
Abstract
Background
In Pseudomonas putida and Pseduomonas aeruginosa, the similar PpuR/RsaL/PpuI and LasR/RsaL/LasI acyl homoserine lactones (AHLs) quorum sensing (QS) systems have been shown to be under considerable regulation by other global regulators. A major regulator is the RsaL protein which strongly directly represses the transcription of the P. putida ppuI and P. aeruginosa lasI AHL synthases. In this study we screened a transposon mutant bank of P. putida in order to identify if any other regulators were involved in negative regulation of AHL QS.
Results
In our screen we identified three Tn5 mutants which displayed overproduction of AHLs in P. putida strain WCS358. Two of the mutants had a Tn5 located in the rsaL gene, whereas in one mutant the transposon was located in the lon protease gene. Lon proteases play important roles in protein quality control via degradation of misfolded proteins. It was determined that in the P. putida lon mutant, AHL levels, PpuR levels and ppuI promoter activity all increased significantly; we therefore postulated that PpuR is a target for Lon. The Lon protease had no effect on AHL production in P. aeruginosa.
Conclusion
The Lon protease is a negative regulator of AHL production in P. putida WCS358. The Lon protease has also been shown by others to influence AHL QS in Vibrio fischeri and Agrobacterium tumefaciens and can thus become an important regulator of AHL QS timing and regulation in bacteria.
Similar content being viewed by others

Background
Quorum sensing (QS) is a common form of gene regulation based on cell-density involving intercellular communication relying on the production and response to signaling molecules [1, 2]. In Gram-negative bacteria, acyl-homoserine lactones (AHLs) are the most common signal molecules which were first described in the marine bacterium Vibrio fischeri as being involved in the cell-density dependent regulation of bioluminescence [1, 3]. The general mechanism of AHL QS relies on two proteins belonging to the LuxI and LuxR protein families. LuxI-family proteins are the major class of AHL synthase enzymes whereas LuxR-family proteins form complexes with AHLs which are then able to bind at specific DNA promoter sequences (called lux-type boxes) of QS regulated genes affecting their expression.
AHL QS has become a paradigm for bacterial communication having the common scheme that AHLs are produced at a basal level at low cell densities. At high cell densities, the concentrations of AHLs surpasses a certain threshold (corresponding to the "quorum" cell density) allowing interaction with the LuxR-family protein and the system then usually undergoes positive feedback through increase of expression of the luxI-family gene resulting in strong sudden activation of AHL QS. This cell-density dependent response has evolved as a means to provide advantages to a community of bacteria by synchronizing group behavior. AHL QS has been studied in several Gram-negative bacteria and the physiological processes controlled by this system are diverse but are often related to virulence in pathogenic organisms [1, 2, 4, 5].
The AHL QS systems are not always rigorously responding to cell-density as they are often integrated with other global regulatory responses and are thus influenced by other environmental factors [6–8]. Several systematic studies in Pseudomonas aeruginosa have shown that AHL QS is a global regulatory network controlling the expression of over 300 genes [9–11]. Recently other regulon studies in P. aeruginosa have demonstrated that the RpoS, VqsR and PprB global regulators are intimately interconnected with AHL QS regulons [8]. This is probably why some AHL QS systems are themselves regulated in response to various stimuli ensuring a timely control at the appropriate environmental conditions. In fact, the regulation of AHL QS has been particularly studied in P. aeruginosa highlighting that the luxI/LuxI and luxR/LuxR family genes/proteins are themselves extensively regulated. Positive regulation of the lasI/R and rhlI/R, the two AHL QS systems homologs of luxI/R present in P. aeruginosa, occurs via transcriptional regulators such as GacA, Vfr and PprB (reviewed by [6–8]). At present however it is not known whether any of these positive regulators are acting directly on the AHL QS genes and the precise stimuli affecting these positive regulatory responses are also not clear. Other regulators repress AHL QS in P. aeruginosa likely to ensure that it is not activated at low cell-densities. Reports of these negative regulators include the H-NS like protein MvaT, the luxR-like orphan QscR, the post-transcriptional regulator RsmA, the alternative sigma factors RpoS and RpoN, and the newly characterized RsaL repressor (reviewed by [6–8]). Of these repressors only RsaL has been shown to regulate directly the AHL QS genes; more precisely it regulates the expression of the lasI AHL synthase by binding to its promoter repressing transcription [12, 13]. Interestingly we also reported that RsaL is a major negative regulator of AHL QS in Pseudomonas putida [14]. Unlike in P. aeruginosa however, in strain WCS358 there is only one AHL QS system, designated PpuI/R, which produces and responds to N-3-oxo-dodecanoyl homoserine lactone (C12-3-oxo-AHL); this system is highly identical to the LasI/R system of P. aeruginosa [13, 14]. Localized in between the ppuI/R genes is the small rsaL repressor which when inactivated results in dramatic increase of ppuI expression and hence AHL production [14]. In P. putida RsaL repression of the AHL synthase gene appears to be much stronger that in P. aeruginosa [12, 14].
