
Abstract
Background
Accumulated evidence suggests that insulin resistance and impairments in cerebral insulin receptor signaling may contribute to age-related cognitive deficits and Alzheimer's disease. The enhancement of insulin receptor signaling is, therefore, a promising strategy for the treatment of age-related cognitive disorders. The mitochondrial respiratory chain, being involved in insulin-stimulated H2O2 production, has been identified recently as a potential target for the enhancement of insulin signaling. The aim of the present study is to examine: (1) whether a specific respiratory substrate, dicholine salt of succinic acid (CS), can enhance insulin-stimulated insulin receptor autophosphorylation in neurons, and (2) whether CS can ameliorate cognitive deficits of various origins in animal models.
Results
In a primary culture of cerebellar granule neurons, CS significantly enhanced insulin-stimulated insulin receptor autophosphorylation. In animal models, CS significantly ameliorated cognitive deficits, when administered intraperitoneally for 7 days. In 16-month-old middle-aged C57Bl/6 mice (a model of normal aging), CS enhanced spatial learning in the Morris water maze, spontaneous locomotor activity, passive avoidance performance, and increased brain N-acetylaspartate/creatine levels, as compared to the age-matched control (saline). In rats with chronic cerebral hypoperfusion, CS enhanced spatial learning, passive avoidance performance, and increased brain N-acetylaspartate/creatine levels, as compared to control rats (saline). In rats with beta-amyloid peptide-(25–35)-induced amnesia, CS enhanced passive avoidance performance and increased activity of brain choline acetyltransferase, as compared to control rats (saline). In all used models, CS effects lasted beyond the seven-day treatment period and were found to be significant about two weeks following the treatment.
Conclusion
The results of the present study suggest that dicholine salt of succinic acid, a novel neuronal insulin sensitizer, ameliorates cognitive deficits and neuronal dysfunctions in animal models relevant to age-related cognitive impairments, vascular dementia, and Alzheimer's disease.
Similar content being viewed by others


Background
A large body of accumulated evidence suggests that insulin resistance and impairments in cerebral insulin receptor signaling may contribute to age-related cognitive deficits and Alzheimer's disease (AD) [1–10]. The enhancement of brain insulin receptor signaling is, therefore, a promising strategy for the treatment of age-related cognitive disorders. Optimal insulin receptor signaling requires hydrogen peroxide (H2O2) generated in cells during insulin stimulation [11–17]. The mitochondrial respiratory chain, being involved in insulin-stimulated H2O2 production, has been identified recently as a potential target for the enhancement of insulin signaling [18]. The rate of insulin-stimulated H2O2 production depends on the concentration of respiratory substrate, succinate [19].
The aim of the present study is to examine: (1) whether a specific respiratory substrate, dicholine salt of succinic acid (CS), can enhance insulin-stimulated insulin receptor autophosphorylation in neurons, and (2) whether CS can ameliorate cognitive deficits of various origins in animal models.
Results
Dicholine salt of succinic acid enhances insulin-stimulated insulin receptor autophosphorylation in neurons
To examine whether CS enhances the insulin-stimulated autophosphorylation of insulin receptor in neurons, we studied an effect of CS on insulin-stimulated insulin receptor autophosphorylation in a primary culture of rat cerebellar granule neurons (CGN). Figure 1 shows that, whereas by itself, 50 μmol/L of CS does not stimulate insulin receptor autophosphorylation significantly (P = 0.065 vs. control), this concentration of CS significantly enhances the effect of suboptimal concentration of 5 nmol/L insulin (P < 0.001 vs. 5 nmol/L insulin). CS significantly enhances insulin-stimulated insulin receptor autophosphorylation in the range of concentrations from 10 to 100 μmol/L (P < 0.05 vs. 5 nmol/L insulin), although no significant difference is observed between the effects of different concentrations of CS. These results suggest that CS is a neuronal insulin sensitizer, which works in concert with insulin to stimulate insulin receptor autophosphorylation in neurons.

Effects of CS on the autophosphorylation of insulin receptor in cerebellar granule neurons. CGN cultures were stimulated with insulin, CS, or combinations of insulin and CS at indicated concentrations for 20 min. Autophosphorylation of insulin receptor was measured as described in Materials and Methods. In each experiment, amount of phosphorylated insulin receptor β-subunit (pYpY-IR) was normalized to total amount of insulin receptor β-subunit and expressed as a percentage of the response produced to 100 nmol/L insulin. Columns represent the means ± SEM of pYpY-IR values obtained from five to nine separate experiments, each performed in duplicate. *P < 0.05 vs. control. †P < 0.05 vs. insulin 100 nmol/L, #P < 0.05 vs. insulin 5 nmol/L.
Dicholine salt of succinic acid improves cognition and neuron functioning in middle-aged mice
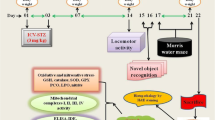
To examine whether dicholine salt of succinic acid can ameliorate age-related cognitive deficits, 16-month-old middle-aged C57Bl/6 mice (mean life span of these mice is 26 to 28 months [20]) were treated with CS (1 to 25 mg/kg, i.p.) or saline (control, i.p.) for seven consecutive days. Then, behavioral tests and measurement of whole-brain N-acetylaspartate/creatine (NAA/Cr) ratio by proton magnetic resonance spectroscopy (1H-MRS) in vivo were carried out on days as indicated in the experimental schedule (Figure 2). 5-Month-old young adult mice were treated with i.p. saline for 7 days.

Experimental schedule of studies in middle-aged C57Bl/6 mice. Mice were treated with CS (i.p. for 7 days) or saline (i.p. for 7 days, control) and then tested. LA, locomotor activity. PA, step-down passive avoidance. MWM, Morris water maze. MRS, 1H-MRS in vivo.
As shown in Figure 3, there was a significant difference in spontaneous locomotor activity in the open field between control middle-aged mice and young adult mice (P < 0.001). CS significantly increased the locomotor activity of middle-aged mice, as compared to age-matched controls, when administered in doses of 1–25 mg/kg (P < 0.05).

Effects of CS on spontaneous locomotor activity in middle-aged mice. Spontaneous locomotor activity in mice was evaluated in the open field test on the day as indicated in the experimental schedule (Figure 2). YNG, young adult mice (i.p. saline for 7 days); MID, middle-aged mice (i.p. saline for 7 days, control); CS1, CS10, and CS25, middle-aged mice treated i.p. for 7 days with CS in doses of 1, 10, or 25 mg/kg respectively. Each group comprised a minimum of eight mice. Columns represent the means ± SEM of locomotor activity counts during a 3-min observation period. *P < 0.05 vs. MID. †P < 0.05 vs. YNG.
As shown in Figure 4, there was no significant difference in step-down latencies in the passive avoidance test between young adult mice, control middle-aged mice, and CS-treated middle-aged mice on a day of acquisition trial. However, 24 hours later, in the retention test, middle-aged mice exhibited a significant decrease in step-down latencies (P < 0.05) as compared to young adult mice, indicating a learning deficit induced by aging. CS increased retention latencies in middle-aged mice, as compared to age-matched controls, when administered in doses of 10 and 25 mg/kg (P < 0.05). These data suggest that CS significantly improves passive avoidance learning in middle-aged mice, as compared to the age-matched controls.

