
Abstract
Background
Kutzneria is a representative of a rarely observed genus of the family Pseudonocardiaceae. Kutzneria species were initially placed in the Streptosporangiaceae genus and later reconsidered to be an independent genus of the Pseudonocardiaceae. Kutzneria albida is one of the eight known members of the genus. This strain is a unique producer of the glycosylated polyole macrolide aculeximycin which is active against both bacteria and fungi. Kutzneria albida genome sequencing and analysis allow a deeper understanding of evolution of this genus of Pseudonocardiaceae, provide new insight in the phylogeny of the genus, as well as decipher the hidden secondary metabolic potential of these rare actinobacteria.
Results
To explore the biosynthetic potential of Kutzneria albida to its full extent, the complete genome was sequenced. With a size of 9,874,926 bp, coding for 8,822 genes, it stands alongside other Pseudonocardiaceae with large circular genomes. Genome analysis revealed 46 gene clusters potentially encoding secondary metabolite biosynthesis pathways. Two large genomic islands were identified, containing regions most enriched with secondary metabolism gene clusters. Large parts of this secondary metabolism “clustome” are dedicated to siderophores production.
Conclusions
Kutzneria albida is the first species of the genus Kutzneria with a completely sequenced genome. Genome sequencing allowed identifying the gene cluster responsible for the biosynthesis of aculeximycin, one of the largest known oligosaccharide-macrolide antibiotics. Moreover, the genome revealed 45 additional putative secondary metabolite gene clusters, suggesting a huge biosynthetic potential, which makes Kutzneria albida a very rich source of natural products. Comparison of the Kutzneria albida genome to genomes of other actinobacteria clearly shows its close relations with Pseudonocardiaceae in line with the taxonomic position of the genus.
Similar content being viewed by others

Background
Bacteria of the Actinomycetales order represent an amazing source of biologically active compounds which are produced as secondary metabolites [1]. The broad spectrum of structural features and wide array of activities of these metabolites attract attention as a limitless source of novel chemical scaffolds as well as new drugs for human and veterinary medicine, and agriculture. With the recent advances in sequencing technologies significant progress in sequencing and analysis of actinobacterial genomes was achieved [2, 3]. Not only genomes of biotechnologically important and typical representatives of the taxonomical order were sequenced, but also strains with interesting and unusual features, leading to deeper insights into phylogeny, genetics, physiology, ecology, and the secondary metabolism biosynthetic potential of these bacteria. One of the major discoveries of the genomic era in actinomycetes research is the presence of multiple gene clusters dedicated to secondary metabolism in one genome. This observation caused an increase of interest in diverse groups of actinomycetes (especially rare representatives of its subgroups) as a possible source of new biologically active metabolites. One of such groups includes species belonging to the Pseudonocardiaceae family. Multiple genome projects dedicated to this group of bacteria are in progress, with seven being completed and published [4, 5]. Here we report the sequencing and analysis of the complete genome of Kutzneria albida DSM 43870T (formerly “Streptosporangium albidum”) [6, 7]. The genus Kutzneria is a minor branch of the Pseudonocardiaceae family currently containing only 8 species [8–10]. To our knowledge, this is the first member of the Kutzneria genus and only the eighth representative of the family Pseudonocardiaceae for which a complete genome sequence is available.
K albida was isolated from soil samples collected in the rocky regions of Gunma Prefecture of Japan [6]. The strain is known as a producer of aculeximycin, a macrolide antibiotic with an interesting chemical structure and activity against Gram-positive bacteria, fungi, and mosquito larvae [11, 12]. Being generally toxic, aculeximycin cannot be used in medicine [13]. However, some structural features, especially the unusual glycoside chain, make it an attractive source of building blocks for use in synthetic biotechnology and combinatorial biosynthesis of secondary metabolites [14, 15]. Sequencing K. albida genome allowed not only to identify genes responsible for aculeximycin biosynthesis, but also to discover an unusual abundance and diversity of secondary metabolites that could potentially be produced by this species. To the best of our knowledge, this genome is the most enriched with clusters involved in secondary metabolites among actinobacterial genomes.
Results and discussion
General features of the genome
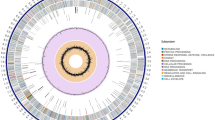
After gaps closure, a single contig with the size of 9,874,926 bp and a 70.6% G + C content was obtained. General features of K. albida genome are summarized in Table 1. The genome of K. albida consists of a single circular replicon; no extrachromosomal replicon was detected. The origin of replication (oriC) was identified as a 1,044 nt non-coding region between the two genes – dnaA, encoding the chromosomal replication initiation factor, and dnaN, coding for the β-subunit of the DNA polymerase III (Figure 1). Interestingly, the oriC has no classic DnaA boxes [TT(G/A)TCCACA], and shows almost no similarity to the oriC of Streptomyces species [16, 17]. On the opposite side of the chromosome from oriC, a putative dif (deletion-induced filamentation, chromosome resolving site) locus was identified. Its sequence is in good accordance with actinobacterial dif sites [18]. Additionally, the obvious cumulative GC skew inversion in the regions of oriC and dif supports our oriC assignment and suggests the existence of two replichores composing the circular K. albida chromosome (Figure 1).

Schematic representation of the Kutzneria albida genome, created with the help of Circos [[19]]; megabases are labeled; smaller ticks correspond to 100 kbp segments. From outside: genes on the forward and the reverse strands (blue: shorter than 900 bp, green: between 900 and 1500 bp long, orange: longer than 1500 bp); 46 secondary metabolite clusters coloured by type (NRPS: blue; siderophore NRPS: lighter blue; PKS: green; hybrid PKS-NRPS: dark purple; terpene: orange; other: yellow); genomic islands; G + C content, 10kbp window (blue colour highlights segments with G + C content <69%); G + C content, 100 kbp window (lighter blue is higher G + C, darker blue is lower G + C content); G + C skew (green: positive; blue: negative); cumulative G + C skew. The oriC is placed at coordinate zero.
Unlike the linear topology of Streptomyces genomes, the chromosome of K. albida is circular (Table 1) [20]. This appears to be a common feature of Pseudonocardiaceae representatives genomes, since the chromosomes of other representatives of this family sequenced so far also have a circular topology [5]. With a total length of 9,874,926 bp, the K. albida chromosome is larger than that of Streptomyces coelicolor A3(2) (8.7 Mbp) [21] and Streptomyces avermitilis MA-4680 (9.0 Mbp) [22], as well as genomes of Pseudonocardiaceae Saccharomonospora viridis P101T (4.3 Mbp) [23], Pseudonocardia dioxanivorans CB1190 (7.1 Mbp) [24], Saccharothrix espanaensis DSM 44229T (9.3 Mbp) [5], and Saccharopolyspora erythraea NRRL 2338 (8.2 Mbp) [25], but slightly smaller than the genome of another Pseudonocardiaceae rifamycin producer Amycolatopsis mediterranei S699 (10.2 Mbp) [26]. The K. albida genome is among the largest actinobacterial genomes sequenced so far.
K. albida chromosome contains 8,822 predicted protein-coding sequences (CDSs) (Table 1). Three ribosomal RNA (rrn) operons were identified in the genome along with 47 tRNA genes (Table 1) [27]. The rrn operons are located on the leading strand of the chromosome proximally to oriC and have remarkably low G + C content in comparison to the genome average.
Functional annotation of K. albida genes within the actNOG (Actinobacteria) subset of the eggNOG database (using protein BLAST with an expectation value cut-off 0.001) [28], showed that 6,648 (75%) out of 8,822 genes had at least one biological function assigned (Table 2), with some of the genes assigned to more than one category. Of the remainder, 960 CDSs (10.8%) had no hits against actNOGs, and 1,591 genes (18%) had hits but were not assigned to functional categories; of the latter, 47 are highly conserved. Among the genes with functional assignment, 2,493 (28%) are implicated in metabolism, including 403 (4.5%) in amino acid metabolism, 519 (5.8%) in carbohydrate metabolism, and 295 (3.3%) participating in secondary metabolism (Table 2). At the same time, 10% of the annotated genes (836) code for proteins involved in transcriptional processes, including 60 sigma factors, 15 anti-sigma factors, 6 anti-anti-sigma factors, and 747 (8.4%) putative DNA binding regulators. The large number of proteins implicated in transcriptional processes suggests a high complexity of K. albida gene expression control. The high proportion of proteins involved in transcriptional regulation is accompanied by a similarly high percentage of proteins involved in signal transduction pathways (350 genes, 4%), which suggests a close connection between extracellular nutrient sensing and transcriptional regulation of uptake and degradation pathways. This feature of the K. albida genome is typical for the majority of known soil living actinobacteria and reflects their life style and ecological niche, providing higher flexibility of the strain adaptation to changes in growth conditions [20]. In addition to this, a large portion of the genome encodes extracellular polymer-degrading enzymes. In total, 94 putative secretory hydrolase encoding genes were found in the K. albida genome (1% of all genes), including 18 glycoside hydrolases and 20 extracellular proteases.
