
Abstract
Background
Development of proximal and distal colorectal cancers involve partly different mechanisms associated with the microsatellite instability (MSI) and the chromosomal instability (CIN) pathways. Colorectal cancers in patients under 50 years of age represent about 5% of the total number of tumors and have been associated with an increased frequency of MSI tumors. However, MSI and CIN may play different roles in the development of colon cancer and rectal cancer, and we have specifically investigated their contribution to the development of rectal cancer at young age.
Methods
Thirty rectal cancers diagnosed before the age of 50 were characterized for DNA-ploidy, MSI, mutations of KRAS and CTNNB1 and immunohistochemical expression of p53, β-catenin and of the mismatch repair (MMR) proteins MLH1 and MSH2.
Results
DNA aneuploidy was detected in 21/30 tumors, KRAS mutations in 6 tumors, no mutations of CTNNB1 were detected but immunohistochemical staining for β-catenin showed nuclear staining in 6 tumors, and immunohistochemical expression of p53 was detected in 18 tumors. MSI was detected in 3/30 tumors, all of which showed and immunohistochemical loss of staining for the MMR protein MSH2, which strongly indicates a phenotype associated with hereditary nonpolyposis colorectal cancer (HNPCC).
Conclusions
MSI occurs only in a small fraction of the tumors from young patients with rectal cancer, but when present it strongly indicates an underlying HNPCC-causing mutation, and other mechanisms than HNPCC thus cause rectal cancer in the majority of young patients.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Background
Colorectal cancer represents 12% of the cancer cases in Sweden with about 5% of the patients being below 50 years of age [1]. Rectal cancer constitutes about 1/3 of the large bowel cancers and 5% of the patients are younger than 50 years of age at diagnosis. Tumor development at young age may be an indicator of hereditary cancer, such as hereditary nonpolyposis colorectal cancer (HNPCC), which is characterized by development of colorectal cancer at a mean age of 43 years and an increased risk also for cancer of the endometrium, ovaries, urothelium and the small intestine [2]. HNPCC has in population-based studies been demonstrated to account for about 2% of colorectal cancer, but has been estimated to cause about 1/3 of the colorectal cancer cases occurring at young age [3, 4]. HNPCC is caused by germline mutations in one of several mismatch-repair (MMR) genes, most commonly in the genes MLH1 and MSH2. Studies of colorectal cancers from young patients have revealed MSI frequencies between 17 and 58% (compared to about 15% in sporadic colorectal cancer), with the higher frequencies found in patients below age 35 at diagnosis [5–10].
Colorectal cancer cells evade growth control through a series of genetic changes, which have been recognized to affect distinctive pathways. The chromosomal instability (CIN) pathway is involved in about 70–80% of the tumors and is characterized by aneuploidy, multiple chromosomal rearrangements, and an accumulation of somatic mutations in oncogenes such as KRAS and β-catenin and tumor suppressor genes such as TP53 and the adenomatous polyposis coli (APC) gene [11]. The microsatellite instability (MSI) pathway characterizes about 15% of the colorectal cancers and is associated with diploid or near-diploid tumors and a defective MMR machinery, which is reflected as MSI in the tumor tissue. MSI tumors are more likely to be right-sided with mucinous histology, poor differentiation and tumor-infiltrating lymphocytes [12]. MSI is only infrequently caused by germline HNPCC-causing MMR gene mutations, but is more commonly due to somatic hypermethylation of the MLH1 promoter [13].
Differences in aetiology, clinical behaviour and histopathology between colon cancer and rectal cancer suggest different preferred tumorigenic pathways in these tumor types. The few molecular genetic studies that have separately analyzed these clinical entities demonstrate a lower frequency of MSI and KRAS mutations and a higher frequency of aneuploid tumors, p53 mutation/overexpression and β-catenin overexpression in rectal cancer. These findings are likely to reflect that CIN and the TP53 pathways contribute more than MSI to rectal cancer development [3, 14–18]. However, the importance of MSI versus CIN in rectal cancer development in young patients has to our knowledge not been characterized. We have therefore studied 30 rectal cancers from patients below 50 years of age, an age limit chosen in accordance with the Amsterdam criteria for the classification of HNPCC [2] with respect to flow cytometric DNA content, MSI, KRAS mutations and immunohistochemical expression of the MMR proteins MLH1 and MSH2 and the tumor suppressor proteins p53 and β-catenin. The aim was to clarify possible differences between colon cancer and rectal cancer in young patients as regards the contribution of the MSI and the CIN pathways to tumor formation and to estimate the proportion of cases attributable to defective MMR.