Effects of CS on learning in the step-down passive avoidance test in middle-aged mice. Step-down latencies in passive avoidance task were measured during acquisition trial and, 24 hours later, during retention trial on days as indicated in the experimental schedule (Figure 2). A: Acquisition trial. B: Retention trial. YNG, young adult mice (i.p. saline for 7 days); MID, middle-aged mice (i.p. saline for 7 days, control); CS1, CS10, and CS25, middle-aged mice treated i.p. for 7 days with CS in doses of 1, 10, or 25 mg/kg respectively. Each group comprised a minimum of eight mice. Columns represent the step-down latencies means ± SEM. *P < 0.05 vs. MID. †P < 0.05 vs. YNG.
As shown in Figure 5, both path length and latency to escape to the hidden platform in the Morris water maze decreased progressively (i.e., learning was progressive) during the 4-day training period in all groups of mice. Two-way ANOVA revealed significant day effect in all experimental groups (P < 0.01). There was a significant difference, however, between middle-aged mice and young adult mice on the third day (P < 0.01 for both measures) and the fourth day (P < 0.001 for both measures) of the training, indicating that spatial learning in middle-aged mice was slower, as compared to young adult mice. Two-way ANOVA revealed significant group effect across days (F1,16 = 26.9, P < 0.01) and group × day interaction (F1,3,70 = 7.51, P < 0.05), when groups of young adult and middle-aged mice were compared. CS significantly decreased the escape latencies in middle-aged mice, as compared to age-matched controls, at the fourth day of the training, when administered in doses of 1–25 mg/kg (P < 0.05). Two-way ANOVA revealed significant effect for CS doses of 1 mg/kg (F1,14 = 8.47, P < 0.05) and 25 mg/kg (F1,15 = 7.26, P < 0.05) across days and significant both group effect (F1,14 = 10.48, P < 0.05) and group × day interaction (F1,1,62 = 6.79, P < 0.05) for dose of 10 mg/kg. CS significantly decreased the path lengths in middle-aged mice, as compared to age-matched controls, on the day 4 of training, when administered in doses of 1–25 mg/kg (P < 0.05). Two-way ANOVA revealed significant group effects for CS doses of 1 mg/kg (F1,14 = 8.01, P < 0.05) and 25 (F1,15 = 6.95 P < 0.05) across days and significant both group effect (F1,14 = 9.84, P < 0.05) and group × day interaction (F1,1,62 = 7.01, P < 0.05) for CS dose of 10 mg/kg. These data suggest that CS significantly improved spatial learning in middle-aged mice, as compared to the age-matched controls.

Effects of CS on spatial learning in the water-maze test in middle-aged mice. Path lengths and latencies to escape to the hidden platform in water maze were measured during the 4-day training period on days as indicated in the experimental schedule (Figure 2). A, B: The day 1 of the trial. C, D: The day 2 of the trial. E, F: The day 3 of the trial. G, H: The day 4 of the trial. YNG, young adult mice (i.p. saline for 7 days); MID, middle-aged mice (i.p. saline for 7 days, control); CS1, CS10, and CS25, middle-aged mice treated i.p. for 7 days with CS in doses of 1, 10, or 25 mg/kg respectively. Each group comprised a minimum of eight mice. Columns represent the path lengths or latencies means ± SEM. *P < 0.05 vs. MID. †P < 0.05 vs. YNG.
N-Acetylaspartate/creatine (NAA/Cr) ratio is widely believed to be a reliable noninvasive marker of neuronal function and viability in the adult brain. As shown in Figure 6, middle-aged mice exhibited a significant 30% drop in whole-brain brain NAA/Cr levels (P < 0.001) by data of 1H-MRS in vivo study, as compared to young adult mice, thus indicating age-related decline in neuronal function. When administered in doses of 10 and 25 mg/kg, CS significantly increased whole-brain NAA/Cr levels in middle-aged mice, as compared to the age-matched controls (P < 0.01). These data suggest that CS significantly improved neuron functioning in middle-aged mice.

Effects of CS on brain NAA/Cr levels in middle-aged mice. NAA/Cr levels in brains of mice were measured by 1H-MRS in vivo on day as indicated in the experimental schedule (Figure 2). A: Brain NAA/Cr levels. YNG, young adult mice (i.p. saline for 7 days); MID, middle-aged mice (i.p. saline for 7 days, control); CS1, CS10, and CS25, middle-aged mice treated i.p. for 7 days with CS in doses of 1, 10, or 25 mg/kg respectively. Each group comprised a minimum of eight mice. Columns represent the NAA/Cr means ± SEM expressed as a percentage of the mean NAA/Cr in brains of young adult mice. *P < 0.05 vs. MID. †P < 0.05 vs. YNG. B: Representative 1H MRS spectra from control young adult mice (YNG), middle-aged mice (MID), and middle-aged mice treated with CS in dose of 25 mg/kg (CS25). Signal assignment: NAA, methyl protons of NAA; Cr, methyl protons of Cr.
Dicholine salt of succinic acid improves cognition and neuron functioning in rats with chronic cerebral hypoperfusion
To examine whether CS can ameliorate cognitive deficits induced by chronic cerebral hypoperfusion, rats with permanent bilateral carotid artery occlusion (two-vessel occlusion, 2VO) were treated with CS (1 to 25 mg/kg, i.p.) or saline (control) for seven consecutive days, and a battery of tests were carried out on days as summarized in the experimental schedule (Figure 7). Sham-operated rats were treated with i.p. saline for 7 days.

Experimental schedule of studies in 2VO rats. Rats were undergone to bilateral occlusion of the common carotid arteries (2VO) and three hours later the treatment was started. Rats received CS or saline (control) i.p. for 7 days and then tested. PA, step-through passive avoidance. MWM, Morris water maze. MRS, 1H-MRS in vivo.
As shown in Figure 8, there was no significant difference in step-through latencies to enter the dark compartment in passive avoidance test between sham-operated rats, control 2VO rats, and CS-treated 2VO rats on the day of acquisition trial. However, 24 hours later, in the retention test, 2VO rats exhibited significant decrease in step-through latencies as compared to sham-operated rats (P < 0.01), indicating a learning deficit induced by chronic cerebral hypoperfusion. CS significantly increased retention latencies in 2VO rats, as compared to control 2VO rats, when administered in doses of 1–25 mg/kg (P < 0.05). These data suggest that CS significantly improved passive avoidance learning in rats with chronic cerebral hypoperfusion.

Effects of CS on learning in the step-through passive avoidance test in 2VO rats. Step-through latencies in passive avoidance task were measured during acquisition trial and, 24 hours later, during retention trial on days as indicated in the experimental schedule (Figure 7). A: Acquisition trial. B: Retention trial. SH, sham-operated rats (i.p. saline for 7 days); CTR, 2VO rats (i.p. saline for 7 days, control); CH, 2VO rats treated i.p. for 7 days with choline chloride in dose of 10 mg/kg; CS1, CS10, and CS25, 2VO rats treated i.p. for 7 days with CS in doses of 1, 10, or 25 mg/kg respectively. Each group comprised a minimum of nine rats. Columns represent the step-through latencies means ± SEM. *P < 0.05 vs. CTR. †P < 0.05 vs. SH.