A region of the chromosome (approximately 4 Mbp) extending either side of oriC appears to contain the majority of the genes predicted to be essential with the highest density in close proximity to oriC (Figure 1). This distribution of housekeeping genes with the notable core region is typical for actinobacterial genomes [5, 21, 25]. Outside this region, the chromosome has apparently undergone major expansion by acquiring novel genetic elements as a result of horizontal gene transfer. This oriC-distal region is enriched with the genetic loci encoding secondary metabolism, many of which have little similarity to other Pseudonocardiaceae secondary metabolism genes. After manual correction of AntiSMASH results [29], approximately 14% of the K. albida chromosome (1.46 Mbp) was found to be involved in secondary metabolites biosynthesis. This includes 852 ORFs, or 9.6% of all genes annotated in the genome. Thus, secondary metabolism genes occupy a significantly bigger proportion of K. albida genome when compared to other actinomycetes, including sequenced genomes of Pseudonocardiaceae. For example, S. coelicolor A3(2) secondary metabolism genes cover only 5% of the chromosome, whereas in S. avermitilis MA-4680 secondary metabolism genes represent 6.6% of the genome length [22].
Another feature of the K. albida chromosome is the presence of numerous genomic islands outside the ancestral core genome. These regions comprise the youngest part of the chromosome acquired as a result of horizontal gene transfer events. They are characterized by lower than average genomic G + C content, biased codon usage, as well as presence of numerous insertion elements and transposon relicts. Among K. albida genomic islands, the most interesting are the two large regions with a length of 204 kbp (4583813–4788042) and 718 kbp (5733548–6452132), respectively (Figure 1). These regions are significantly distinct from the rest of the genome. Additional synteny analysis between K. albida and draft genome sequence of K. spp. 744 (SRX005446, SRX005562) clearly showed that the longer genomic island is acquired [30]. We were unable to determine the origin of the 204 kbp genomic island, but in the case of the 718 kbp region there were clearly no counterparts identified in sequence of K. spp. 744 genome, even though the regions around this portion of the K. albida chromosome are similar in both species. Both regions designated as genomic island are surrounded by the long inverted repeats (432 bp, 69% identical bp in the case of 204 kbp island, and 323 bp, 70% identical bp in the case of 718 kbp island) that might point on their origin from the large plasmids that were inserted into the chromosome. However, other possibilities cannot be excluded. Interestingly, the location of the predicted genomic islands corresponds to the locations of the regions with high density of secondary metabolism gene clusters indicating their possible origin (Figure 1).
Phylogenetic and orthologous analysis
K. albida was originally classified as “Streptosporangium albidum” in 1967 [6]. However, later it was renamed to Kutzneria albida due to the primary structures of 5S rRNA and 16 s rRNA, as well as to chemotaxonomic properties of the strain and the electrophoretic mobility of its ribosomal proteins [10]. Consequently, the genus was transferred to the family Pseudonocardiaceae, due to the range of taxonomic features that were found to be more similar to the typical strain of the family, Saccharothrix australiensis ATCC 31497T[10, 31]. An unsupervised nucleotide BLAST analysis of the DNA sequence corresponding to the 16S rRNA gene from K. albida with the 16S rRNA of different actinobacteria and E. coli as out-group was performed to determine phylogenetic relationships of the strain within the taxon (Figure 2). This analysis clearly showed that K. albida is distinct from the genus Streptosporangium and Streptomyces, and closer related to representatives of Pseudonocardiaceae. The highest similarity was observed between 16S rRNA of K. albida and Lentzea albida, Actinosynnema mirum, Saccharothrix algeriensis, Streptoalloteichus tenebrarius, Saccharopolyspora erythraea and Amycolatopsys alba, all of which belong to the Pseudonocardiacea family. At the same time, it is clear that the closest relatives of K. albida inside Pseudonocardiaceae are Streptoalloteichus tenebrarius, Lentzea albida, Actinosynnema mirum, and Saccharothrix algeriensis (Figure 2). These species are forming a branch distinct from other representatives of the family.

The 16S rRNA phylogram of representative Actinobacteria strains, with an outgroup containing E. coli as non-related strains. Ribosomal RNA sequences for all strains but S. albus were obtained from the Silva rRNA database [32]. Bootstrap value 1000.
The molecular phylogeny fully corresponds to and supports the data obtained during comparison of 16S rRNA. As expected, K. albida shares the highest number of orthologous genes (reciprocal best-hit pairs in pairwise protein BLAST searches) with the Pseudonocardiaceae and less with the Streptosporangiaceae (Table 3). Ten genomes of actinobacteria were used in this analysis: Kutzneria albida DSM 43870T, Actinosynnema mirum DSM 43827T, Amycolatopsis mediterranei S699, Kitasatospora setae NBRC 14216T, Saccharopolyspora erythrea NRRL 2338, Saccharotrix espanaensis DSM 44229T, Streptomyces avermitilis MA-4680, Streptomyces coelicolor A3(2), Streptomyces griseus subsp. griseus NBRC 13350, Streptosporangium roseum DSM 43021T. As expected, K. albida shares the highest number of orthologs with A. mediterranei (3,726), followed by Saccharotrix espanaensis (3,585 genes), Actinosynnema mirum, and S. erythraea (3,311 and 3,260 genes, respectively). In comparison, the number of genes shared with Streptosporangium roseum is 3,144. In contrast, the K. albida genome shares less orthologous genes with all the species from the genus Streptomyces tested: S. coelicolor – 3,017, S. avermitilis – 3,031, S. griseus – 2,935. Furthermore, when comparing all analyzed genomes, the number of orthologs drops to 1,766 genes, defining the non-strict minimal core genome shared between the analyzed species. This number is bigger than the previously predicted core genome for the family [5] because of a less strict method of calculating orthologs used in this study. Our result should not be regarded as a disagreement with earlier results, because our goal is only to provide additional support for the proper placement of Kutzneria albida among other Pseudonocardiaceae – we are not aiming at the identification of the 10-species core genome. Most of these 1,766 genes are located around oriC, while genes unique to K. albida or conserved in only one species are located further away from the oriC.
Aculeximycin biosynthesis gene cluster
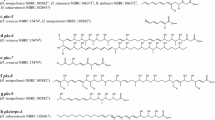
The only characterized secondary metabolite produced by K. albida is aculeximycin (Figure 3) [12, 15]. This compound is particularly interesting due to its activity against a broad range of Gram-positive bacteria, as well as against fungi and mosquitoes [12]. Aculeximycin, like similar metabolite streptoviridin from Kutzneria viridogrisea, exerts strong general toxicity caused by uncoupling of oxidative phosphorylation in mitochondria [13, 33]. On the other hand, this compound has an intriguing chemical structure with five sugars attached to the macrolactone [14, 15, 34]. Analysis of the K. albida genome sequence revealed the entire set of genes required for the aculeximycin molecule assembly clustered in a region of the chromosome approximately 2.3 Mbp away from the oriC. The acu cluster is 141.5 kbp long and contains 34 ORFs (Figure 3; all acu genes with predicted function are listed in Additional file 1: Table S1). The core of the cluster comprises eight genes encoding type I polyketide synthases (ORFs KALB_6560 – KALB_6567) with 21 modules in total, each containing different sets of reductase and acyltransferase domains as predicted by antiSMASH and SEARCHPKS (Figure 3, Additional file 1: Table S1) [29, 35]. Since biosynthesis of the aculeximycin polyketide scaffold is predicted to require 20 condensation steps including loading it makes us believe that one module is skipped during elongation process or alternate utilization of two modules takes place [36]. The first module encoded by AcuAI (KALB_6567), a three-modular synthase, contains a full set of ketoreduction domains that corresponds to the first condensation step of biosynthesis. However, neither the loading module nor the other four first elongation steps could be predicted based on a collinear logic of polyketide assembly, since the sets of ketoreduction domains in the second and third modules of AcuAI (KALB_6567) and three modules of AcuAII (KALB_6566) did not correspond to the primary structure of the aculeximycin polyketide [15, 37]. The last module encoded by acuAVIII (KALB_6560), contains a thioesterase domain (TE) similar to TE domains of erythromycin and pikromycin synthases that are catalysing lactonization of the matured chain [38, 39]. Malonate, methylmalonate, and ethylmalonate are used as precursors in polyketide chain extension. The hydroxyl group at C14 position is most probably incorporated as post-PKS oxygenation, and the gene encoding a hydroxylase, acuO2 (KALB_6568), is present within the cluster (Figure 3, Additional file 1: Table S1).