In this study we were interested to investigate whether in P. putida WCS358 other negative ppuI/R (or PpuI/R) AHL QS regulators, either acting in concert with RsaL or independently, are present. By screening a Tn5 genomic mutant bank of strain WCS358 a negative regulatory mutant of AHL QS was identified and characterized to be inactivating a gene encoding for a Lon-like protease. It was demonstrated that this protease targeted PpuR thus affecting PpuR protein levels indicating that it is involved in regulating the AHL QS system.
Results and Discussion
Identification and characterization of AHL-overproducing mutants of P. putida WCS358
In order to establish whether, besides RsaL, there were other negative regulators of the ppuI/R system, we screened P. putida WCS358 Tn5 genomic mutants for AHL overproduction. The genetic screen we employed here has been previously described relying on the AHL biosensor C. violaceum CVO26 [14, 15]. P. putida WCS358 promotes little pigment formation in the AHL biosensor strain CVO26 because it produces very low quantities of AHLs as well as producing AHLs which have low specificity towards CviR of strain CVO26. In order to identify AHL-overproducing mutants we spread between 1000–2000 CVO26 cfu (c olony f orming u nits) and 300–500 P. putida WCS358 Tn5 mutant cfu from a Tn5 mutant bank on one plate and screened for strong purple loci. After screening 25,000 WCS358 Tn5 mutants, three mutants were scored which significantly induced violacein purple pigment production in CVO26. Two mutants were localized in the rsaL gene which as previously reported is an important negative regulator of ppuI expression and its inactivation leads to a dramatic increase of AHL production [14]. One mutant did not map in the rsaL gene and had the Tn5 inserted in an ORF of 2397 nucleotides encoding a protein of 798 amino acids (Figure 1). This ORF displayed high homology to the Lon proteases of several bacteria; over 90 % identity with proteins form several Pseudomonas sp. including the Lon protease of P. aeruginosa ([16]; PA1803). The Lon protein belongs to the family of ATP-dependent proteases which is well conserved in prokaryotes and eukaryotes and has been associated with various cellular activities [17]. Lon proteases play important roles in protein quality control via degradation of misfolded proteins. Lon proteolysis can also be crucial for controlling the protein levels of regulatory proteins thus affecting programs of gene expression [17].

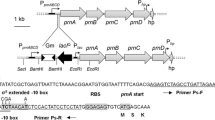
Physical map of the 4.4 kb EcoRI DNA fragment containing the lon protease cloned from the chromosome of P. putida WCS358. Position of the first and last nucleotide of the lon gene are shown together with the position of the Tn5 insertion. The ORF or part of ORFs downstream the lon gene display 88% and 92% similarity to the pp_2303 and pp_2304 ORFs of P. putida KT2440. In the upstream region of the lon gene, part of clpX ORF is localized.
This lon protease mutant identified here was designated P. putida IBE4 and produced three times more C12-3-oxo-AHL as determined through AHL quantification experiments as well as through TLC analysis (Figure 2 and Figure 4). This increase of AHL production was also justified by higher ppuI promoter activity; ppuI encodes for PpuI the AHL synthase enzyme (Figure 3A). As previously reported [14]ppuI expression does not increase with cell-density and is low in the wild-type as the QS system is strongly negatively regulated by RsaL. In fact in P. putida RsaL appears to be the on/off switch of the system. It not known yet what is the stimulus which leads to RsaL de-repression. It is clear however form our data that absence of Lon increases significantly ppuI transcription. Similarly, also the rsaL quorum sensing PpuR-regulated promoter displayed significantly higher activities in the lon protease mutant (Figure 3B). Importantly, by providing the lon protease in trans via plasmid pBBRlon, which carries the cloned genomic locus harbouring the lon protease gene expressed from its own promoter, C12-3-oxo-AHL levels, and ppuI and rsaL promoter activities were restored to wild-type levels (Figures 2, 3 and 4). From the TLC analysis it was actually observed that extra copies of lon gene carried in a plasmid dramatically reduced AHL production (Figure 4). It was therefore postulated that Lon could target some component(s) of the PpuI/RsaL/PpuI AHL system being therefore involved in QS regulation in P. putida.