Figure 9 shows that path length to escape to the hidden platform in Morris water maze task decreased during the 2-day training period, in all groups of rats. There was, however, a significant difference between control 2VO rats and sham-operated rats on the first day and the second day of training (P < 0.01), indicating impairments in spatial learning induced by chronic cerebral hypoperfusion. CS significantly decreased the path lengths in 2VO rats, as compared to control 2VO rats, on day 1 and, particularly, on day 2 of training, when administered in doses of 1–25 mg/kg (P < 0.01). These data suggest that CS significantly improved spatial learning in rats with chronic cerebral hypoperfusion.

Effects of CS on spatial learning in the water-maze test in 2VO rats. Path lengths to escape to the hidden platform in water maze were measured during the 2-day training period on days as indicated in the experimental schedule (Figure 7). A: The day 1 of the trial. B: The day 2 of the trial. SH, sham-operated rats (i.p. saline for 7 days); CTR, 2VO rats (i.p. saline for 7 days, control); CH, 2VO rats treated i.p. for 7 days with choline chloride in dose of 10 mg/kg; CS1, CS10, and CS25, 2VO rats treated i.p. for 7 days with CS in doses of 1, 10, or 25 mg/kg respectively. Each group comprised a minimum of eight rats. Columns represent the path lengths means ± SEM. *P < 0.05 vs. CTR. †P < 0.05 vs. SH.
As shown in Figure 10, 2VO rats exhibited a significant 22% decrease in whole-brain NAA/Cr levels, as compared to sham-operated rats (P < 0.001), indicating impairments in neuronal function induced by chronic cerebral hypoperfusion. CS significantly increased whole-brain NAA/Cr levels in 2VO rats, as compared to controls, when administered in doses of 1–25 mg/kg (P < 0.05).

Effects of CS on brain NAA/Cr ratios in 2VO rats. NAA/Cr levels in brains of rats were measured by 1H-MRS in vivo on day as indicated in the experimental schedule (Figure 7). SH, sham-operated rats (i.p. saline for 7 days); CTR, 2VO rats (i.p. saline for 7 days, control); CH, 2VO rats treated i.p. for 7 days with choline chloride in dose of 10 mg/kg; CS1, CS10, and CS25, 2VO rats treated i.p. for 7 days with CS in doses of 1, 10, or 25 mg/kg respectively. Each group comprised from five to nine animals. Columns represent the NAA/Cr means ± SEM expressed as a percentage of the mean NAA/Cr in brains of sham-operated rats. *P < 0.05 vs. CTR. †P < 0.05 vs. SH.
There was no significant difference between 2VO rats treated with choline chloride (10 mg/kg, i.p. for 7 days) and control 2VO rats (saline, i.p. for 7 days) in passive avoidance performance (P = 0.10) and in spatial learning (P = 0.11). Hence, choline chloride, a reference choline compound, showed no significant therapeutic effect on cognitive deficits in 2VO rats. However, MRS data indicate a role of choline in normalization of NAA/Cr deficits.
Dicholine salt of succinic acid improves learning and increases activity of brain choline acetyltransferase in rats with β-amyloid peptide-(25–35)-induced amnesia
To examine whether CS can ameliorate cognitive deficits induced in rats by a single injection of β-amyloid peptide-(25–35) into the brain nucleus basalis magnocellularis (NBM), the β-amyloid peptide-(25–35)-induced rats were treated with CS (1 to 25 mg/kg, i.p.) or saline (control) for seven consecutive days. Sham-operated rats induced by a single injection of saline into the brain NBM were treated with i.p. saline for 7 days. Step-through passive avoidance test and measurement of activity of choline acetyltransferase (ChAT) in brain cortex homogenates were carried out on days, as indicated in the experimental schedule (Figure 11).

Experimental schedule of studies in β-amyloid peptide-(25–35)-induced rats. Rats were undergone to a single injection of β-amyloid peptide-(25–35) into brain NBM. Two weeks later, rats were treated with CS or saline (control) i.p. for 7 days and then tested. PA, step-through passive avoidance. ChAT, choline acetyltransferase measurement.
As shown in Figure 12, there was a significant difference in step-through latencies to enter the dark compartment in passive avoidance test between sham-operated rats and control β-amyloid peptide-(25–35)-induced rats, on the day of acquisition (P < 0.05), and 24 hours later, on the day of retention trials (P < 0.01). On the day of acquisition trials, CS significantly decreased step-through latencies, as compared to the controls, when administered in doses of 10 and 25 mg/kg (P < 0.01). On the day of retention trials, CS significantly increased step-through latencies, as compared to the control, when administered in doses of 10 and 25 mg/kg (P < 0.01). These data suggest that CS significantly improves passive avoidance learning in rats with β-amyloid peptide-(25–35)-induced amnesia.

Effects of CS on learning in the step-through passive avoidance test in rats with β-amyloid peptide-(25–35)-induced amnesia. Step-through latencies in passive avoidance task were measured during acquisition trial and, 24 hours later, during retention trial on days as indicated in the experimental schedule (Figure 11). A: Acquisition trial. B: Retention trial. SH, sham-operated rats (i.p. saline for 7 days); CTR, β-amyloid peptide-(25–35)-induced rats (i.p. saline for 7 days, control); CH, β-amyloid peptide-(25–35)-induced rats treated i.p. for 7 days with choline chloride in dose of 10 mg/kg; CS1, CS10, and CS25, β-amyloid peptide-(25–35)-induced rats treated i.p. for 7 days with CS in doses of 1, 10, or 25 mg/kg respectively. Each group comprised eight rats. Columns represent the step-through latencies means ± SEM. *P < 0.05 vs. CTR. †P < 0.05 vs. SH.
As shown in Table 1, there was a significant 27% decrease in ChAT activity in the control β-amyloid peptide-(25–35)-induced rats, as compared to sham-operated rats (P < 0.001), indicating cholinergic dysfunction induced by β-amyloid peptide-(25–35) toxicity. CS significantly increased ChAT activity in rats with β-amyloid peptide-(25–35)-induced amnesia, when administered in the highest studied dose of 25 mg/kg (P < 0.05 vs. control).
Choline chloride (10 mg/kg, i.p.), the reference choline compound, showed no significant effect on passive avoidance learning and cerebral ChAT activity in rats with β-amyloid peptide-(25–35)-induced amnesia, when administered for 7 days.
Discussion
In the present study we identified a highly effective treatment of cognitive deficits of various origins with dicholine salt of succinic acid, the neuronal insulin sensitizer.
Initially, we examined whether CS can work as an insulin sensitizer in the brain. It is generally accepted that insulin signaling requires autophosphorylation of the insulin receptor kinase at tyrosine residues in the activation loop of the kinase domain [21–27]. Upon autophosphorylation, the receptor undergoes a major conformational change, resulting in unrestricted access of protein substrates and ATP to the kinase active site and an approximate two-order increase in the kinase activity [28–30]. In the present study, we demonstrate that, although CS alone does not stimulate insulin receptor autophosphorylation significantly, it significantly enhances the response to the suboptimal insulin concentration. The effect of the combination of insulin and CS is much greater than sum of effects of insulin and CS taken alone. These results suggest that CS is a neuronal insulin sensitizer, which works in concert with insulin to stimulate insulin receptor autophosphorylation in neurons.