Chemical structure of aculeximycin and genetic organization of the acu biosynthesis gene cluster. PKS domain prediction was performed with antiSMASH and SEARCHPKS tools [29, 35]. Domain abbreviations: DD – docking domain, KS – ketosynthase, AT – acyltransferase, DH – dehydratase, ER – enoylreductase, KR – ketoreductase, ACP – acyl carrying protein, TE – thioesterase.
Aculeximycin is a highly glycosylated compound (Figure 3). Two sugars – D-mannose and L-vancosamine – are attached at positions C23 and C37, respectively [15, 34]. A short oligosaccharide chain named aculexitriose (O-6-deoxy-beta-D-glucopyranosyl-(1–2)-O-[3-amino-2,3,6-trideoxy-beta-D-arabino-hexopyranosyl-(1–3)]-6-deoxy-D-glucopyranose) is attached at position C11. D-mannose is supplied from the primary metabolism, as in the case of biosynthesis of other antibiotics containing this moiety [40]. Deoxysugar biosynthesis genes are present within the cluster: The genes acuS6 (KALB_6571) and acuS2 (KALB_6585) encode glucose-1-phosphate thymidylyltransferase and dTDP-glucose 4,6-dehydratase, respectively – enzymes participating in the initial common steps in deoxysugars biosynthesis [41, 42]. Genes for downstream enzymes leading to the production of each individual sugar from NDP-4-keto-6-deoxy-D-glucose are also present within the cluster. L-vancosamine is built by the sequential action of AcuS1 (NDP-hexose-C3-methyltransferase, KALB_6588), AcuS3 (NDP-4-dehydrorhamnose-3,5-epimerase, OF6583), AcuS4 (NDP-hexose-2,3-dehydratase, KALB_6582), AcuS5 (NDP-hexose-4-ketoreductase, KALB_6576), and AcuN1 (KALB_6580) or AcuN2 (KALB_6578) aminotransferases, similar to what had been described for the glycopeptide antibiotic chloroeremomycin [43]; 6-deoxy-glucopyranose – AcuS5; 3-amino-2,3,6-trideoxy-beta-D-arabino-hexopyranose – AcuS4, AcuS5, AcuN1 or AcuN2 (Additional file 1: Table S1). Eight putative glycosyltransferase genes (acuGT1-8; KALB_6584, 6581, 6579, 6575, 6574, 6570, 6569, 6559) were found within the aculeximycin biosynthesis gene cluster. The redundant number of GTs can be explained by the fact that some of them might participate in self-resistance mechanism. AcuGT3 (KALB_6579) has a high degree of similarity to numerous glycosyltransferases involved in resistance to macrolides, including one involved in self-resistance of oleandomycin producer [44, 45]. Another component of this resistance complex might be the gene product of acuH (KALB_6577) encoding a β-hexosaminidase homologue. This family of enzymes is involved in the cleavage of GalNAc residues from oligosaccharides. The acu cluster also contains acuW (KALB_6572) gene encoding type III ABC transporter protein containing both transmembrane and ATP binding domains within one polypeptide [46, 47]. In addition, 5 putative regulatory genes were found within the acu cluster. One of them, AcuR5 (KALB_6573), belongs to the TetR family of transcription regulators and is located next to the acuW gene. Four other regulators (AcuR1-4: OF6591, 6590, 6589, 6586) belong to the LuxR family and might participate in control and fine tuning of the aculeximycin production (Figure 3, Additional file 1: Table S1) [48].
Secondary metabolites production potential
Besides aculeximycin, no other secondary metabolites are known to be produced by K. albida. However, genome analysis using antiSMASH revealed 47 gene clusters potentially involved in secondary metabolism, including the acu cluster [29]. Manual correction of the obtained data resulted in 46 gene clusters related to secondary metabolism (Table 4). This makes us think that the K. albida genome is one of the richest in terms of secondary metabolism genes reported till now. General features of the secondary metabolism gene clusters of K. albida are summarized in Additional file 1: Table S2.
The most represented type of secondary metabolite biosynthesis genes within the K. albida genome are non-ribosomal peptide synthases (NRPS) (16 out of 46; Table 4, Additional file 1: Table S2). Core genes in clusters kal 1, 9, 14, 16, 30, and 35 encode NRPS proteins with only three domains: adenylation (A), peptidyl-carrier protein (PCP), and an approximately 700 aa long N-terminal domain that is proposed to act as condensation domain (C) [49]. NRPS genes in these clusters are accompanied by genes usually found in the biosynthesis of siderophores [50–53]. Several other NRPS clusters in the genome could be designated as siderophores producing due to the presence of genes encoding ornithine N-monooxygenase (kal22) or isochorismate (salicylate) synthase (kal27, kal32, kal44) known to be involved in biosynthesis of precursors [52–54], or conserved Fe3+-siderophore ABC transport system genes (kal27 and kal44) [55]. Besides NRPS produced siderophores, kal23 cluster is predicted to be responsible for biosynthesis of the aerobactin-like Fe3+-chelating compound found in different species of Corynebacterium and Bacillus[56].
The second most abundant K. albida secondary metabolism genes are encoding polyketide biosynthesis. Analysis of the genome revealed 6 type I PKS (kal 3, 6, 28, 39, 40, and 46) and 2 type II PKS (kal21 and 45) gene clusters (Table 4, Additional file 1: Table S2). The kal3 cluster consists of one gene encoding single module PKS (KALB_1318) with a full set of ketoreductase domains. The closest homologues of this enzyme encode mycocerosic acid synthase in Stigmatella aurantiaca and in several Mycobacterium species involved in the production of the multimethyl-branched fatty acids by the elongation of fatty acids with 4 units of methyl-malonate [57, 58]. The core genes in the cluster kal28 (KALB_5021, 5022, 5023) are similar to genes encoding omega-3-polyunsaturated fatty acid synthases. These unusual fatty acids are known to accumulate in the membranes of cold- and high pressure-resistant marine bacteria [59, 60]. Other type I PKS gene clusters (kal 6, 39, 40 and 46) have no homologues in another sequenced bacterial genomes and their products could be just partially predicted from the genes organization. Two other PKS clusters encode type II PKS systems that show some similarity to whiE clusters involved in spore pigment production in different streptomycetes (kal21) [61, 62] and to genes involved in biosynthesis of the aromatic polyketides mithramycin [63] and nogalamycin [64] (kal45).
Seven K. albida gene clusters combine type I PKS and NRPS biosynthetic pathways (Table 4, Additional file 1: Table S2) [65]. Gene clusters kal4 and kal10 encode type I PKSs and a NRPSs with a single condensation domain. The PKS portion of kal10 resembles the KS involved in production of phenolic glycolipids in Mycobacteria, another Fe3+-chelating compound [66]. The NRPS part might act at the initiation step by direct loading of the starting unit into the ACP of PKS, similar to the mechanism proposed for biosynthesis of mycobactins, mycobacterial siderophores of the phenolic glycolipids family [67, 68].
5 gene clusters for terpenoid biosynthesis could be found in the K. albida genome (Table 4, Additional file 1: Table S2). The C5 precursors for these secondary metabolites are provided by the non-mevalonate (MEV/DOXP) pathway, genes for which are present in the genome [69, 70]. At the same time we were unable to identify any genes encoding enzymes of the mevalonate pathway. The kal2 and kal25 gene clusters a predicted to be involved in the production of the earthy flavored sesquiterpene geosmin and moldy-smelling monoterpene 2-methylisoborneol (2-MIB) respectively [71, 72]. Core genes of both of them are highly conserved among different actinomycetes. Interestingly, there is one more germacradienol synthase gene (KALB_0677, geo2) located upstream from geo1 (KALB_0676, geo1) in kal2 cluster. Its function is not clear. kal5 cluster is predicted to be involved in the biosynthesis of carotenoids based on the key gene CtrB5 (KALB_2031) similarity to the known phytoene synthases [73]. Similar, kal20 gene cluster is suggested to be involved in the biosynthesis of hopanoids, bacterial pentacyclic triterpenoids [74].