C12-3-oxo-AHL measurement produced by P. putida WCS358, by lon protease mutant derivative P. putida IBE4 and lon mutant containing a plasmid expressing the Lon protease, P. putida IBE4 (pBBRlon). C12-3-oxo-AHL was extracted from spent supernatants, AHL levels were measured with P. putida SM17 (prsaL220) with a volume of extract corresponding to an amount of 5 × 108 cfu as described in the Methods section. C12-3-oxo-AHL levels are proportional to β-galactosidase activity (Miller Units). Standard deviation bars are given on the mean value of three independent cultures. Statistical analysis was performed with Anova resulting in a significant main effect of the mutation with F(2,12) = 20.45 and p < .001. The same measurement was also performed using 1 μM of synthetic C12-3-oxo-AHL (obtained from P. Williams, University of Nottingham, UK).

Gene promoter activities of ppuI and rsaL in P. putida WCS358 and lon mutant derivatives. A. Promoter activities of the ppuI C12-3-oxo-AHL synthase in P. putida WCS358 (pPPUI220), in the lon protease mutant derivative P. putida IBE4 (pPPUI220) and lon mutant containing a plasmid expressing the Lon protease, P. putida IBE4 (pPPUI220)(pBBRlon). The ppuI promoter activities are expressed in β-galactosidase activity (Miller Units) as the plasmid construct pPPUI220 contains the ppuI promoter transcriptionally fused to a promoterless lacZ gene (see text for all details). Standard deviation bars are given on the mean value of three independent experiments. Logarithmic phase (Log.) corresponds to 8 hours of growth, stationary phase (Stat.) to 16 hrs and late stationary (Late stat.) to 48 hours of growth. Statistical analysis was performed with Anova resulting in a significant main effect of the mutation with F(1,14) = 18.90 and p < .001. Statistical analysis including the age of the cultures results in a significant difference between the wild type and the IBE4 mutant in the Logarithmic phase of growth [F(1,14) = 28.70 p < .001]. B. Promoter activities of the rsaL in P. putida WCS358 (pRSA220), in the lon protease mutant derivative P. putida IBE4 (pRSA220) and lon mutant containing a plasmid expressing the Lon protease, P. putida IBE4 (pRSA220)(pBBRlon). The ppuI promoter activities are expressed in β-galactosidase activity (Miller Units) as the plasmid construct pRSA220 contains the rsaL promoter transcriptionally fused to a promoterless lacZ gene. Standard deviation bars are given on the mean value of three independent experiments. Logarithmic phase (Log.) corresponds to 8 hours of growth, stationary phase (Stat.) to 16 hrs and late stationary (Late stat.) to 48 hours of growth. Statistical analysis was performed with Anova resulting in a significant main effect of the mutation with F(1,18) = 126.12 and p < .000001. Statistical analysis including the age of the cultures results in a significant difference between the wild type and the IBE4 mutant in the late stationary phase of growth [F(1,18) = 18.79 p < .001].

TLC analysis of C12-3-oxo-AHL produced by parent strain P. putida WCS358 (lane 1), by lon mutant derivative P. putida IBE4 (lane 2) and lon mutant derivative carrying a plasmid expressing Lon P. putida IBE4 (pBBRlon) (lane 3). Lane 4 contains the standards C10-3-oxo-AHL and C12-3-oxo-AHL. The AHLs were visualized with AHL-sensor strain E. coli (pSB1075); a volume corresponding to 5 × 108 cfu was loaded on the TLC assay.
The P. putida Lon protease affects PpuR protein levels
Having established that the lon knock-out mutant P. putida IBE4 resulted in increased ppuI promoter activity and hence higher AHL production it was of interest to determine if the possible target of the Lon protease was the PpuR protein. The reason being that as PpuR positively regulates ppuI and rsaL expression in a positive induction loop typical of LuxI/R-type QS systems, altering PpuR levels could therefore affect AHL production through ppuI and rsaL promoter activities. In order to determine if PpuR was a target of the Lon protease we raised polyclonal antibodies against PpuR in order to visualize PpuR protein levels. We therefore examined levels of PpuR in 16-h-old stationary phase cultures of P. putida WCS358 and lon protease knock-out mutant P. putida IBE4 using anti-PpuR antibodies. The levels of PpuR protein were found to be very low and hardly detectable in strain WCS358; this also reflected the very low ppuR promoter activities which were previously observed [14]. In order to be able to better visualize PpuR levels we introduced in both the wild type and mutant derivative IBE4, the plasmid pBBRppuR which carries the ppuR gene expressed from the lac promoter (Figure 1; Table 1). As depicted in Figure 5, PpuR levels in P. putida IBE4 were detected as being significantly higher than those in the wild type parent strain indicating that absence of the Lon protease resulted in larger amounts of PpuR present in the cell. This experiment was reproducible being repeated three times obtaining similar results also loading different amounts of