Dicholine salt of succinic acid is a chemical substance, but its components, choline and succinate, are naturally occurring metabolites widely distributed in mammalian tissues. Average concentrations of succinate and choline in the blood of healthy humans at rest are 1–9 μmol/L [31] and 7–10 μmol/L [32] respectively. During physical exercises or severe hypoxia, levels of succinate and choline increase markedly [33, 34]. For example, breath-hold dives for 1 min and treadmill running increases venous succinate in humans to 125 and 93 μmol/L respectively [33]. This means that CS is an effective insulin sensitizer at physiologically occurring concentrations, typically observed in mammals during hypoxia or apneic work.
We further investigated the effects of intraperitoneal administration of CS on cognition and neuronal function in animals with cognitive deficits of various origins.
To examine whether CS, the neuronal insulin sensitizer, can ameliorate cognitive deficits induced by normal aging, experiments with middle-aged C57Bl/6 mice were carried out. These mice represent a model of a mild cognitive deficit associated with age-related changes in the expression of genes, including genes with possible roles in synaptic plasticity and learning [35], while neuronal loss with aging is relatively rare in mice of this strain [20]. We demonstrate that CS significantly improves passive avoidance learning, spontaneous locomotor activity, and spatial learning in middle-aged mice, as compared to age-matched controls. To test whether CS can ameliorate neuronal dysfunction in middle-aged mice, 1H-MRS in vivo study was carried out in addition to tests listed above. It is generally accepted that the brain level of N-acetylaspartate (NAA), frequently expressed as N-acetylaspartate/Creatine ratio, is a reliable noninvasive marker of neuronal integrity and functioning, since NAA is localized almost exclusively to neurons in the adult brain and NAA decrease is closely correlated with neuronal loss or neuronal dysfunction [36–41]. Reversible NAA deficits reflect a reversible state of neuronal dysfunction preceding neuronal loss [42–45]. NAA quantification is, therefore, considered to be a valuable tool for assessing the effects of potential neuroprotective therapies. In this study, age-related NAA deficits in middle-aged mice were reversed after the treatment with CS, indicating the neuroprotective effect of CS. Taken together, results in middle-aged mice demonstrate that CS ameliorates both age-related cognitive deficits and neuronal dysfunction caused by normal aging. These CS effects were long-lasting beyond the seven-day treatment period and were found to be significant about two weeks following the treatment.
To determine whether CS, the neuronal insulin sensitizer, can ameliorate cognitive deficits induced by chronic cerebral hypoperfusion, experiments with 2VO rats were carried out. Although originally described as a rat model for vascular dementia, the chronic cerebral hypoperfusion induced by permanent bilateral carotid artery occlusion is relevant to Alzheimer's disease, since there is also a slowly developing reduction in cerebral blood flow in this condition [46]. The two-vessel occlusion causes a chronic decrease in the cerebral blood flow to 52–64% of the original level [47] and induces progressive and long-lasting cognitive deficit, cholinergic dysfunction, and progressive neuronal damage in the brain [48, 49]. In this study, we found that CS significantly improved passive avoidance learning and spatial learning in 2VO rats, as compared to the control 2VO rats. NAA deficits in brains of 2VO rats were also reversible on the treatment with CS. Thus, CS ameliorates both cognitive deficits and neuronal dysfunction in 2VO rats. These CS effects were long-lasting, extending beyond the seven-day treatment period, and were found to be significant ten days following the CS treatment.
There is evidence that β-amyloid, a hallmark of Alzheimer's disease, can interfere with insulin receptor signaling [50], whereas insulin stimulates clearance and degradation of β-amyloid and thus prevents β-amyloid accumulation in the brain [51–53]. Activity of choline acetyltransferase, a key enzyme of acetylcholine biosynthesis, is reduced in insulin receptor-positive neurons in Alzheimer's disease and expression of choline acetyltransferase is increased with insulin stimulation [54]. These findings provide a rationale for the use of insulin and insulin sensitizers in the treatment of cognitive deficits caused by β-amyloid toxicity. In the present study, we examined the effect of CS, the neuronal insulin sensitizer, on cognitive deficits induced in rats by a single injection of β-amyloid peptide-(25–35) into brain nucleus basalis magnocellulari s. Such injection causes behavioral dysfunctions, impairs learning and memory, and disrupts cortical cholinergic innervations, thus modeling Alzheimer's disease [55]. Also, β-amyloid peptide-(25–35) decreases activity of brain choline acetyltransferase, a key enzyme of acetylcholine biosynthesis [56]. In the present study, we demonstrate that CS significantly improves passive avoidance learning and increases ChAT activity in the brains of β-amyloid peptide-(25–35)-induced rats. The CS effects lasted beyond the seven-day treatment period and were found to be significant two weeks following the treatment.
Choline chloride, the reference choline compound, was used in the present study to discriminate the effects of choline and succinate moieties in the CS molecule. As compared to highly effective CS treatment, choline chloride demonstrated no significant effects on cognitive performance in 2VO rats and rats with β-amyloid peptide-(25–35)-induced amnesia. However, MRS data reveal a role for choline in normalization of NAA/Cr deficits in brain of 2VO rats. This indicates that although CS effects relates mainly to the action of succinate, the neuroprotective effect of CS is caused, at least in part, by the presence of choline moiety in the CS molecule.
Earlier, several lines of evidence have suggested that treating insulin resistance might improve cognitive function. It has been reported that rosiglitazone, a peripheral insulin sensitizer and potent PPAR full agonist, attenuates learning and memory deficits in Tg2576 Alzheimer mice [57]. In a small pilot study, rosiglitazone appeared to significantly improve the cognitive ability of AD patients [58]. In a Phase II clinical study, rosiglitazone was found to improve the cognitive ability of mild to moderate AD patients [59]. It has been proposed that PPARγ agonism induces neuronal mitochondrial biogenesis and improves glucose utilization leading to improved cellular function and provides mechanistic support for the improvement in cognition observed in treatment of Alzheimer's patients with rosiglitazone [60].
In general, the results of the present study support the idea that targeting insulin receptor signaling in neurons may help to reduce both cognitive deficits and neuronal dysfunctions associated with aging and Alzheimer's disease.
Conclusion
The results of the present study suggest that dicholine salt of succinic acid, the novel neuronal insulin sensitizer, ameliorates cognitive deficits and neuronal dysfunctions in animal models relevant to age-related cognitive impairments, vascular dementia, and Alzheimer's disease.
Methods
Materials
Dicholine succinate salt (2:1), formula [(CH3)2NCH2CH2OH]2+·-OOCCH2CH2COO-, was prepared by a reaction of succinic acid with choline base in the Russian Scientific Center on Drug Safety (Staraya Kupavna, Moscow region). PhosphoDetect™ Insulin Receptor (pTyr1162/1163) ELISA kit and Insulin Receptor (β-Subunit) ELISA Kit were from Calbiochem. Other materials were purchased from Sigma, ICN, Gibco, Biosource, Molecular Probes, or Acros.
Animals
Male Wistar rats and C57Bl/6 mice were from the Laboratory of Biological Trials of the Pushchino Branch of Shemyakin-Ovchinnikov Institute of Bioorganic Chemistry (Pushchino, Moscow Region). Animals were housed in groups of 4 per cage at a constant temperature 21°C in a light-controlled room at 14/10 light-dark cycle. Food and water were freely available. All animal studies were carried out in accordance with the requirements of our institutional committees for the keeping and use of laboratory animals and in accordance with the "Principles of Laboratory Animal Care" formulated by the National Institutes of Health. All animals were allocated to experimental groups randomly, using computer-generated random numbers.