Several other types of secondary metabolites could be potentially produced by K. albida (Table 4, Additional file 1: Table S2). The gene cluster kal8 encodes 4 enzymes involved in the biosynthesis of compatible solutes ectoine and 5-hydroxyectoine [75, 76]. The kal11 cluster is similar to genes involved in biosynthesis of indolocarbazole group of secondary metabolites [77, 78]. Several lantibiotics biosynthesis gene clusters could be found in the genome of K. albida as well (kal12, 13, 29, 41, 42). Interestingly, ORFs 3600 and 3601 from kal13 are coding for YcaO domain-containing proteins involved in the formation of thiazole/oxazole, another post-translational modifications observed in ribosomally synthesized natural products [79, 80]. The cluster kal24 contains only one gene (KALB_4567), which encodes a 13 kDa protein with the high degrees of similarity to bacteriocins of the linocin M18 family [81]. Linocin M18 was first isolated from Brevibacterium linens due to its ability to inhibit the growth of several Listeria species, and similar genes were later discovered in other bacterial genomes. Recent findings indicate that linocins might play a role in the compartmentalization of oxidative-stress response processes in bacterial cells [82]. At the same time, the product of KALB_4567 is two times shorter than typical linocin M18.
Testing secondary metabolism potential
The genome sequence of K. albida unveiled enormous secondary metabolites biosynthetic potential of this bacterium (Additional file 1: Table S2). However, so far only aculeximycin is known to be produced by this strain [11, 12]. To further elucidate the biosynthetic potential of K. albida the strain was grown in different media and extra- and intra-cellular accumulated metabolites were tested using high resolution LC-MS (Additional file 1: Figures S1, S2). After 7 days of growth aculeximycin and its aglycone production was observed only in two out of six used media. At the same time, multiple secondary metabolites accumulation was observed in all cases. The DNP analysis [83] of obtained data led to idea that majority of compounds accumulated by K. albida are not described yet. Furthermore, extracts were found to be active against Bacillus subtilis, even those that did not contain aculeximycin. These facts are making further analysis of secondary metabolites produced by K. albida especially interesting.
As expected from the secondary metabolism genes analysis many of the metabolites produced by K. albida are acting as siderophores (Additional file 1: Table S2; Additional file 1: Figure S3). A siderophores accumulation test using a modified CAS assay clearly showed that K. albida produced significant amounts of Fe3+-chelating compounds during growth on different media when compared to S. coelicolor and S. albus (Additional file 1: Figure S3) [84]. Additionally, extracts from cultures grown on tested media including liquid and solid CAS media were separated by TLC and overlaid with CAS agarose for siderophores detection (Additional file 1: Figure S3). As a result, K. albida was found to accumulate from one to at least 5 Fe3+-chelating compounds with different physicochemical properties when growing at different conditions. Any of them could be predicted based on information available in DNP [83]. This finding supports our prediction of evolving of the K. albida genome in directions leading to adaptation to iron deficient environment, where the strain was isolated from [6, 12].
One of the previously described compounds that were identified within the extracts from K. albida is a cyclic leucilphenylalanine (cFL) (Additional file 1: Figure S2). It was produced in 4 out of 7 tested media. This compound accumulation could be directly linked to the cyclodipeptide synthase gene (KALB_7471) found in the kal43 cluster. These proteins comprise an interesting class of enzymes utilizing amino-acyl tRNA as a substrate to produce diketopiperazine containing cyclic dipeptides [85–87]. In many cases the cyclodipeptide synthase products are further modified by the decorating enzymes. No genes possibly involved in post-processing of cyclic peptides were found in close proximity to the KALB_7471. The number of recently discovered cyclodipeptide synthases is expanding. In many cases these enzymes are not strictly specific for some particular substrates and can produce several types of cyclic peptides [85, 87]. However, we were not able to identify any other possible products of KALB_7471 in extracts of K. albida. In order to test the enzyme specificity the gene was cloned and expressed in E. coli. Comparison of extracts from E. coli containing KALB_7471 expression construct and empty vector control led to identification of four new compounds (Figure 4). One of them, as was predicted from analysis of K. albida extracts, was cFL. Three other compounds were identified as cFM, cFY and cFF based on exact mass and fragmentation patterns (Figure 4). This finding led us to the conclusion that the K. albida enzyme as the first substrate prefers phenylalanine, but can also utilize other amino acids as a second substrate. Re-examination of K. albida extracts led to identification of cFF and cFY, however not cFM.

HPLC-MS analysis of extracts from E. coli expressing KALB_7471. A. UV–vis traces that correspond to the KALB_7471 expressing strain (red) and control strain (blue) are shown. Cyclic dipeptides are marked and their masses are indicated. B. MS2-fragmentation spectra of KALB_7471 products. The characteristic neutral losses of 28 and 45 Da, resulting in the detection of the respective ammonium ions are shown.
Conclusions
The complete genome of Kutzneria albida, the first representative of Kutzneria genus was sequenced, annotated and analyzed. The genome of this strain is one of the biggest circular actinobacterial genomes sequenced thus far. The phylogenetic and orthology analyses clearly distinguish Kutzneria albida from Streptosporangiaceae thereby providing the first genomic evidence for transferring the genus into the Pseudonocardiaceae family.
Two large genomic islands are present in the K. albida genome. Localization of these islands corresponds to regions of a high density of genes involved in secondary metabolism providing clues into the origin of a large part of the strain’s auxiliary metabolism. In general, about 14% of the chromosome is occupied with the secondary metabolism gene clusters, including cluster predicted to be involved in aculeximycin biosynthesis.
Aculeximycin and its aglycone accumulation were observed during growth of the strain in several media. However, a vast majority of compounds produced by the strain were not found in available secondary metabolites databases. As predicted from the genome analysis and confirmed experimentally, a large proportion of secondary metabolism of K. albida is devoted to siderophores production. On the other hand, cyclic dipeptides were found in the extract of the strain.
In summary, sequencing of the K. albida genome provides new insights into understanding the evolution of minor groups of actinobacteria and will attract more attention to these fascinating bacteria as an inexhaustible source of novel biologically active secondary metabolites. The large diversity of secondary metabolism gene clusters in the genome of K. albida is reflected in metabolites produced. Furthermore, isolation, structural and biological characterization of secondary metabolites produced by this strain might lead to discovery of new interesting biological activities as well as new chemical scaffolds thus proving the concept of genome mining of minor groups of actinobacteria for new secondary metabolites discovery.
Methods
Sequencing of Kutzneria albidagenome
The type strain of Kutzneria albida (DSM 43870T) was obtained as a lyophilized culture from DSMZ (Braunschweig, Germany). Genomic DNA was isolated from 30 ml cultures grown in tryptone soy broth (TSB) [88] at 28°C for 24 hours. Total DNA isolation was performed according to the salting out procedure followed by RNase treatment [88]. The obtained DNA was used to construct both a 12 k PE and a WGS library for pyrosequencing on a Genome Sequencer FLX (Roche Applied Science), using the Titanium chemistry to reduce problems with high G + C regions [89]. Assembly of the shotgun reads was performed with the GS Assembler software (version 2.3). A total of 491,980 reads (170,469,390 bp) were assembled into 197 contigs in 1 scaffold.
Completion of the draft sequence
For finishing of the genome sequence, the CONSED software package was used [90]. Of the 197 gaps, 57 could be closed in silico as these gaps were caused by repetitive elements. For gap closure and assembly validation, the remaining genomic contigs were bridged by 140 PCR products.
Gaps between contigs of the whole genome shotgun assembly were closed by sequencing PCR products carried out by IIT GmbH (Bielefeld, Germany) on ABI 377 sequencing machines. To obtain a high quality genome sequence and to correct for homopolymer errors common in pyrosequencing, additional Illumina GAIIx data was used. A total of 5,064,677 reads of 50 bp length was mapped on the genome, resulting in a 25.6x coverage. A total of 19 SNPs, 50 single nucleotide insertions and 49 single nucleotide deletions were found and corrected.
Genome analysis and annotation
In the first step, gene finding was done using GISMO [91] followed by GenDB 2.0 automatic annotation [92]. In the second annotation step, all predicted ORFs were manually re-inspected to correct start codon and function assignments. Intergenic regions were checked for ORFs missed by the automatic annotation using BLAST [93].
