, Watson AM, Mattson MP, Blouin RA . Cytochrome P450 and antioxidant activity in interleukin-6 knockout mice after induction of the acute-phase response. J Interferon Cytokine Res 2001; 21: 821–826.
Waxman DJ, Holloway MG . Sex differences in the expression of hepatic drug metabolizing enzymes. Mol Pharmacol 2009; 76: 215–228.
Waxman DJ, O'Connor C . Growth hormone regulation of sex-dependent liver gene expression. Mol Endocrinol 2006; 20: 2613–2629.
Acknowledgements
This work was supported in part by grants from the National Institutes of Health (U01 GM092676 and GM092666, P01 GM32165 and R01 GM094418) and the Japan Society for the Promotion of Science Postdoctoral Fellowship for Research Abroad (H23-694) and The Uehara Memorial Foundation Research Fellowship (201340221) to Dr Shirasaka. We thank the Hartwell Center at St Jude Children’s Research Hospital for sequencing DNA for this study.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies the paper on the The Pharmacogenomics Journal website
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-ShareAlike 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-sa/4.0/
About this article

Cite this article
Shirasaka, Y., Chaudhry, A., McDonald, M. et al. Interindividual variability of CYP2C19-catalyzed drug metabolism due to differences in gene diplotypes and cytochrome P450 oxidoreductase content. Pharmacogenomics J 16, 375–387 (2016). https://doi.org/10.1038/tpj.2015.58
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/tpj.2015.58
- Springer Nature Limited
This article is cited by
-
Development of Physiology Based Pharmacokinetic Model to Predict the Drug Interactions of Voriconazole and Venetoclax
Pharmaceutical Research (2022)
-
A double-Flp-in method for stable overexpression of two genes
Scientific Reports (2020)
-
Variability of voriconazole concentrations in patients with hematopoietic stem cell transplantation and hematological malignancies: influence of loading dose, procalcitonin, and pregnane X receptor polymorphisms
European Journal of Clinical Pharmacology (2020)
-
A Physiologically Based Pharmacokinetic Model of Voriconazole Integrating Time-Dependent Inhibition of CYP3A4, Genetic Polymorphisms of CYP2C19 and Predictions of Drug–Drug Interactions
Clinical Pharmacokinetics (2020)
-
Personalized Drug Dosage – Closing the Loop
Pharmaceutical Research (2017)