Methods
Patients
Thirty patients diagnosed with histologically verified adenocarcinomas of the rectum before age 50 were recruited for the study (Table 1). Tumor stage was Dukes' A in 6 patients, B in 6, C in 12 and D in 6 patients. Tumor location was low (<5 cm from the anal verge) in 5 patients, middle rectum (5–10 cm) in 13, and high (10–15 cm) in 12 patients. Tumor histopathology was reviewed by a gastrointestinal pathologist (B.H.) and differentiation was classified as moderate in 23 tumors and mucinous /signet ring cell in 5 tumors, as well differentiated in 1 tumor and as poor in 1 tumor. The patients had a mean age of 42 (range 25–49) years and the material included 9 women and 21 men. Family histories of cancer were available from 28 of the 30 patients; 20 had no history of familial colorectal and/or endometrial cancer, 6 had one or more relatives with colorectal and/or endometrial cancer at any age and 2 patients fulfilled the Amsterdam II criteria for the classification of HNPCC, whereas data on family history was not available from 2 patients (Table 1) [2]. Adjuvant treatment with 5-fluorouracil (5-FU) and leucovorin (in one case combined with oxaliplatin) was given to 16 patients. Median follow-up for the survivors was 46 (range 19–85) months. Six patients had metastasis at diagnosis and 9 patients died from tumor median 21 (range 3–40) months after diagnosis (Table 1). Paraffin embedded tumor specimens were retrieved, and were sectioned and stained with Hematoxylin & Eosin to verify that representative tumor tissue was present in the sample. Three 10 μM paraffin sections were used for DNA extraction (as described below) and 3 μM sections were used for immunohistochemical staining. Permission for the study was granted by the Lund university ethics committee.
DNA-extraction, PCR amplification and DNA sequencing
DNA was extracted from three 10-μM sections of paraffin embedded tumor tissue using proteinase-K incubation for 2–12 hours at 65°C. After incubation, the samples were boiled for 10 min to inactivate the enzyme, quickly centrifuged and 1 μL was used for the PCR amplifications. PCR was performed using AmpliTaq Gould (Perkin Elmer – Roche, Branchburg, NJ, USA) and the reagents supplied according to the PCR programme: initial denaturation (94°C, 7 min) followed by 35 cycles of denaturation (94°C, 15 sec), annealing (45–50°C, 15 sec) and extension (72°C, 15 sec) whereafter a final elongation step at 72°C for 10 min was added followed by cooling at 4°C. All primer sequences and exact PCR conditions are available from the authors upon request. DNA sequencing was performed on an Applied Biosystems 373 sequencer using cycle sequencing with AmpliTaq polymerase FS and the reagents supplied by Applied Biosystems (Foster City, CA, USA).
MSI-analysis
MSI was determined using the mononucleotide markers BAT25, BAT26, BAT34C4, BAT40 and the dinucleotide markers D2S123 and D17S250. All these MSI markers are among those recommended in the NCI reference panel for the classification of MSI [19]. All primer sequences are available from the authors upon request. Fluorescently labelled forward primers were used for the PCR reactions: Tet (green) for BAT25, Fam (blue) for BAT26, BAT34C4 and D2S123 and Hex (yellow) for BAT40 and D17S250. The PCR products were mixed with formamide and GeneScan 500 TAMRA size standard (Applied Biosystems), denatured and separated in Performance Optimized Polymer-4 (POP-4™) on a ABI PRISM™ 310 Genetic Analyzer (Applied Biosystems). The tumors were classified as MSI-high if at least 2/5 markers displayed MSI, as MSI-low if instability was present in 1/5 markers and as MSS if none of the 5 markers showed instability.