total proteins (data not shown). We verified that ppuR promoter activity in the lon mutant was not affected (data not shown) in order to exclude that Lon might have been acting on other proteins components affecting ppuR transcription. This provides indirect evidence that the Lon protease targets PpuR thus being able to regulate quorum sensing in P. putida. Other LuxR-family quorum sensing regulators have also been shown to be targeted by the Lon protease. The TraR protein of Agrobacterium tumefaciens was shown to be susceptible to Lon proteolysis when free of AHL-ligand [18]. Similarly, LuxR of Vibrio fischeri complexes in vivo with Lon degrading it; in a Lon mutant it was also observed that LuxR accumulates at higher levels [19]. It therefore appears that many members of this family could be targets of the Lon protease in order to control their levels; Lon can consequently be regarded as a regulator of quorum sensing ensuring the correct timing of the response. At present we do not know whether the lon/Lon protease is itself regulated thus adding a further element(s) of control of QS in P. putida. Regulation of QS in Pseudomonas has been shown to be very complex and intricate involving a myriad of global regulators; this indicates that timing and response of the QS system is important and the Lon protease must now be added to this regulatory circuit. Other proteases have also been implicated in targeting members of the LuxR-protein family [18]. Lon has also been reported to affect the accumulation of several other types of transcriptional regulators in bacteria thus affecting transcription of important global regulatory systems [17, 20–22]. For example, Lon influences the regulation of antibiotic production in Pseudomonas fluorescens Pf-5 through what is postulated to be degradation of of a positive regulator [21]. In P. aeruginosa, the Lon protease was found to be important for biofilm formation and motility [20] and pathogenicity and type III secretion in Pseudomonas syringae are regulated by Lon via an effect on the stability of transcriptional regulators [19].

Western hybridization performed using anti-PpuR antibody on total cellular protein extracts. Total protein extracts were performed using different protein amounts of either parent strain P. putida WCS358 or lon mutant derivative P. putida IBE4 each carrying a plasmid expressing PpuR (pBBRppuR). Lane 1 corresponds to 10 μg of purified PpuR-6His; lane 2 and lane 3 correspond to total proteins from 2 × 107 cfu of WCS358(pBBRppuR) and IBE4(pBBRppuR) respectively; lanes 4 and 5 correspond to total proteins from 107 cfu. For lanes 4 and 5 the images were scanned using a Versadoc (Biorad) and the QuantityOne software; results of this quantification are displayed as a histogram. See text for all details.
The P. aeruginosa Lon protease does not affect C12-3-oxo-AHL production
The Lon protease of P. putida WCS358 displayed almost 90% amino acid identity over the entire length of the protein with the Lon protease of P. aeruginosa (PA1803; [16]). In addition, the PpuI/RsaL/PpuR AHL QS system of strain WCS358 is highly similar to the LasI/RsaL/LasR of P. aeruginosa; they both produce and respond to C12-3-oxo-AHL and the three proteins are highly identical [12–14]. Since the two species are very close phylogenetically and the two AHL QS systems are orthologs with PpuR and LasR being highly identical, it was of interest to determine if also in P. aeruginosa the Lon protease played a role in AHL QS regulation. We therefore determined C12-3-oxo-AHL levels in wild-type P. aeruginosa PAO1 and lon protease mutant P. aeruginosa 18577. Differently to what occurs in P. putida WCS358, the lon protease knock-out mutant of P. aeruginosa produced comparable amounts of C12-3-oxo-AHL to wild-type parent strain PAO1 (Figure 6). This indicated that unlike what occurred in P. putida WCS358, in P. aeruginosa, Lon most probably does not target proteins of the LasI/RsaL/LasR AHL QS system. It was observed however that in the P. aeruginosa PAO1 genome, an ORF of 795 amino acids (PA0779) displayed 40% identity with the Lon protease is present thus it cannot be excluded that other similar proteases are present which can target protein(s) of AHL QS systems.

C12-3-oxo-AHL measurement produced by of P. aeruginosa PAO1 and by lon protease mutant derivative P. aeruginosa 18577. C12-3-oxo-AHL was extracted from spent supernatants, AHL levels were measured with P. putida SM17 (prsaL220) with a volume of extract corresponding to an amount of 5 × 108 cfu as described in the Methods section. C12-3-oxo-AHL levels is proportional to β-galactosidase activity (Miller Units). Standard deviation bars are given on the mean value of three independent cultures. Differences between PAO1 and PAO1 ID18577, analysed using T-test for independent groups were not significant (t = 2.2; p = .06; df = 6;t(6) = 2.2). The same measurement was also performed using 1 μM of synthetic C12-3-oxo-AHL (obtained from P. Williams, University of Nottingham, UK).