Neuronal culture
Cerebellar granule neurons (CGN) were prepared from 7- to 8-day-old Wistar rats as described [61, 62]. Cerebellum was dissected and placed in ice-cold Ca2+/Mg2+-free Hanks' buffered saline (HBSS) without Phenol Red (Gibco). After mincing the tissue with fine scissors, the tissue was placed in Ca2+/Mg2+-free HBSS with Phenol Red and 0.1% trypsin for 15 min at 36°C. Trypsin was inactivated by washing with normal HBSS. Cells were dissociated by trituration and pelleted in HBSS. Then, the cells were resuspended in Neurobasal Medium (Gibco) supplemented with B-27 Supplement (Gibco), 20 mmol/L KCl, GlutaMax (Gibco) and penicillin/streptomycin and plated with density 5 × 106 cells/ml onto 35 mm × 10 mm sterile cell culture dishes which had been previously coated with poly-D-lysine. The cultures were maintained at 36°C in a humidified atmosphere of 5% CO2 and 95% air and fed with supplemented Neurobasal Medium. Cultures were treated on day 3 with 10 μmol/L cytosine arabinoside (Sigma) for 24 h to prevent glial proliferation. CGN at 7 to 8 days were used for experiments.
Insulin receptor phosphorylation assay
Amounts of double phosphorylated β-subunit of insulin receptor (pYpY-IR) were measured by PhosphoDetect™ insulin receptor (pTyr1162/1163) ELISA kit (Calbiochem) suitable for studies with rat insulin receptor. CGN cultures were incubated in Hepes-buffered salt solution (145 mmol/L NaCl, 5.6 mmol/L KCl, 1.8 mmol/L CaCl2, 1 mmol/L MgCl2, 20 mmol/L HEPES, and 5 mmol/L glucose) at pH 7.4 for 30 min, followed by exposure to vehicle, insulin, CS, or combinations of insulin with CS for 20 min. The experiment was terminated by removing the medium, washing with ice-cold PBS, and adding 120 μL per dish cell lysis buffer (Biosource) supplemented with 1 mmol/L PMSF, 50 mmol/L protease inhibitor set III (Sigma), and 2 mmol/L sodium ortovanadate as the inhibitor of tyrosine phosphatases at 4°C for 20 min. Lysates were centrifuged at 12,000 rpm at 4°C for 12 min. In each CGN lysate, pYpY-IR amounts were measured as described by the manufacturer's manual. Obtained values were normalized to total amounts of insulin receptor β-subunit (IR) measured by insulin receptor (β-subunit) ELISA kit (Calbiochem). The results are expressed as a percentage of the response produced to 100 nmol/L insulin.
Two-vessel occlusion
Experimental cerebral hypoperfusion was induced in rats by permanent bilateral occlusion of the common carotid arteries (two-vessel occlusion, 2VO) as described [47]. The rats were anesthetized with sodium pentobarbital (40 mg/kg, i.p.) and the common carotid arteries of the rat were separated from the cervical sympathetic and vagal nerves through a ventral cervical incision. Then, the arteries were ligated with silk sutures. The same surgical procedure was performed in the sham-operated rats but without the actual ligation.
β-Amyloid peptide-(25–35) injection
β-Amyloid peptide-(25–35)-induced amnesia in rats was induced as described previously [55]. The rats were anesthetized with sodium pentobarbital (30 mg/kg, i.p.) and β-amyloid peptide-(25–35) was injected bilaterally into nucleus basalis magnocellularis of rat brain as a sterile solution of 2 μg per 1 μL of saline per side through the guide cannula with Hamilton microsyringe according to stereotaxic coordinates: AP -1.5, DL ± 2.7, and H 8.1 [63]. Sham-operated rats were injected bilaterally into NBM with 1 μL of saline.
Behavioral tests
Spontaneous locomotor activity of mice was evaluated in open field tests in an automated mode using the Opto-Varimex-3 (Columbus Instruments, OH) photocell-based activity monitor. The Opto-Varimex-3 animal activity monitor employs a photocell beam grid. Animals were placed individually into the activity monitor and spontaneous locomotor activity (total accumulated counts of a horizontal single photocell interruption) was collected for a 3-min period.
The apparatus for step-down passive avoidance test consisted of a box (22 × 24 × 27 cm) with a stainless-steel grid floor. A circular Plexiglas platform (diameter, 8 cm; height, 2 cm) was fixed at the center of the box. During the training, each mouse was placed individually on the platform. When the mouse stepped down from the platform and placed its four paws on the grid floor, an electric shock 1.0 mA was delivered for 3 s. The retention trial was carried out twenty-four hours after the training session in a manner similar to the training except that no electric shock was delivered via grid floor. Each mouse was placed again on the platform, and step-down latency was recorded until 180 s had elapsed.
A step-through box for passive avoidance test consisted of a light compartment connected to a dark compartment by a controllable door. This test consisted of two trials. In the acquisition trial, the rats were individually placed into the light compartment, the door to a dark compartment was opened, and the latency until the rat entered the dark compartment was recorded. After the rat had stepped through the door, the door was closed and an electric shock 0.8 mA was delivered for 1 s via the grid floor. After receiving the footshock, the rat was returned to a home cage. The retention trial was carried out twenty-four hours after the acquisition trial. In the retention trial, each animal was placed into the light compartment, and the step-through latency was recorded until 180 s had elapsed.
The water maze test was performed as described by Morris [64]. The experimental apparatus consisted of circular water pool (diameter, 120 cm; height, 60 cm) containing water at 24°C to a depth of 40 cm and rendered opaque by adding milk. A Plexiglas escape platform (8 × 8 cm for mice or 10 × 10 cm for rats) was submerged 1.5 cm (for mice) or 2 cm (for rats) below the water surface and placed at the midpoint of one quadrant. The location of the platform remained the same throughout the training period. The pool was located in a test room containing various prominent visual cues. Six training trials per day were conducted with an inter-trial interval of 2 min. Animals were placed in the pool at one of six starting positions. In each training trial, the time and path length required to escape onto the hidden platform was recorded. Results of six training trials were averaged to obtain a single representative value, and the averages were used for final statistics. Animals that found the platform were allowed to remain on the platform for 30 s and were then returned to the home cage during the inter-trial interval. Animals that did not find the platform within 120 s were softly guided to the platform for 30 s at the end of the trial.
All behavioral experiments were carried out by investigators blinded to treatment groups.
1H-MRS measurements in vivo
Spectra were recorded at 1H frequency 400 MHz using the Bruker AM-400 WB spectrometer (Bruker, Reinsretten, Germany) with a vertical magnet equipped with a home-build probe of outer diameter 70 mm. An animal under pentobarbital sodium anesthesia (40 mg/kg, i.p.) was fixed in the probe head, the surface coil (6 mm in diameter for mice or 14 mm in diameter for rats) being positioned directly onto the skull at animal's sinciput. Magnetic field homogeneity was optimized by the water signal. Line widths of 40 to 90 Hz were routinely obtained. The 1H-MRS spectra were accumulated and processed as described [65, 66]. Metabolite ratios of NAA/Cr were calculated from the relative peak height measurements using the spectrometer's software. All 1H-MRS measurements were carried out by investigator blinded to treatment groups.