Phylogenetic analysis
19 rDNA sequences were aligned using MAFFT v7.017 (gap open penalty 1.53, offset value 0.123, scoring matrix 200PAM/k = 2, algorithm: auto). Dendrogram was built using Geneious [94] (Tamura-Nei genetic distance model, neighbor-joining method, E. coli as an outgroup, bootstrap value 1000, consensus tree with 50% support threshold). Ribosomal RNA sequences for all strains but S. albus were obtained from the Silva rRNA database [32].
Orthology analysis
10 genomes were used for the analysis of the numbers of orthologous genes between genome pairs (GenBank accession version is indicated in parenthesis): Actinosynnema mirum DSM 43827T (CP001630.1), Amycolatopsis mediterranei S699 (CP003729.1), Kitasatospora setae NBRC 14216T (AP010968.1), Kutzneria albida DSM 43870T (deposited to GenBank with accession CP007155), Saccharopolyspora erythraea NRRL 2338 (AM420293.1), Saccharotrix espanaensis DSM 44229T (HE804045.1), Streptomyces avermitilis MA-4680 (BA000030.3), Streptomyces coelicolor A3(2) (AL645882.2), Streptomyces griseus subsp. griseus NBRC 13350 (AP009493.1), Streptosporangium roseum DSM 43021T (CP001814.1). In the first step, 45 pairwise genome-wide reciprocal best-hit protein BLAST searches were performed on 10 genomes, using InParanoid [95] (configured as follows: two-pass BLAST, with bootstrapping, not using an outgroup, matrix BLOSUM45, minimal BLAST bit score 40, sequence overlap cut-off 0.5, segment coverage overlap 0.25). Pseudo genes were excluded from this step. In the next step, MultiParanoid [96] was applied (with default parameters – genes clustered twice were not removed) to generate single file of orthologous gene clusters. The file contains a total of 8,745 orthologous gene clusters with 65,033 genes. Finally, this file was parsed, returning – for each analyzed pair of genomes – the number of orthologous gene clusters which contained genes from both of these genomes. This number was then reported. To find the number of genes common to all 10 genomes, we identified the number of orthologous gene clusters containing 10 unique genome identifiers.
Analysis of secondary metabolite clusters
For the identification of secondary metabolite clusters, the genome of K. albida was scanned for homologues to known secondary metabolite synthases via BLAST search. These manual investigations were supported by antiSMASH [29]. A set of genes was considered to be a cluster, when there was at least one gene encoding a secondary metabolite synthase. Consequently, a locus possessing a gene with only a single domain, for example an A domain, was not considered to be a cluster. The boundaries of the clusters were defined by the last gene upstream and downstream of a secondary metabolite synthase with homology to a gene encoding a regulator, transporter or tailoring enzyme. In cases where this gene was part of a putative operon, the whole operon was included into the cluster. The modular organization of the type I polyketide and nonribosomal peptide megasynthases were determined using web tools [97, 98].
Secondary metabolites production and LC-MS analysis
K. albida was grown in 20 ml of TSB media for 4 days. 2 ml of pre-culture was inoculated into 50 ml of production media. Six different medias were used: TSB, NL5 (NaCl 1 g/l, KH2PO4 1 g/l, MgSO4x7H2O 0.5 g/l, Trace elements solution 2 ml/l, Glycerol 25 g/l, L-glutamine 5.84 g/l), NL19 (Soy flour 20 g/l, Mannitol 20 g/l), NL111 (Meat extract 20 g/l, Maltose extract 10 g/l, CaCO3 10 g/l), CAS [84] and SG [99]. Strain was grown for 7 days at 30°C and 250 rpm. Metabolites were extracted with ethyl acetate from supernatant and acetone-methanol (1:1) mixture from biomass. Extracts were evaporated, dissolved in 100 μl of methanol and samples from biomass and supernatant were combined. 1 μl of each sample were separated on an Ultimate 3000 HPLC (Dionex) using C18 column (Affymetrix) and linear gradient of acetonitrile against 0.1% ammonium formate solution in water. Samples were analyzed on ultrahigh resolution mass spectrometer system maXis (Bruker Daltonics).
Cloning and expression of KALB_7471
KALB_7471 was amplified from the genome of K. albida using KOD polymerase (Novagen) and primers Kal7474LETNcF (TACCATGGTGTTGACCAC GAGCCCATT) and Kal7474ETBR (CGGATCCCGCACGGCCAGCGATTCGG) and cloned as NcoI/BamHI into pET28b. Obtained plasmid was introduced into E. coli BL21(DE3). Strain was grown in 50 ml of LB media till OD600 0.4 and expression was induced with 0.5 mM IPTG. Accumulation of KALB_7471 protein was tested after 8 hours of growth at 30°C by SDS PAGE. Metabolites were extracted with ethyl acetate, evaporated, dissolved in 200 μl of methanol and analyzed as described above. E. coli BL21(DE3) containing empty pET28b was used as control.
Nucleotide sequence accession numbers
The genome of Kutzneria albida was deposited to GenBank with accession number CP007155.
Supporting information
Data sets supporting the results of this article are included within the article and its Additional file 1.
References
Demain AL, Adrio JL: Contributions of microorganisms to industrial biology. Mol Biotechnol. 2008, 38 (1): 41-55.
Ventura M, Canchaya C, Tauch A, Chandra G, Fitzgerald GF, Chater KF, Van Sinderen D: Genomics of Actinobacteria: tracing the evolutionary history of an ancient phylum. Microbiol Mol Biol Rev. 2007, 71 (3): 495-548.
Paradkar A, Trefzer A, Chakraburtty R, Stassi D: Streptomyces genetics: a genomic perspective. Crit Rev Biotechnol. 2003, 23 (1): 1-27.
Pagani I, Liolios K, Jansson J, Chen IM, Smirnova T, Nosrat B, Markowitz VM, Kyrpides NC: The Genomes OnLine Database (GOLD) v.4: status of genomic and metagenomic projects and their associated metadata. Nucleic Acids Res. 2012, 40: D571-D579.
Strobel T, Al-Dilaimi A, Blom J, Gessner A, Kalinowski J, Luzhetska M, Pühler A, Szczepanowski R, Bechthold A, Rückert C: Complete genome sequence of Saccharothrix espanaensis DSM 44229T and comparison to the other completely sequenced Pseudonocardiaceae. BMC Genomics. 2012, 13: 465-
Furumai T, Ogawa H, Okuda T: Taxonomic study on Streptosporangium albidum nov. sp. J Antibiot (Tokyo). 1968, 21 (3): 179-181.
Labeda DP, Kroppenstedt RM: Phylogenetic analysis of Saccharothrix and related taxa: proposal for Actinosynnemataceae fam. nov. Int J Syst Evol Microbiol. 2000, 50 (1): 331-336.
Suriyachadkun C, Ngaemthao W, Chunhametha S, Tamura T, Sanglier JJ: Kutzneria buriramensis sp. nov., isolated from soil, and emended description of the genus Kutzneria. Int J Syst Evol Microbiol. 2013, 63 (1): 47-52.
Pohanka A, Menkis A, Levenfors J, Broberg A: Low-abundance kutznerides from Kutzneria sp. 744. J Nat Prod. 2006, 69 (12): 1776-1781.
Stackebrandt E, Kroppenstedt RM, Jahnke KD, Kemmerling C, Gurtler H: Transfer of Streptosporangium-Viridogriseum (Okuda Et-Al 1966), Streptosporangium-Viridogriseum Subsp Kofuense (Nonomura and Ohara 1969), and Streptosporangium-Albidum (Furumai Et-Al 1968) to Kutzneria Gen-Nov as Kutzneria-Viridogrisea Comb-Nov, Kutzneria-Kofuensis Comb-Nov, and Kutzneria-Albida Comb-Nov, Respectively, and Emendation of the Genus Streptosporangium. Int J Syst Bacteriol. 1994, 44 (2): 265-269.
Ikemoto T, Matsunaga H, Konishi K, Okazaki T, Enokita R, Torikata A: Aculeximycin, a new antibiotic from Streptosporangium albidum. I. Taxonomy of producing organism and fermentation. J Antibiot (Tokyo). 1983, 36 (9): 1093-1096.
Ikemoto T, Katayama T, Shiraishi A, Haneishi T: Aculeximycin, a new antibiotic from Streptosporangium albidum. II. Isolation, physicochemical and biological properties. J Antibiot (Tokyo). 1983, 36 (9): 1097-1100.
Miyoshi H, Tamaki M, Murata H, Ikemoto T, Shibuya T, Harada KI, Suzuki M, Iwamura H: Uncoupling mechanism of glycoside antibiotic aculeximycin in isolated rat-liver mitochondria. J Biochem. 1996, 119 (2): 274-280.