Conclusion
In this study we determined that the Lon protease is a negative regulator of AHL production in P. putida WCS358. AHL production and response in WCS358 occurs via the PpuR/RsaL/PpuI AHL QS system; this system is highly homologous to the LasR/RsaL/LasI system of P. aeruginosa. It was observed that in a Lon mutant, C12-3-oxo-AHL levels, PpuR levels and ppuI promoter activity all increase significantly; we therefore postulated that PpuR is a target for Lon. Unlike what occurs in P. putida WCS358, in P. aeruginosa Lon has no effect on AHL production. It was however observed that P. aeruginosa possesses in its genome another ORF with very similar features to Lon thus it cannot be excluded that other proteases could be involved in AHL QS regulation. As two other LuxR family regulators have been shown to be targeted by Lon, it is concluded that proteases could play an important role in AHL QS timing and regulation.
Methods
Bacterial strains, plasmids and media
The bacterial strains and plasmids used in this study are listed in Table 1. Pseudomonas putida WCS358 is a plant growth-promoting strain isolated from the rhizosphere of potato roots.Chromobacterium violaceum CVO26 is a double mini-Tn5 mutant derived from ATCC31532, this mutant is non-pigmented and production of the purple pigment can be induced by providing exogenous AHL inducer molecules [23]. Escherichia coli DH5α [24]and C. violaceum CV026 were grown in LB medium [25] at 37°C and 30°C respectively. Pseudomonas was grown in LB medium or M9 minimal medium [25] at 30°C. The following antibiotic concentrations were used: ampicillin (Amp) 100 μg/ml; kanamycin (Km) 100 μg/ml; nalidixic acid (Nx) 25 μg/ml; tetracycline (Tc) 10 μg/ml (E. coli), 40 μg/ml (Pseudomonas); chloramphenicol (Cm) 25 μg/ml (E. coli), 250 μg/ml (Pseudomonas); gentamycin (Gm) 10 μg/ml (E. coli), 40 μg/ml (Pseudomonas).
Recombinant DNA techniques
DNA manipulation as digestion with restriction enzymes, agarose gel electrophoresis, purification of DNA fragments, ligations with T4 ligase, end-filling with Klenow enzyme, hybridization, radioactive labeling by random priming and transformation of E. coli, were performed as described previously [25]. Southern hybridizations were performed using N+Hybond membrane (Amersham Biosciences); plasmids were purified using the Jet star colums (Genomed, GmbH, Germany) or with the alkaline lysis method [26]; total DNA from Pseudomonas was isolated by Sarkosyl/Pronase lysis as described previously [27]. Triparental matings between E. coli and P. putida were carried out with the helper strain E. coli DH5α (pRK2013) [28].
Isolation of an AHL over-expressing genomic mutant and cloning of a lon-like protease of P. putida WCS358
In order to identify P. putida WCS358 mutants that overproduce AHL, a Tn5 genomic mutant library was screened against the AHL biosensor strain C. violaceum CVO26. P. putida WCS358 induced only slightly pigmentation when streaked in close proximity to strain C. violaceum CVO26. This screening for AHL over-producers used here was previously employed to isolate rsaL mutants on P. putida WCS358 [14]. 25,000 Tn5 genomic mutants of P. putida WCS358 were screened against CVO26 for AHL overproducer mutants as previously described [14, 15]. This led to the identification of one Tn5 mutant (designated P. putida IBE4), of which the Tn5 was not located in the rsaL gene and could induce strongly pigmentation of CVO26. The position of the Tn5 was determined after cloning from the mutant IBE4 chromosome a 4.2 kb BamHI-StuI fragment in pBluescript KS yielding pSCHIA5, which contained part of Tn5 (including the Km resistance gene) and part of adjacent WCS358 DNA. The adjacent DNA to the Tn5 was sequenced using as primer the oligonucleotide sequence Tn5ext (see Table 1) which was designed against the border of the Tn5 DNA sequence. The 4.2 kb BamHI-StuI fragment was then used as a probe to clone a 4.5 kb EcoRI from the chromosome of parent strain WCS358 in pBluescript KS yielding pB1A. This latter EcoRI fragment was sequenced (sequencing service, CRIBI, University of Padua, Padua, I) revealing that the Tn5 was positioned within an ORF of 2397 nucleotides encoding for a putative Lon-like protease of 799 amino acids (Figure 1).
Reporter gene fusion assay
β-galactosidase activities were determined during growth in LB medium essentially as described by Miller et al., [29] with the modifications of Stachel et al. [30]. All experiments were performed in triplicate and the mean value is given. The growth curves of all mutants were comparable to the one obtained for the parent strain.