Choline acetyltransferase assay
Rats were decapitated under sodium pentobarbital (40 mg/kg, i.p.) anesthesia. Brains were quickly removed and homogenized. ChAT activity in cerebral cortex homogenates was measured by the method described by Fonnum [67]. All ChAT measurements were carried out by investigator blinded to treatment groups.
Statistics
Data were analyzed for statistical significance by one-way analysis of variance (ANOVA). Data of the water maze test were analyzed for statistical significance by two-way ANOVA. Values are given as means ± SEM. Differences were considered significant at P < 0.05.
Abbreviations
- AD:
-
Alzheimer's disease
- ANOVA:
-
analysis of variance
- ChAT:
-
choline acetyltransferase
- CGN:
-
cerebellar granule neurons
- Cr:
-
creatine
- CH:
-
choline chloride
- CS:
-
dicholine salt of succinic acid
- HBSS:
-
Hanks' buffered salt solution
- HEPES:
-
4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid
- 1H-MRS:
-
proton magnetic resonance spectroscopy
- i.p.:
-
intraperitoneal
- NAA:
-
N-acetylaspartate
- NBM:
-
nucleus basalis magnocellularis
- PBS:
-
phosphate-buffered saline
- PMSF:
-
phenylmethylsulfonyl fluoride
- SEM:
-
standard error of mean
- 2VO:
-
two-vessel occlusion.
References
Craft S: Insulin resistance syndrome and Alzheimer's disease: age- and obesity-related effects on memory, amyloid, and inflammation. Neurobiol Aging. 2005, 26 (Suppl 1): 65-9. 10.1016/j.neurobiolaging.2005.08.021.
Geroldi C, Frisoni GB, Paolisso G, Bandinelli S, Lamponi M, Abbatecola AM, Zanetti O, Guralnik JM, Ferrucci L: Insulin resistance in cognitive impairment: the InCHIANTI study. Arch Neurol. 2005, 62 (7): 1067-72. 10.1001/archneur.62.7.1067.
Stolk RP, Breteler MM, Ott A, Pols HA, Lamberts SW, Grobbee DE, Hofman A: Insulin and cognitive function in an elderly population. The Rotterdam Study. Diabetes Care. 1997, 20 (5): 792-5. 10.2337/diacare.20.5.792.
Watson GS, Craft S: Modulation of memory by insulin and glucose: neuropsychological observations in Alzheimer's disease. Eur J Pharmacol. 2004, 490 (1–3): 97-113. 10.1016/j.ejphar.2004.02.048.
Hoyer S: Glucose metabolism and insulin receptor signal transduction in Alzheimer disease. Eur J Pharmacol. 2004, 490 (1–3): 115-25. 10.1016/j.ejphar.2004.02.049.
Frolich L, Blum-Degen D, Bernstein HG, Engelsberger S, Humrich J, Laufer S, Muschner D, Thalheimer A, Turk A, Hoyer S, Zochling R, Boissl KW, Jellinger K, Riederer P: Brain insulin and insulin receptors in aging and sporadic Alzheimer's disease. J Neural Transm. 1998, 105: 423-438. 10.1007/s007020050068.
Frolich L, Blum-Degen D, Riederer P, Hoyer S: A disturbance in the neuronal insulin receptor signal transduction in sporadic Alzheimer's disease. Ann N Y Acad Sci. 1999, 893: 290-293. 10.1111/j.1749-6632.1999.tb07839.x.
de la Monte SM, Wands JR: Review of insulin and insulin-like growth factor expression, signaling, and malfunction in the central nervous system: Relevance to Alzheimer's disease. J Alzheimers Dis. 2005, 7: 45-61.
Steen E, Terry BM, Rivera EJ, Cannon JL, Neely TR, Tavares R, Xu XJ, Wands JR, de la Monte SM: Impaired insulin and insulin-like growth factor expression and signaling mechanisms in Alzheimer's disease – is this type 3 diabetes?. J Alzheimers Dis. 2005, 7: 63-80.
Carantoni M, Zuliani G, Munari MR, D'Elia K, Palmieri E, Fellin R: Alzheimer disease and vascular dementia: relationships with fasting glucose and insulin levels. Dement Geriatr Cogn Disord. 2000, 11: 176-80. 10.1159/000017232.
May JM, de Haen C: Insulin-stimulated intracellular hydrogen peroxide production in rat epididymal fat cells. J Biol Chem. 1979, 254 (7): 2214-20.
Mahadev K, Zilbering A, Zhu L, Goldstein BJ: Insulin-stimulated hydrogen peroxide reversibly inhibits protein-tyrosine phosphatase 1b in vivo and enhances the early insulin action cascade. J Biol Chem. 2001, 276 (24): 21938-42. 10.1074/jbc.C100109200.
Mahadev K, Wu X, Zilbering A, Zhu L, Lawrence JT, Goldstein BJ: Hydrogen peroxide generated during cellular insulin stimulation is integral to activation of the distal insulin signaling cascade in 3T3-L1 adipocytes. J Biol Chem. 2001, 276 (52): 48662-9. 10.1074/jbc.M105061200.
Meng T-C, Buckley DA, Galic S, Tiganis T, Tonks NK: Regulation of insulin signaling through reversible oxidation of the protein-tyrosine phosphatases TC45 and PTP1B. J Biol Chem. 2004, 279 (36): 37716-37725. 10.1074/jbc.M404606200.
Goldstein BJ, Mahadev K, Wu X, Zhu L, Motoshima H: Role of insulin-induced reactive oxygen species in the insulin signaling pathway. Antioxid Redox Signal. 2005, 7 (7–8): 1021-31. 10.1089/ars.2005.7.1021.
Goldstein BJ, Mahadev K, Wu X: Redox paradox: insulin action is facilitated by insulin-stimulated reactive oxygen species with multiple potential signaling targets. Diabetes. 2005, 54 (2): 311-21. 10.2337/diabetes.54.2.311.
Seo JH, Ahn Y, Lee SR, Yeol Yeo C, Chung Hur K: The major target of the endogenously generated reactive oxygen species in response to insulin stimulation is phosphatase and tensin homolog and not phosphoinositide-3 kinase (PI-3 kinase) in the PI-3 kinase/Akt pathway. Mol Biol Cell. 2005, 16 (1): 348-57. 10.1091/mbc.E04-05-0369.
Pomytkin IA, Kolesova OE: Key role of succinate dehydrogenase in insulin-induced inactivation of protein tyrosine phosphatases. Bull Exp Biol Med. 2002, 133 (6): 568-70. 10.1023/A:1020229724717.
Pomytkin IA, Kolesova OE: Effect of insulin on the rate of hydrogen peroxide generation in mitochondria. Bull Exp Biol Med. 2003, 135 (6): 541-2. 10.1023/A:1025412915297.
Jucker M, Ingram DK: Murine models of brain aging and age-related neurodegenerative diseases. Behav Brain Res. 1997, 85 (1): 1-26. 10.1016/S0166-4328(96)02243-7.