Murata H, Harada K, Suzuki M, Ikemoto T, Shibuya T, Haneishi T, Torikata A: Structural elucidation of aculeximycin. II. Structures of carbohydrate moieties. J Antibiot (Tokyo). 1989, 42 (5): 701-710.
Murata H, Ohama I, Harada K, Suzuki M, Ikemoto T, Shibuya T, Haneishi T, Torikata A, Itezono Y, Nakayama N: Structural elucidation of aculeximycin. IV. Absolute structure of aculeximycin, belonging to a new class of macrolide antibiotics. J Antibiot (Tokyo). 1995, 48 (8): 850-862.
Zawilak-Pawlik A, Kois A, Majka J, Jakimowicz D, Smulczyk-Krawczyszyn A, Messer W, Zakrzewska-Czerwinska J: Architecture of bacterial replication initiation complexes: orisomes from four unrelated bacteria. Biochem J. 2005, 389 (Pt 2): 471-481.
Smulczyk-Krawczyszyn A, Jakimowicz D, Ruban-Osmialowska B, Zawilak-Pawlik A, Majka J, Chater K, Zakrzewska-Czerwinska J: Cluster of DnaA boxes involved in regulation of Streptomyces chromosome replication: from in silico to in vivo studies. J Bacteriol. 2006, 188 (17): 6184-6194.
Kono N, Arakawa K, Tomita M: Comprehensive prediction of chromosome dimer resolution sites in bacterial genomes. BMC Genomics. 2011, 12: 19-
Krzywinski M, Schein J, Birol I, Connors J, Gascoyne R, Horsman D, Jones SJ, Marra MA: Circos: an information aesthetic for comparative genomics. Genome Res. 2009, 19 (9): 1639-1645.
Kirby R: Chromosome diversity and similarity within the Actinomycetales. FEMS Microbiol Lett. 2011, 319 (1): 1-10.
Bentley SD, Chater KF, Cerdeno-Tarraga AM, Challis GL, Thomson NR, James KD, Harris DE, Quail MA, Kieser H, Harper D, Bateman A, Brown S, Chandra G, Chen CW, Collins M, Cronin A, Fraser A, Goble A, Hidalgo J, Hornsby T, Howarth S, Huang CH, Kieser T, Larke L, Murphy L, Oliver K, O'Neil S, Rabbinowitsch E, Rajandream MA, Rutherford K, et al: Complete genome sequence of the model actinomycete Streptomyces coelicolor A3(2). Nature. 2002, 417 (6885): 141-147.
Ikeda H, Ishikawa J, Hanamoto A, Shinose M, Kikuchi H, Shiba T, Sakaki Y, Hattori M, Omura S: Complete genome sequence and comparative analysis of the industrial microorganism Streptomyces avermitilis. Nat Biotechnol. 2003, 21 (5): 526-531.
Pati A, Sikorski J, Nolan M, Lapidus A, Copeland A, Glavina Del Rio T, Lucas S, Chen F, Tice H, Pitluck S, Cheng JF, Chertkov O, Brettin T, Han C, Detter JC, Kuske C, Bruce D, Goodwin L, Chain P, D'Haeseleer P, Chen A, Palaniappan K, Ivanova N, Mavromatis K, Mikhailova N, Rohde M, Tindall BJ, Goker M, Bristow J, Eisen JA, et al: Complete genome sequence of Saccharomonospora viridis type strain (P101). Stand Genomic Sci. 2009, 1 (2): 141-149.
Sales CM, Mahendra S, Grostern A, Parales RE, Goodwin LA, Woyke T, Nolan M, Lapidus A, Chertkov O, Ovchinnikova G, Sczyrba A, Alvarez-Cohen L: Genome sequence of the 1,4-dioxane-degrading Pseudonocardia dioxanivorans strain CB1190. J Bacteriol. 2011, 193 (17): 4549-4550.
Oliynyk M, Samborskyy M, Lester JB, Mironenko T, Scott N, Dickens S, Haydock SF, Leadlay PF: Complete genome sequence of the erythromycin-producing bacterium Saccharopolyspora erythraea NRRL23338. Nat Biotechnol. 2007, 25 (4): 447-453.
Tang B, Zhao W, Zheng H, Zhuo Y, Zhang L, Zhao GP: Complete genome sequence of Amycolatopsis mediterranei S699 based on de novo assembly via a combinatorial sequencing strategy. J Bacteriol. 2012, 194 (20): 5699-5700.
Schattner P, Brooks AN, Lowe TM: The tRNAscan-SE, snoscan and snoGPS web servers for the detection of tRNAs and snoRNAs. Nucleic Acids Res. 2005, 33: W686-W689.
Powell S, Szklarczyk D, Trachana K, Roth A, Kuhn M, Muller J, Arnold R, Rattei T, Letunic I, Doerks T, Jensen LJ, von Mering C, Bork P: eggNOG v3.0: orthologous groups covering 1133 organisms at 41 different taxonomic ranges. Nucleic Acids Res. 2012, 40 (D1): D284-D289.
Medema MH, Blin K, Cimermancic P, De Jager V, Zakrzewski P, Fischbach MA, Weber T, Takano E, Breitling R: antiSMASH: rapid identification, annotation and analysis of secondary metabolite biosynthesis gene clusters in bacterial and fungal genome sequences. Nucleic Acids Res. 2011, 39: W339-W346.
Merkl R: SIGI: score-based identification of genomic islands. BMC Bioinformatics. 2004, 5: 22-
Whitham TS, Athalye M, Minnikin DE, Goodfellow M: Numerical and Chemical Classification of Streptosporangium and Some Related Actinomycetes. Anton Leeuw Int J Gen Mol Microbiol. 1993, 64 (3–4): 387-429.
Quast C, Pruesse E, Yilmaz P, Gerken J, Schweer T, Yarza P, Peplies J, Glockner FO: The SILVA ribosomal RNA gene database project: improved data processing and web-based tools. Nucleic Acids Res. 2013, 41 (Database issue): D590-D596.
Miyoshi H, Tamaki M, Harada K, Murata H, Suzuki M, Iwamura H: Uncoupling action of antibiotic sporaviridins with rat-liver mitochondria. Biosci Biotechnol Biochem. 1992, 56 (11): 1776-1779.
Murata H, Suzuki K, Tabayashi T, Hattori C, Takada Y, Harada K, Suzuki M, Ikemoto T, Shibuya T, Haneishi T, Torikata A, Itezono Y: Structural elucidation of aculeximycin. III. Planar structure of aculeximycin, belonging to a new class of macrolide antibiotics. J Antibiot (Tokyo). 1995, 48 (8): 838-849.
Yadav G, Gokhale RS, Mohanty D: SEARCHPKS: A program for detection and analysis of polyketide synthase domains. Nucleic Acids Res. 2003, 31 (13): 3654-3658.
Moss SJ, Martin CJ, Wilkinson B: Loss of co-linearity by modular polyketide synthases: a mechanism for the evolution of chemical diversity. Nat Prod Rep. 2004, 21 (5): 575-593.
Fischbach MA, Walsh CT: Assembly-line enzymology for polyketide and nonribosomal Peptide antibiotics: logic, machinery, and mechanisms. Chem Rev. 2006, 106 (8): 3468-3496.
Akey DL, Kittendorf JD, Giraldes JW, Fecik RA, Sherman DH, Smith JL: Structural basis for macrolactonization by the pikromycin thioesterase. Nat Chem Biol. 2006, 2 (10): 537-542.
Pinto A, Wang M, Horsman M, Boddy CN: 6-Deoxyerythronolide B synthase thioesterase-catalyzed macrocyclization is highly stereoselective. Org Lett. 2012, 14 (9): 2278-2281.
Magarvey NA, Haltli B, He M, Greenstein M, Hucul JA: Biosynthetic pathway for mannopeptimycins, lipoglycopeptide antibiotics active against drug-resistant gram-positive pathogens. Antimicrob Agents Chemother. 2006, 50 (6): 2167-2177.
Nedal A, Zotchev SB: Biosynthesis of deoxyaminosugars in antibiotic-producing bacteria. Appl Microbiol Biotechnol. 2004, 64 (1): 7-15.
Salas JA, Mendez C: Biosynthesis pathways for deoxysugars in antibiotic-producing actinomycetes: isolation, characterization and generation of novel glycosylated derivatives. J Mol Microbiol Biotechnol. 2005, 9 (2): 77-85.