Purification and quantification of C12-3-oxo-AHL
The purification, detection and characterization of AHLs were performed as previously described [23, 31]. Pseudomonas strains were grown O/N in M9 minimal medium supplemented with citric acid and the OD600 was measured. The spent culture supernatants were extracted two times with the same culture volume of ethyl acetate-0.1% acetic acid. The extracts were then dried and resuspended in ethyl acetate with an amount which resulted in 1 μl of final extract corresponding to 2 × 107 cells of the original culture. The quantity of C12-3-oxo-AHL in the extracts was determined using the specific 3-oxo-C12-AHL sensor P. putida SM17 (prsal220) as previously described [13]. Briefly, P. putida SM17 is a double mutant of ppuI and rsaL genes consequently it does not produce the RsaL repressor and 3-oxo-C12-AHL. Adding exogenous 3-oxo-C12-AHL is quantified through β-galactosidase activity by using stain SM17 harboring (prsal220); this plasmid contains the PpuR-3-oxo-C12-AHL regulated rsaL promoter fused to promoterless lacZ gene. Overnight cultures of SM17(prsal220) were diluted in 10 ml of LB medium to an A660 of 0.1; the AHL extract to be quantified was then added and after 6 hours of growth β-galactosidase activity was determined. This 3-oxo-C12-AHL bacterial sensor has a linear dose response between 0.1 μM to 5 μM of 3-oxo-C12-AHL. Synthetic 3-oxo-C12-AHL was used as standard molecules (obtained from P. Williams, University of Nottingham, UK). The detection of the AHLs on the TLC plate was obtained overlaying the TLC plate with a thin layer of LB top-agar seeded with E. coli (pSB1075) as previously described [14, 31, 32].
PpuR antibodies and protein anaylsis
Antibodies against PpuR of P. putida were generated by injecting purified protein into rabbits. P. putida PpuR was purified as PpuR-His6 using expression plasmid pET28b (Novagen); ppuR was cloned as a 729 PCR fragment originated from primers FW121 and FW122 using P. putida WC358 genomic DNA as template and was cloned as a NcoI-HindIII fragment in pET28b yielding pPPUR3586H. This latter plasmid was introduced in E. coli BL21(DE3)pLys (Novagen) which then resulted in the isopropyl-β-D-thiogalactopyranoside-induced over-expression of PpuR tagged with six histidines at the C- terminus. The purification of PpuR-His6 was then carried out by Ni 2+ affinity under denaturing conditions according to the standard procedure suggested by the column manufacturer (Sigma-Aldrich, St. Louis, Mo.).
Proteins were transferred onto PVDF membrane (Immobilon-P; Millipore) using a tank system according to the manufacturer's instruction. The membrane was subjected to Western analysis using anti-PpuR polyclonal antibodies raised in rabbits and polyclonal goat anti rabbit immunoglobulins HRP (DakoCytomation, Glostrup, DK) and developed using Immun-Star HRP Substrate kit (BioRad laboratories, Hercules CA, USA). No significant cross-reaction of the polyclonal antibody against other P. putida WCS358 proteins was observed in this study.
DNA sequencing and nucleotide sequence accession numbers
All DNA sequences were performed at the CRIBI center (University of Padua, Italy) and the nucleotide sequence of the 4.414 EcoRI fragment has been deposited in GenBank/EMBL/DDBJ under the following accession number AM690373.
References
Fuqua C, Greenberg EP: Listening in on bacteria: acyl-homoserine lactone signalling. Nat Rev Mol Cell Biol. 2002, 3 (9): 685-695. 10.1038/nrm907.
Waters CM, Bassler BL: Quorum sensing: cell-to-cell communication in bacteria. Annu Rev Cell Dev Biol. 2005, 21: 319-346. 10.1146/annurev.cellbio.21.012704.131001.
Ruby EG: Lessons from a cooperative, bacterial-animal association: the Vibrio fischeri-Euprymna scolopes light organ symbiosis. Annu Rev Microbiol. 1996, 50: 591-624. 10.1146/annurev.micro.50.1.591.
Camara M, Williams P, Hardman A: Controlling infection by tuning in and turning down the volume of bacterial small-talk. Lancet Infect Dis. 2002, 2 (11): 667-676. 10.1016/S1473-3099(02)00447-4.
Von Bodman SB, Bauer WD, Coplin DL: Quorum sensing in plant-pathogenic bacteria. Annu Rev Phytopathol. 2003, 41: 455-482. 10.1146/annurev.phyto.41.052002.095652.
Juhas M, Eberl L, Tummler B: Quorum sensing: the power of cooperation in the world of Pseudomonas. Environ Microbiol. 2005, 7 (4): 459-471. 10.1111/j.1462-2920.2005.00769.x.
Lazdunski AM, Ventre I, Sturgis JN: Regulatory circuits and communication in Gram-negative bacteria. Nat Rev Microbiol. 2004, 2 (7): 581-592. 10.1038/nrmicro924.
Venturi V: Regulation of quorum sensing in Pseudomonas. FEMS Microbiol Rev. 2006, 30 (2): 274-291. 10.1111/j.1574-6976.2005.00012.x.