Rosen OM, Herrera R, Olowe Y, Petruzzelli LM, Cobb MH: Phosphorylation activates the insulin receptor tyrosine protein kinase. Proc Natl Acad Sci USA. 1983, 80 (11): 3237-40. 10.1073/pnas.80.11.3237.
Tornqvist HE, Pierce MW, Frackelton AR, Nemenoff RA, Avruch J: Identification of insulin receptor tyrosine residues autophosphorylated in vitro. J Biol Chem. 1987, 262 (21): 10212-9.
White MF, Shoelson SE, Keutmann H, Kahn CR: A cascade of tyrosine autophosphorylation in the β-subunit activates the phosphotransferase of the insulin receptor. J Biol Chem. 1988, 263: 2969-2980.
Zhang B, Tavare JM, Ellis L, Roth RA: The regulatory role of known tyrosine autophosphorylation sites of the insulin receptor kinase domain. An assessment by replacement with neutral and negatively charged amino acids. J Biol Chem. 1991, 266 (2): 990-996.
Ellis L, Clauser E, Morgan DO, Edery M, Roth RA, Rutter WJ: Replacement of insulin receptor tyrosine residues 1162 and 1163 compromises insulin-stimulated kinase activity and uptake of 2-deoxyglucose. Cell. 1986, 45 (5): 721-32. 10.1016/0092-8674(86)90786-5.
Tornqvist HE, Gunsalus JR, Nemenoff RA, Frackelton AR, Pierce MW, Avruch J: Identification of the insulin receptor tyrosine residues undergoing insulin-stimulated phosphorylation in intact rat hepatoma cells. J Biol Chem. 1988, 263 (1): 350-359.
Issad T, Tavare JM, Denton RM: Analysis of insulin receptor phosphorylation sites in intact rat liver cells by two-dimensional phosphopeptide mapping. Predominance of the trisphosphorylated form of the kinase domain after stimulation by insulin. Biochem J. 1991, 275 (Pt 1): 15-21.
Hubbard SR, Wei L, Ellis L, Hendrickson WA: Crystal structure of the tyrosine kinase domain of the human insulin receptor. Nature. 1994, 372 (6508): 746-54. 10.1038/372746a0.
Hubbard SR: Crystal structure of the activated insulin receptor tyrosine kinase in complex with peptide substrate and ATP analog. EMBO J. 1997, 16 (18): 5572-81. 10.1093/emboj/16.18.5572.
Wie L, Hubbard SR, Hendrickson WA, Ellis L: Expression, Characterization, and Crystallization of the Catalytic Core of the Human Insulin Receptor Protein-tyrosine Kinase Domain. J Biol Chem. 1995, 270 (14): 8122-8130. 10.1074/jbc.270.14.8122.
Komaromy-Hiller G, Sundquist PD, Jacobsen LJ, Nuttall KL: Serum succinate by capillary zone electrophoresis: marker candidate for hypoxia. Ann Clin Lab Sci. 1997, 27 (2): 163-8.
Zeisel SH: Choline: an essential nutrient for humans. Nutrition. 2000, 16: 669-671. 10.1016/S0899-9007(00)00349-X.
Hochachka PW, Dressendorfer RH: Succinate accumulation in man during exercise. Eur J Appl Physiol Occup Physiol. 1976, 35 (4): 235-42. 10.1007/BF00423282.
Van Cappellen van Walsum AM, Jongsma HW, Wevers RA, Nijhuis JG, Crevels J, Engelke UFH, Moolenaar SH, Oeseburg B, Nijland R: 1H-NMR spectroscopy of cerebrospinal fluid of fetal sheep during hypoxia-induced academia and recovery. Pediatr Res. 2002, 52 (1): 56-63. 10.1203/00006450-200207000-00012.
Verbitsky M, Yonan AL, Malleret G, Kandel ER, Gilliam TC, Pavlidis P: Altered hippocampal transcript profile accompanies an age-related spatial memory deficit in mice. Learn Mem. 2004, 11 (3): 253-60. 10.1101/lm.68204.
Simmons ML, Frondoza CG, Coyle JT: Immunocytochemical localization of N-acetyl-aspartate with monoclonal antibodies. Neuroscience. 1991, 45: 37-45. 10.1016/0306-4522(91)90101-S.
Moffett JR, Namboodiri MA, Cangro CB, Neale JH: Immunohistochemical localization of N-acetylaspartate in rat brain. Neuroreport. 1991, 2: 131-134. 10.1097/00001756-199103000-00005.
Guimaraes AR, Schwartz P, Prakash MR, Carr CA, Berger UV, Jenkins BG, Coyle JT, Gonzalez RG: Quantitative in vivo 1H nuclear magnetic resonance spectroscopic imaging of neuronal loss in rat brain. Neuroscience. 1995, 69 (4): 1095-1101. 10.1016/0306-4522(95)00300-8.
Strauss I, Williamson JM, Bertram EH, Lothman EW, Fernandez EJ: Histological and 1H magnetic resonance spectroscopic imaging analysis of quinolinic acid-induced damage to the rat striatum. Magn Reson Med. 1997, 37 (1): 24-33. 10.1002/mrm.1910370106.
Barker PB: N-acetyl aspartate – a neuronal marker?. Ann Neurol. 2001, 49 (4): 423-4. 10.1002/ana.90.
Demougeot C, Marie C, Giroud M, Beley A: N-acetylaspartate: a literature review of animal research on brain ischaemia. J Neurochem. 2004, 90 (4): 776-83. 10.1111/j.1471-4159.2004.02583.x.
Dautry C, Vaufrey F, Brouillet E, Bizat N, Henry PG, Conde F, Bloch G, Hantraye P: Early N-acetylaspartate depletion is a marker of neuronal dysfunction in rats and primates chronically treated with the mitochondrial toxin 3-nitropropionic acid. J Cereb Blood Flow Metab. 2000, 20 (5): 789-99.
De Stefano N, Matthews PM, Arnold DL: Reversible decreases in N-acetylaspartate after acute brain injury. Magn Reson Med. 1995, 34 (5): 721-7. 10.1002/mrm.1910340511.
Demougeot C, Garnier P, Mossiat C, Bertrand N, Giroud M, Beley A, Marie C: N-Acetylaspartate, a marker of both cellular dysfunction and neuronal loss: its relevance to studies of acute brain injury. J Neurochem. 2001, 77 (2): 408-15. 10.1046/j.1471-4159.2001.00285.x.
Ellis CM, Lemmens G, Williams SC, Simmons A, Dawson J, Leigh PN, Chaudhuri KR: Changes in putamen N-acetylaspartate and choline ratios in untreated and levodopa-treated Parkinson's disease: a proton magnetic resonance spectroscopy study. Neurology. 1997, 49 (2): 438-44.
Grammas P: A damaged microcirculation contributes to neuronal cell death in Alzheimer's disease. Neurobiol Aging. 2000, 21 (2): 199-205. 10.1016/S0197-4580(00)00102-0.
Tomimoto H, Ihara M, Wakita H, Ohtani R, Lin JX, Akiguchi I, Kinoshita M, Shibasaki H: Chronic cerebral hypoperfusion induces white matter lesions and loss of oligodendroglia with DNA fragmentation in the rat. Acta Neuropathol (Berl). 2003, 106 (6): 527-34. 10.1007/s00401-003-0749-3.