Chen H, Thomas MG, Hubbard BK, Losey HC, Walsh CT, Burkart MD: Deoxysugars in glycopeptide antibiotics: enzymatic synthesis of TDP-L-epivancosamine in chloroeremomycin biosynthesis. Proc Natl Acad Sci U S A. 2000, 97 (22): 11942-11947.
Salas JA, Hernandez C, Mendez C, Olano C, Quiros LM, Rodriguez AM, Vilches C: Intracellular glycosylation and active efflux as mechanisms for resistance to oleandomycin in Streptomyces antibioticus, the producer organism. Microbiologia. 1994, 10 (1–2): 37-48.
Vilches C, Hernandez C, Mendez C, Salas JA: Role of glycosylation and deglycosylation in biosynthesis of and resistance to oleandomycin in the producer organism, Streptomyces antibioticus. J Bacteriol. 1992, 174 (1): 161-165.
Mendez C, Salas JA: The role of ABC transporters in antibiotic-producing organisms: drug secretion and resistance mechanisms. Res Microbiol. 2001, 152 (3–4): 341-350.
Mendez C, Salas JA: ABC transporters in antibiotic-producing actinomycetes. FEMS Microbiol Lett. 1998, 158 (1): 1-8.
Chen J, Xie J: Role and regulation of bacterial LuxR-like regulators. J Cell Biochem. 2011, 112 (10): 2694-2702.
Marchler-Bauer A, Zheng C, Chitsaz F, Derbyshire MK, Geer LY, Geer RC, Gonzales NR, Gwadz M, Hurwitz DI, Lanczycki CJ, Lu F, Lu S, Marchler GH, Song JS, Thanki N, Yamashita RA, Zhang D, Bryant SH: CDD: conserved domains and protein three-dimensional structure. Nucleic Acids Res. 2013, 41 (D1): D348-D352.
Vandenende CS, Vlasschaert M, Seah SY: Functional characterization of an aminotransferase required for pyoverdine siderophore biosynthesis in Pseudomonas aeruginosa PAO1. J Bacteriol. 2004, 186 (17): 5596-5602.
Ikai H, Yamamoto S: Identification and analysis of a gene encoding L-2,4-diaminobutyrate:2-ketoglutarate 4-aminotransferase involved in the 1,3-diaminopropane production pathway in Acinetobacter baumannii. J Bacteriol. 1997, 179 (16): 5118-5125.
Pelludat C, Brem D, Heesemann J: Irp9, encoded by the high-pathogenicity island of Yersinia enterocolitica, is able to convert chorismate into salicylate, the precursor of the siderophore yersiniabactin. J Bacteriol. 2003, 185 (18): 5648-5653.
Harrison AJ, Yu M, Gardenborg T, Middleditch M, Ramsay RJ, Baker EN, Lott JS: The structure of MbtI from Mycobacterium tuberculosis, the first enzyme in the biosynthesis of the siderophore mycobactin, reveals it to be a salicylate synthase. J Bacteriol. 2006, 188 (17): 6081-6091.
Ge L, Seah SY: Heterologous expression, purification, and characterization of an l-ornithine N(5)-hydroxylase involved in pyoverdine siderophore biosynthesis in Pseudomonas aeruginosa. J Bacteriol. 2006, 188 (20): 7205-7210.
Bunet R, Brock A, Rexer HU, Takano E: Identification of genes involved in siderophore transport in Streptomyces coelicolor A3(2). FEMS Microbiol Lett. 2006, 262 (1): 57-64.
Oves-Costales D, Kadi N, Challis GL: The long-overlooked enzymology of a nonribosomal peptide synthetase-independent pathway for virulence-conferring siderophore biosynthesis. Chem Commun (Camb). 2009, 43: 6530-6541.
Kolattukudy PE, Fernandes ND, Azad AK, Fitzmaurice AM, Sirakova TD: Biochemistry and molecular genetics of cell-wall lipid biosynthesis in mycobacteria. Mol Microbiol. 1997, 24 (2): 263-270.
Mathur M, Kolattukudy PE: Molecular cloning and sequencing of the gene for mycocerosic acid synthase, a novel fatty acid elongating multifunctional enzyme, from Mycobacterium tuberculosis var. bovis Bacillus Calmette-Guerin. J Biol Chem. 1992, 267 (27): 19388-19395.
Allen EE, Bartlett DH: Structure and regulation of the omega-3 polyunsaturated fatty acid synthase genes from the deep-sea bacterium Photobacterium profundum strain SS9. Microbiology. 2002, 148 (Pt 6): 1903-1913.
Wallis JG, Watts JL, Browse J: Polyunsaturated fatty acid synthesis: what will they think of next?. Trends Biochem Sci. 2002, 27 (9): 467-
Blanco G, Brian P, Pereda A, Mendez C, Salas JA, Chater KF: Hybridization and DNA sequence analyses suggest an early evolutionary divergence of related biosynthetic gene sets encoding polyketide antibiotics and spore pigments in Streptomyces spp. Gene. 1993, 130 (1): 107-116.
Davis NK, Chater KF: Spore colour in Streptomyces coelicolor A3(2) involves the developmentally regulated synthesis of a compound biosynthetically related to polyketide antibiotics. Mol Microbiol. 1990, 4 (10): 1679-1691.
Lombo F, Brana AF, Mendez C, Salas JA: The mithramycin gene cluster of Streptomyces argillaceus contains a positive regulatory gene and two repeated DNA sequences that are located at both ends of the cluster. J Bacteriol. 1999, 181 (2): 642-647.
Torkkell S, Kunnari T, Palmu K, Mantsala P, Hakala J, Ylihonko K: The entire nogalamycin biosynthetic gene cluster of Streptomyces nogalater: characterization of a 20-kb DNA region and generation of hybrid structures. Mol Genet Genomics. 2001, 266 (2): 276-288.
Du L, Shen B: Biosynthesis of hybrid peptide-polyketide natural products. Curr Opin Drug Discov Devel. 2001, 4 (2): 215-228.
Trivedi OA, Arora P, Vats A, Ansari MZ, Tickoo R, Sridharan V, Mohanty D, Gokhale RS: Dissecting the mechanism and assembly of a complex virulence mycobacterial lipid. Mol Cell. 2005, 17 (5): 631-643.
Ferreras JA, Stirrett KL, Lu X, Ryu JS, Soll CE, Tan DS, Quadri LE: Mycobacterial phenolic glycolipid virulence factor biosynthesis: mechanism and small-molecule inhibition of polyketide chain initiation. Chem Biol. 2008, 15 (1): 51-61.
Rodriguez GM: Control of iron metabolism in Mycobacterium tuberculosis. Trends Microbiol. 2006, 14 (7): 320-327.
Kuzuyama T, Seto H: Two distinct pathways for essential metabolic precursors for isoprenoid biosynthesis. Proc Jpn Acad Ser B Phys Biol Sci. 2012, 88 (3): 41-52.
Wanke M, Skorupinska-Tudek K, Swiezewska E: Isoprenoid biosynthesis via 1-deoxy-D-xylulose 5-phosphate/2-C-methyl-D-erythritol 4-phosphate (DOXP/MEP) pathway. Acta Biochim Pol. 2001, 48 (3): 663-672.
Cane DE, Ikeda H: Exploration and mining of the bacterial terpenome. Acc Chem Res. 2012, 45 (3): 463-472.
Komatsu M, Tsuda M, Omura S, Oikawa H, Ikeda H: Identification and functional analysis of genes controlling biosynthesis of 2-methylisoborneol. Proc Natl Acad Sci U S A. 2008, 105 (21): 7422-7427.
Cheng Q: Structural diversity and functional novelty of new carotenoid biosynthesis genes. J Ind Microbiol Biotechnol. 2006, 33 (7): 552-559.
Kannenberg EL, Poralla K: Hopanoid biosynthesis and function in bacteria. Naturwissenschaften. 1999, 86 (4): 168-176.
Bursy J, Kuhlmann AU, Pittelkow M, Hartmann H, Jebbar M, Pierik AJ, Bremer E: Synthesis and uptake of the compatible solutes ectoine and 5-hydroxyectoine by Streptomyces coelicolor A3(2) in response to salt and heat stresses. Appl Environ Microbiol. 2008, 74 (23): 7286-7296.
Kol S, Merlo ME, Scheltema RA, De Vries M, Vonk RJ, Kikkert NA, Dijkhuizen L, Breitling R, Takano E: Metabolomic characterization of the salt stress response in Streptomyces coelicolor. Appl Environ Microbiol. 2010, 76 (8): 2574-2581.