Hentzer M, Wu H, Andersen JB, Riedel K, Rasmussen TB, Bagge N, Kumar N, Schembri MA, Song Z, Kristoffersen P, Manefield M, Costerton JW, Molin S, Eberl L, Steinberg P, Kjelleberg S, Hoiby N, Givskov M: Attenuation of Pseudomonas aeruginosa virulence by quorum sensing inhibitors. Embo J. 2003, 22 (15): 3803-3815. 10.1093/emboj/cdg366.
Schuster M, Lostroh CP, Ogi T, Greenberg EP: Identification, timing, and signal specificity of Pseudomonas aeruginosa quorum-controlled genes: a transcriptome analysis. J Bacteriol. 2003, 185 (7): 2066-2079. 10.1128/JB.185.7.2066-2079.2003.
Wagner VE, Bushnell D, Passador L, Brooks AI, Iglewski BH: Microarray analysis of Pseudomonas aeruginosa quorum-sensing regulons: effects of growth phase and environment. J Bacteriol. 2003, 185 (7): 2080-2095. 10.1128/JB.185.7.2080-2095.2003.
Rampioni G, Bertani I, Zennaro E, Polticelli F, Venturi V, Leoni L: The quorum-sensing negative regulator RsaL of Pseudomonas aeruginosa binds to the lasI promoter. J Bacteriol. 2006, 188 (2): 815-819. 10.1128/JB.188.2.815-819.2006.
Rampioni G, Polticelli F, Bertani I, Righetti K, Venturi V, Zennaro E, Leoni L: The Pseudomonas Quorum-Sensing Regulator RsaL Belongs to the Tetrahelical Superclass of H-T-H Proteins. J Bacteriol. 2007, 189 (5): 1922-1930. 10.1128/JB.01552-06.
Bertani I, Venturi V: Regulation of the N-Acyl Homoserine Lactone-Dependent Quorum-Sensing System in Rhizosphere Pseudomonas putida WCS358 and Cross-Talk with the Stationary-Phase RpoS Sigma Factor and the Global Regulator GacA. Appl Environ Microbiol. 2004, 70 (9): 5493-5502. 10.1128/AEM.70.9.5493-5502.2004.
Yao F, Zhou H, Lessie TG: Characterization of N-acyl homoserine lactone overproducing mutants of Burkholderia multivorans ATCC 17616. FEMS Microbiol Lett. 2002, 206 (2): 201-207. 10.1111/j.1574-6968.2002.tb11010.x.
http://www.pseudomonas.com: .
Tsilibaris V, Maenhaut-Michel G, Van Melderen L: Biological roles of the Lon ATP-dependent protease. Res Microbiol. 2006, 157 (8): 701-713. 10.1016/j.resmic.2006.05.004.
Zhu J, Winans SC: The quorum-sensing transcriptional regulator TraR requires its cognate signaling ligand for protein folding, protease resistance, and dimerization. Proc Natl Acad Sci U S A. 2001, 98 (4): 1507-1512. 10.1073/pnas.98.4.1507.
Manukhov IV, Kotova VY, Zavilgelsky GB: GroEL/GroES chaperone and Lon protease regualte expression of the Vibrio fischeri lux operon in Escherichia coli. Molec Biol. 2006, 40: 277-283.
Bretz J, Losada L, Lisboa K, Hutcheson SW: Lon protease functions as a negative regulator of type III protein secretion in Pseudomonas syringae. Mol Microbiol. 2002, 45 (2): 397-409. 10.1046/j.1365-2958.2002.03008.x.
Marr AK, Overhage J, Bains M, Hancock RE: The Lon protease of Pseudomonas aeruginosa is induced by aminoglycosides and is involved in biofilm formation and motility. Microbiology. 2007, 153 (Pt 2): 474-482. 10.1099/mic.0.2006/002519-0.
Whistler CA, Stockwell VO, Loper JE: Lon protease influences antibiotic production and UV tolerance of Pseudomonas fluorescens Pf-5. Appl Environ Microbiol. 2000, 66 (7): 2718-2725. 10.1128/AEM.66.7.2718-2725.2000.
McClean KH, Winson MK, Fish L, Taylor A, Chhabra SR, Camara M, Daykin M, Lamb JH, Swift S, Bycroft BW, Stewart GS, Williams P: Quorum sensing and Chromobacterium violaceum: exploitation of violacein production and inhibition for the detection of N-acylhomoserine lactones. Microbiology. 1997, 143 ( Pt 12): 3703-3711.
Hanahan D: Studies on transformation of Escherichia coli with plasmids. J Mol Biol. 1983, 166 (4): 557-580. 10.1016/S0022-2836(83)80284-8.