Ni JW, Ohta H, Matsumoto K, Watanabe H: Progressive cognitive impairment following chronic cerebral hypoperfusion induced by permanent occlusion of bilateral carotid arteries in rats. Brain Res. 1994, 653 (1–2): 231-6. 10.1016/0006-8993(94)90394-8.
Ni JW, Matsumoto K, Li HB, Murakami Y, Watanabe H: Neuronal damage and decrease of central acetylcholine level following permanent occlusion of bilateral common carotid arteries in rat. Brain Res. 1995, 673 (2): 290-6. 10.1016/0006-8993(94)01436-L.
Xie L, Helmerhorst E, Taddei K, Plewright B, Van Bronswijk W, Martins R: Alzheimer's beta-amyloid peptides compete for insulin binding to the insulin receptor. J Neurosci. 2002, 22 (10): RC221:1-5.
Solano DC, Sironi M, Bonfini C, Solerte SB, Govoni S, Racchi M: Insulin regulates soluble amyloid precursor protein release via phosphatidyl inositol 3 kinase-dependent pathway. FASEB J. 2000, 14 (7): 1015-22.
Zhao L, Teter B, Morihara T, Lim GP, Ambegaokar SS, Ubeda OJ, Frautschy SA, Cole GM: Insulin-degrading enzyme as a downstream target of insulin receptor signaling cascade: implications for Alzheimer's disease intervention. J Neurosci. 2004, 24 (49): 11120-6. 10.1523/JNEUROSCI.2860-04.2004.
Gasparini L, Gouras GK, Wang R, Gross RS, Beal MF, Greengard P, Xu H: Stimulation of beta-amyloid precursor protein trafficking by insulin reduces intraneuronal beta-amyloid and requires mitogen-activated protein kinase signaling. J Neurosci. 2001, 21 (8): 2561-70.
Rivera EJ, Goldin A, Fulmer N, Tavares R, Wands JR, de la Monte SM: Insulin and insulin-like growth factor expression and function deteriorate with progression of Alzheimer's disease: link to brain reductions in acetylcholine. J Alzheimers Dis. 2005, 8 (3): 247-68.
Harkany T, O'Mahony S, Kelly JP, Soos K, Toro I, Penke B, Luiten PG, Nyakas C, Gulya K, Leonard BE: Beta-amyloid(Phe(SO3H)24)25–35 in rat nucleus basalis induces behavioral dysfunctions, impairs learning and memory and disrupts cortical cholinergic innervation. Behav Brain Res. 1998, 90 (2): 133-45. 10.1016/S0166-4328(97)00091-0.
Yamaguchi Y, Kawashima S: Effects of amyloid-beta-(25–35) on passive avoidance, radial-arm maze learning and choline acetyltransferase activity in the rat. Eur J Pharmacol. 2001, 412 (3): 265-72. 10.1016/S0014-2999(01)00730-0.
Pedersen WA, McMillan PJ, Kulstad JJ, Leverenz JB, Craft S, Haynatzki GR: Rosiglitazone attenuates learning and memory deficits in Tg2576 Alzheimer mice. Exp Neurol. 2006, 199 (2): 245-8. 10.1016/j.expneurol.2006.01.018.
Watson GS, Cholerton BA, Reger MA, Baker LD, Plymate SR, Asthana S, Fishel MA, Kulstad JJ, Green PS, Cook DG, Kahn SE, Keeling ML, Craft S: Preserved cognition in patients with early Alzheimer disease and amnestic mild cognitive impairment during treatment with rosiglitazone: a preliminary study. Am J Geriatr Psychiatry. 2005, 13 (11): 950-8. 10.1176/appi.ajgp.13.11.950.
Risner ME, Saunders AM, Altman JF, Ormandy GC, Craft S, Foley IM, Zvartau-Hind ME, Hosford DA, Roses AD: Efficacy of rosiglitazone in a genetically defined population with mild-to-moderate Alzheimer's disease. Pharmacogenomics J. 2006, 6 (4): 246-54.
Strum JC, Shehee R, Virley D, Richardson J, Mattie M, Selley P, Ghosh S, Nock C, Saunders A, Roses A: Rosiglitazone induces mitochondrial biogenesis in mouse brain. J Alzheimers Dis. 2007, 11 (1): 45-51.
Andreeva N, Khodorov B, Stelmashook E, Cragoe E, Victorov I: Inhibition of Na+/Ca2+ exchange enhances delayed neuronal death elicited by glutamate in cerebellar granule cell cultures. Brain Res. 1991, 548 (1–2): 322-5. 10.1016/0006-8993(91)91141-M.
Kiedrowski L, Brooker G, Costa E, Wroblewski JT: Glutamate impairs neuronal calcium extrusion while reducing sodium gradient. Neuron. 1994, 12 (2): 295-300. 10.1016/0896-6273(94)90272-0.
Paxinos G, Watson CH: The rat brain in stereotaxic coordinates. 1998, Academic Press
Morris R: Development of a water-maze procedure for studying spatial leaning in the rat. J Neurosci Methods. 1984, 11: 47-60. 10.1016/0165-0270(84)90007-4.
Gadian DG, Frackoviak RSJ, Crockard HA, Proctor E, Allen K, Williams SR, Ross Russel RW: Acute cerebral ischemia: concurrent changes in cerebral blood flow, energy metabolites, pH, and lactate measured with hydrogen clearance and 31P and 1H nuclear magnetic resonance spectroscopy. I. Methodology. J Cerebr Blood Flow Metab. 1987, 7: 199-206.
Williams SR, Gadian DG, Proctor E: A method for lactate detection in vivo by spectral editing without the need for double irradiation. J Magn Reson. 1986, 66: 562-567.
Fonnum F: A radiochemical method for the estimation of choline acetyltransferase. Biochem J. 1966, 100 (2): 479-484.
Acknowledgements
This work was funded by Biosignal Ltd., Moscow, Russia.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The author(s) declare that they have no competing interests.
Authors' contributions
ZIS carried out surgical operations, behavioral tests, and the data analysis in animal studies. ATP carried out surgical operations and behavioral tests in animal studies. VVS participated in the design of behavioral studies, critical intellectual discussion, and manuscript evaluation/critique. TPS carried out the in vitro studies with CGN cultures and data analysis. YES carried out the in vitro studies with CGN cultures and data analysis. NAP carried out the in vitro studies with CGN cultures and data analysis. VGP participated in the design of the in vitro studies with CGN cultures, critical intellectual discussion, and manuscript evaluation/critique. NAS carried out the 1H NMR study in vivo and the data analysis. EIZ carried out the measurement of ChAT activity and the data analysis. IAP conceived, designed and coordinated the study, and drafted the manuscript. All authors read and approved the final manuscript.
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
Open Access This article is published under license to BioMed Central Ltd. This is an Open Access article is distributed under the terms of the Creative Commons Attribution License ( https://creativecommons.org/licenses/by/2.0 ), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Storozheva, Z.I., Proshin, A.T., Sherstnev, V.V. et al. Dicholine salt of succinic acid, a neuronal insulin sensitizer, ameliorates cognitive deficits in rodent models of normal aging, chronic cerebral hypoperfusion, and beta-amyloid peptide-(25–35)-induced amnesia. BMC Pharmacol 8, 1 (2008). https://doi.org/10.1186/1471-2210-8-1
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1471-2210-8-1