CTNNB1mutations
Exon 3 of the CTNNB1 gene, which codes for β-catenin, was analysed using DNA sequencing and a PCR-based assay designed to detect larger deletions of this region. Exon 3 was sequenced using the primers 5'-TGTAAAACGACGGCCAGTCGTATTTATAGCTGATTTGA-3' (forward) and 5'-TACTCTTACCAGCTACTTGT-3' (reverse). Genomic deletions involving exon 3 were analyzed by PCR amplifiction using the primers 5'-CCAGCGTGGACAATGGCTAC-3' (forward) and 5'-TGAGCTCGAGTCATTGCATAC-3' (reverse), followed by size separation in acrylamide gels.
KRASmutations
KRAS was analyzed for point mutations in codons 12, 13 and 61 by sequencing. PCR amplification was performed in two fragments using the primer pairs K1 (covering codons 12–13) 5'-GGCCTGCTGAAAATGACTGA-3' (forward) and 5'-GTCCTGCACCAGTAATATGC-3' (reverse) and K2 (covering codon 61) 5'-TTCCTACAGGAAGCAAGTAG-3' (forward) and 5'-CACAAAGAAAGCCCTCCCCA-3' (reverse).
Immunohistochemistry for TP53, β-catenin, MLH1 and MSH2
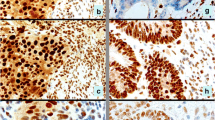
Fresh 5-μM sections from paraffin embedded tumor blocks were transferred to glass slides (DAKO ChemMate capillary gap microscope slides, 75 μM, DAKO A/S, Denmark). Following deparaffinisation in xylene and rehydration through graded ethanol, the sections were microwave treated at 750 W in citrate or EDTA buffer (Table 2) for 15 min to achieve antigen retrieval. Staining was performed in an automated immunostainer (TechMate™ 500 Plus, DAKO A/S, Denmark) using the biotin-streptavidin-peroxidase procedure with diaminobenzidine as the chromogen. Endogenous peroxidase activity was blocked by incubation in H2O2. All antibodies used were commercially available mouse monoclonal IgG's (Table 2). After counterstaining with hematoxylin the slides were dehydrated in ascending concentrations of alcohol and mounted. The immunohistochemical staining (Fig. 1) was independently evaluated by two of the investigators (E.F. and M.N.). The staining for MLH1 and MSH2 was nuclear and was classified as present or absent in the tumor tissue, and retained expression was in all tumors identified in surrounding normal crypts and in the tumor stroma. Nuclear p53 staining was evaluated in 5 high power fields (HPFs) (x40) and was classified as positive if >5% of the tumor cell nuclei were stained. Immunohistochemical staining for β-catenin normally results in a membraneous and/or a weak cytoplasmic staining, whereas a mutation leading to β-catenin stabilization is reflected as an increased cytoplasmic or nuclear staining. We classified the predominant β-catenin staining pattern in 5 HPFs according to cellular localization as membraneous, cytoplasmic or nuclear.

Immunohistochemical staining of tumor 10 a) normal nuclear staining for MLH1 in the tumor as well as in stromal tissue, b) loss of nuclear expression of MSH2 in tumor tissue but retained staining in stromal cells, c) nuclear staining for p53, and d) membraneous and intense cytoplasmic staining for β-catenin.
DNA flow cytometry
50-μM sections from paraffin embedded tumor tissue were used for flow cytometric DNA analysis. The nuclear suspension was stained with propidium iodide for the DNA quantification in an Ortho-Cytoron Absolute flow cytometer (Ortho Diagnostics, Raritan, NJ, USA). Up to 20,000 nuclei were analyzed per sample, which created a histogram profile with 256 channel resolution. Chicken and trout red blood cells were used as internal controls. A cell population was defined as a G0/G1 peak with a corresponding G2 peak. Tumors with a single G0/G1 peak were classified as DNA diploid (DNA-index of 1.0). If more than one cell population was present the sample was classified as aneudiploid. The degree of DNA aneuploidy was expressed as the DNA index (DI) which represents the position of the aneuploid peak in relation to the diploid reference cells. ModFitLT 3.0 software from Verity Software House Inc. (Topsham, ME, USA) was used for the analysis of the DNA histograms.