Nakano H, Omura S: Chemical biology of natural indolocarbazole products: 30 years since the discovery of staurosporine. J Antibiot (Tokyo). 2009, 62 (1): 17-26.
Sanchez C, Mendez C, Salas JA: Indolocarbazole natural products: occurrence, biosynthesis, and biological activity. Nat Prod Rep. 2006, 23 (6): 1007-1045.
Dunbar KL, Melby JO, Mitchell DA: YcaO domains use ATP to activate amide backbones during peptide cyclodehydrations. Nat Chem Biol. 2012, 8 (6): 569-575.
Melby JO, Nard NJ, Mitchell DA: Thiazole/oxazole-modified microcins: complex natural products from ribosomal templates. Curr Opin Chem Biol. 2011, 15 (3): 369-378.
Valdes-Stauber N, Scherer S: Isolation and characterization of Linocin M18, a bacteriocin produced by Brevibacterium linens. Appl Environ Microbiol. 1994, 60 (10): 3809-3814.
Sutter M, Boehringer D, Gutmann S, Gunther S, Prangishvili D, Loessner MJ, Stetter KO, Weber-Ban E, Ban N: Structural basis of enzyme encapsulation into a bacterial nanocompartment. Nat Struct Mol Biol. 2008, 15 (9): 939-947.
Whittle M, Willett P, Klaffke W, Van Noort P: Evaluation of similarity measures for searching the Dictionary of Natural Products database. J Chem Inf Comput Sci. 2003, 43 (2): 449-457.
Perez-Miranda S, Cabirol N, George-Tellez R, Zamudio-Rivera LS, Fernandez FJ: O-CAS, a fast and universal method for siderophore detection. J Microbiol Methods. 2007, 70 (1): 127-131.
Giessen TW, Von Tesmar AM, Marahiel MA: Insights into the Generation of Structural Diversity in a tRNA-Dependent Pathway for Highly Modified Bioactive Cyclic Dipeptides. Chem Biol. 2013, 20 (6): 828-838.
Belin P, Moutiez M, Lautru S, Seguin J, Pernodet JL, Gondry M: The nonribosomal synthesis of diketopiperazines in tRNA-dependent cyclodipeptide synthase pathways. Nat Prod Rep. 2012, 29 (9): 961-979.
Gondry M, Sauguet L, Belin P, Thai R, Amouroux R, Tellier C, Tuphile K, Jacquet M, Braud S, Courcon M, Masson C, Dubois S, Lautru S, Lecoq A, Hashimoto S, Genet R, Pernodet JL: Cyclodipeptide synthases are a family of tRNA-dependent peptide bond-forming enzymes. Nat Chem Biol. 2009, 5 (6): 414-420.
Kieser TBMJ, Buttner MJ, Charter KF, Hopwood D: Practical Streptomyces Genetics. 2000, Norwich, United Kingdom: John Innes Foundation
Schwientek P, Szczepanowski R, Rückert C, Stoye J, Puhler A: Sequencing of high G + C microbial genomes using the ultrafast pyrosequencing technology. J Biotechnol. 2011, 155 (1): 68-77.
Gordon D, Abajian C, Green P: Consed: A graphical tool for sequence finishing. Genome Res. 1998, 8 (3): 195-202.
Krause L, McHardy AC, Nattkemper TW, Puhler A, Stoye J, Meyer F: GISMO - gene identification using a support vector machine for ORF classification. Nucleic Acids Res. 2007, 35 (2): 540-549.
Meyer F, Goesmann A, McHardy AC, Bartels D, Bekel T, Clausen J, Kalinowski J, Linke B, Rupp O, Giegerich R, Puhler A: GenDB - an open source genome annotation system for prokaryote genomes. Nucleic Acids Res. 2003, 31 (8): 2187-2195.
Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ: Basic Local Alignment Search Tool. J Mol Biol. 1990, 215 (3): 403-410.
Kearse M, Moir R, Wilson A, Stones-Havas S, Cheung M, Sturrock S, Buxton S, Cooper A, Markowitz S, Duran C, Thierer T, Ashton B, Meintjes P, Drummond A: Geneious Basic: an integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics. 2012, 28 (12): 1647-1649.
Ostlund G, Schmitt T, Forslund K, Kostler T, Messina DN, Roopra S, Frings O, Sonnhammer ELL: InParanoid 7: new algorithms and tools for eukaryotic orthology analysis. Nucleic Acids Res. 2010, 38: D196-D203.
Alexeyenko A, Tamas I, Liu G, Sonnhammer ELL: Automatic clustering of orthologs and inparalogs shared by multiple proteomes. Bioinformatics. 2006, 22 (14): E9-E15.
Anand S, Prasad MV, Yadav G, Kumar N, Shehara J, Ansari MZ, Mohanty D: SBSPKS: structure based sequence analysis of polyketide synthases. Nucleic Acids Res. 2010, 38: W487-W496.
Rottig M, Medema MH, Blin K, Weber T, Rausch C, Kohlbacher O: NRPSpredictor2--a web server for predicting NRPS adenylation domain specificity. Nucleic Acids Res. 2011, 39: W362-W367.
Rebets Y, Ostash B, Luzhetskyy A, Hoffmeister D, Brana A, Mendez C, Salas JA, Bechtold A, Fedorenko V: Production of landomycins in Streptomyces globisporus 1912 and S cyanogenus S136 is regulated by genes encoding putative transcriptional activators. FEMS Microbiol Lett. 2003, 222 (1): 149-153.
Acknowledgements
This work was supported through funding from the BMBF grant (GenomikPlus) to JK and AL and from the ERC starting grant EXPLOGEN No. 281623 to AL.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
YR performed manual cluster and gene annotation, analyzed and described organization of the biosynthetic clusters, prepared figures, tables, supplementary materials and wrote the manuscript. BT performed orthology and eggNOG analyses, prepared GenBank submission, figures, and data for tables. CR performed sequencing, assembly, finishing and mapping/polishing. IL performed phylogeny analysis. JK, NZ and AB analyzed the sequence data and helped with the manuscript writing. AL proposed the study, participated in its design and coordination and has given the final approval of the manuscript. All authors read and approved the final manuscript.
Yuriy Rebets, Bogdan Tokovenko contributed equally to this work.
Electronic supplementary material
12864_2014_6593_MOESM1_ESM.pdf
Additional file 1: Table S1: Annotation and prediction of gene functions of aculeximycin biosynthesis gene cluster. Locus number is the numeric part of the locus_tag (e.g. locus number 6558 refers to gene KALB_6558). Table S2. Classification and characterization of secondary metabolism (SM) gene clusters identified in Kutzneria albida DSM43870 genome. Figure S1. HPLC-MS analysis of extracts from K. albida culture grown in different media. UV–vis traces are shown. Masses of some compound are marked. Compounds that have hits in DNP are also indicated. Figure S2. HPLC-MS analysis of extracts from K. albida culture grown in different media. Base peak chromatogram traces are shown. Masses of some compound are marked. Aculeximycin and its aglycone are marked as Acu and AcuA respectively. Cyclic dipeptides are marked as T1 (cFL), T2 (cFY) and T3 (cFF). Figure S3. A. Modified CAS siderophore production test. Test was performed as described in [1]. Change in color was monitored after 3 hours. Orange color indicates siderophores accumulation. B. TLC analysis of extracts from K. albida cultures grown in different media (1 – TSB supernatant, 2 – TSB biomass, 3 – NL5 supernatant, 4 – NL5 biomass, 5 – NL19 supernatant, 6 – NL19 biomass, 7 – NL111 supernatant, 8 – NL111 biomass, 9 – CAS supernatant, 10 – CAS biomass, 11 – SG supernatant). The solvent phase was acetone-methanol 9:1. Plate was overlaid with 0.8% CAS agar. Changes in color were monitored after 30 minutes of incubation. (PDF 1 MB)
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
This article is published under an open access license. Please check the 'Copyright Information' section either on this page or in the PDF for details of this license and what re-use is permitted. If your intended use exceeds what is permitted by the license or if you are unable to locate the licence and re-use information, please contact the Rights and Permissions team.
About this article

Cite this article
Rebets, Y., Tokovenko, B., Lushchyk, I. et al. Complete genome sequence of producer of the glycopeptide antibiotic Aculeximycin Kutzneria albida DSM 43870T, a representative of minor genus of Pseudonocardiaceae. BMC Genomics 15, 885 (2014). https://doi.org/10.1186/1471-2164-15-885
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1471-2164-15-885