Sambrook J, Fritsch EF, Maniatis T: Molecular cloning: a laboratory manual, 2nd ed. Edited by: Press CSHL. 1989, Cold Spring Harbor, N.Y.
Birnboim HC: A rapid alkaline extraction method for the isolation of plasmid DNA. Methods Enzymol. 1983, 100: 243-255.
Better M, Lewis B, Corbin D, Ditta G, Helinski DR: Structural relationships among Rhizobium meliloti symbiotic promoters. Cell. 1983, 35 (2 Pt 1): 479-485. 10.1016/0092-8674(83)90181-2.
Figurski DH, Helinski DR: Replication of an origin-containing derivative of plasmid RK2 dependent on a plasmid function provided in trans. Proc Natl Acad Sci U S A. 1979, 76 (4): 1648-1652. 10.1073/pnas.76.4.1648.
Miller JH: Experiments in molecular genetics. Edited by: Laboratory CSH. 1972, Cold Spring Harbor, N.Y.
Stachel SE, An G, Flores C, Nester EW: A Tn3 lacZ transposon for the random generation of b-galactosidase gene fusions: application to the analysis of gene expression in Agrobacterium. Embo J. 1985, 4 (4): 891-898.
Winson MK, Swift S, Fish L, Throup JP, Jorgensen F, Chhabra SR, Bycroft BW, Williams P, Stewart GS: Construction and analysis of luxCDABE-based plasmid sensors for investigating N-acyl homoserine lactone-mediated quorum sensing. FEMS Microbiol Lett. 1998, 163 (2): 185-192. 10.1111/j.1574-6968.1998.tb13044.x.
Shaw PD, Ping G, Daly SL, Cha C, Cronan JE, Rinehart KL, Farrand SK: Detecting and characterizing N-acyl-homoserine lactone signal molecules by thin-layer chromatography. Proc Natl Acad Sci U S A. 1997, 94 (12): 6036-6041. 10.1073/pnas.94.12.6036.
Geels FP, Schippers B: Reduction in yield depression in high frequency potato cropping soil after seed tuber treatments with antagonist fluorescent Pseudomonas spp. Phytopathol Z. 1983, 108: 207-221.
Jacobs MA, Alwood A, Thaipisuttikul I, Spencer D, Haugen E, Ernst S, Will O, Kaul R, Raymond C, Levy R, Chun-Rong L, Guenthner D, Bovee D, Olson MV, Manoil C: Comprehensive transposon mutant library of Pseudomonas aeruginosa. Proc Natl Acad Sci U S A. 2003, 100 (24): 14339-14344. 10.1073/pnas.2036282100.
Kovach ME, Elzer PH, Hill DS, Robertson GT, Farris MA, Roop RM, Peterson KM: Four new derivatives of the broad-host-range cloning vector pBBR1MCS, carrying different antibiotic-resistance cassettes. Gene. 1995, 166 (1): 175-176. 10.1016/0378-1119(95)00584-1.
O'Toole GA, Kolter R: Initiation of biofilm formation in Pseudomonas fluorescens WCS365 proceeds via multiple, convergent signalling pathways: a genetic analysis. Mol Microbiol. 1998, 28 (3): 449-461. 10.1046/j.1365-2958.1998.00797.x.
Acknowledgements
IB was supported by a fellowship from the Italian Cystic Fibrosis Research Foundation (Project FFC9/2005 financed by the Delegazione FFC di Vicenza). This work was in part supported by a grant from ISPESL, Rome, Italy (contract B/98-2DIPIA/03). This work was also supported by a grant from the Italian Ministry of University and Research PRIN-2006 entitled "Basi genetiche e molecolari della patogenicita' batterica". We thank the ICGEB staff of the animal house in their assistance in raising polyclonal PpuR antibodies and to Sara Ferluga for assistance in artwork.
Author information
Authors and Affiliations
Corresponding author
Additional information
Authors' contributions
IB performed the screening of the Tn5 mutant bank, lon gene cloning and sequencing, AHL quantifications, gene promoter assays and western analysis. GR cloned ppuR in the expression vector and purified PpuR for raising antibodies in rabbits. LL was involved in supervision of GR work and discussions and planning with VV. VV participated in experimental design and data analysis, coordinated the study and drafted the manuscript. All authors read and approved the manuscript.
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
Open Access This article is published under license to BioMed Central Ltd. This is an Open Access article is distributed under the terms of the Creative Commons Attribution License ( https://creativecommons.org/licenses/by/2.0 ), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Bertani, I., Rampioni, G., Leoni, L. et al. The Pseudomonas putida Lon protease is involved in N-acyl homoserine lactone quorum sensing regulation. BMC Microbiol 7, 71 (2007). https://doi.org/10.1186/1471-2180-7-71
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1471-2180-7-71