Results
The results of the molecular genetic characterization of the tumors are presented in Table 3. In summary, aneuploidy was found in 21/30 tumors (7 of which were near-diploid with a DI of 1.1–1.2) whereas 9 tumors (including the 3 MSI tumors) were diploid. No tumor displayed any mutation involving exon 3 of CTNNB1, but immunohistochemical staining for β-catenin revealed a membraneous staining in 5 tumors, a cytoplasmic staining in 19 tumors and a nuclear staining for β-catenin in 6 tumors. p53 was immunohistochemically expressed in 18/30 tumors; strong wide-spread nuclear staining was found in 16 tumors and a weaker staining in >5% of the tumor cells in 2 tumors. KRAS mutations were identified in 6 patients (20%); all mutations were missense mutations, 5 mutations occurred in codon 12 and one in codon 13, whereas no mutation was detected in codon 61. All 6 tumors with KRAS mutations displayed immunohistochemical p53 protein expression. MSI was found in 3 tumors and the remaining 27 tumors were MSS. All 30 tumors displayed normal expression of MLH1, whereas loss of expression for MSH2 was detected in 3 tumors. A family history of colorectal cancer (in 2 patients in combination with endometrial cancer) was reported in 8/28 evaluable patients, 2 of which also fulfilled the Amsterdam II criteria for the diagnosis of HNPCC.
Discussion
We characterized 30 rectal cancers from patients younger than 50 years at diagnosis with respect to tumor biologic phenomena associated with the MSI and the CIN pathways for colorectal tumor development. Rectal cancer has been associated with a higher frequency of aneuploidy, MSS tumors, mutations in KRAS and TP53 and nuclear accumulation of β-catenin compared to colon cancer, which probably reflects different preferred pathways in the development of these tumor types [3, 12, 14–18]. Our findings of high frequencies of MSS (27/30) tumors, aneuploidy (21/30), immunohistochemical p53 expression (18/30) and increased β-catenin expression (25/30) (Table 3) is in line with previous reports of genetic alterations in rectal cancer, but support such a model for the development of rectal cancer also in the majority of young patients.
Of the 30 rectal cancers investigated herein, 3 showed MSI, which is higher than the 1–2% MSI detected in larger series of rectal cancers, but lower than the 15% MSI tumors observed among sporadic colorectal cancers. This finding indicates that the frequency of MSI in young patients with rectal cancer is considerably lower than the 17–58% MSI tumors and 20–40% HNPCC-causing mutations reported in mixed series of colorectal cancers from young patients [5–10, 14, 18]. All three MSI tumors identified in our study displayed immunohistochemical loss of MSH2 expression. Whereas immunohistochemical loss of MLH1 may reflect either a germline mutation or somatic hypermethylation of the MLH1 promoter, loss of MSH2 expression is almost exclusively caused by a germline mutation of the MSH2 gene [12, 19, 20]. Thus, hypermethylation of MLH1 plays a limited role in rectal cancers from young patients, which is in accordance with an association between MLH1 hypermethylation and proximal tumors, older patients and female sex [13]. Consequently, the finding of MSI in a rectal cancer is, together with a family history of the disease, a strong indicator of HNPCC. Although HNPCC-associated tumors show a prediliction for the proximal colon about 40% of HNPCC-associated tumors will develop in the distal colon or within the rectum.
Mutations in codons 12 and 13 of KRAS are found in about 1/3 of colorectal tumors [21, 22]. These mutations develop early in colorectal tumor progression [23]. The prognostic value of KRAS mutations is somewhat unclear, but a recent study compiling data from over 3000 patients with colorectal cancer, did in a multivariate analysis demonstrate a significant impact on survival by the codon 12 glycine → alanine mutation, which is found in about 9% of the tumors [24]. We identified KRAS mutations in 6/30 rectal cancers (Table 3). All mutations were heterozygous missense mutations of types that have previously been described, and one patient displayed the codon 12 glycine → alanine change. A lower frequency of KRAS mutations has been found in distal tumors and an inverse relationship between MSI and alterations of KRAS and TP53 mutations has been described [22], a finding that is supported by our data with KRAS mutations in 6/30 tumors and nuclear expression of p53 in 18/30 rectal cancers.
β-catenin is involved in cell-cell adhesion as well as in signal transduction. Free β-catenin is bound to the adenomatosis polyposis coli (APC) protein and to glycogen synthase kinase 3B (GSK-3B), which phosphorylate serine/threonine residues in β-catenin and thereby destabilizes the protein, which is degraded by the ubiquitin-proteasome pathway. Mutations of APC or of the regulatory domain of CTNNB1 has been demonstrated in colorectal cancer and lead to stabilization and cytoplasmic accumulation of free β-catenin [25, 26]. Increased cytoplasmic levels of β-catenin allows the protein to enter the nucleus where it promotes transcriptional activation through the TCF/LEF transcription factors. We found no mutations in the regulatory domain (exon 3) of the CTNNB1 gene, but immunohistochemitry for β-catenin revealed an increased cytoplasmic expression or a nuclear accumulation in 25/30 tumors. Nuclear accumulation of β-catenin has been reported to occur at a higher frequency in rectal cancer than in colon cancer, whereas mutations affecting the CTNNB1 gene, have been reported to occur at a higher frequency in MMR-deficient tumors [26, 27].
Conclusions
We conclude that rectal cancer, also when developing in young patients, is frequently associated with DNA-aneuploidy, increased expression of p53 and β-catenin, whereas MSI and mutations in KRAS occur at a low frequency.
In this study only 10% of the rectal cancers from young patients were MSI-high, but all of these tumors showed loss of expression for MSH2, which suggests an underlying HNPCC-causing mutation. Thus, other mechanisms than defective MMR and HNPCC probably cause rectal cancer development in the majority of young patients.
References
The National Board of Health and Welfare: Cancer Incidence in Sweden 1999. Stockholm. 1999
Vasen HF, Watson P, Mecklin JP, Lynch HT: New clinical criteria for hereditary nonpolyposis colorectal cancer (HNPCC, Lynch syndrome) proposed by the International Collaborative group on HNPCC. Gastroenterology. 1999, 116: 1453-56.
Aaltonen LA, Salovaara R, Kristo P, Canzian F, Hemminki A, Peltomäki P, Chadwick RB, Käärinen H, Eskelinen M, Järvinen H, et al: Incidence of hereditary nonpolyposis colorectal cancer and the feasibility of molecular screening for the disease. N Engl J Med. 1998, 338: 1481-87. 10.1056/NEJM199805213382101.
Samowitz WS, Curtin K, Lin HH, Robertson MA, Schaffer D, Nichols M, Gruenthal K, Leppert MF, Slattery ML: The colon cancer burden of genetically defined hereditary nonpolyposis colorectal cancer. Gastroenterology. 2001, 121: 830-838.
Liu B, Farrington SM, Petersen GM, Hamilton SR, Parsons R, Papadopoulos N, Fujiwara T, Jen J, Kinzler KW, Wyllie AH, et al: Genetic instability occurs in the majority of young patients with colorectal cancer. Nat Med. 1995, 1: 348-52.
Brassett C, Joyce JA, Froggatt NJ, Williams G, Furniss D, Walsh S, Millar R, Evans DG, Maher ER: Microsatellite instability in early onset and familial colorectal cancer. J Med Genet. 1996, 33: 981-985.
Capozzi E, Della Puppa L, Fornasarig M, Pedroni M, Boiocchi M, Viel A: Evaluation of the replication error phenotype in relation to molecular and clinicopathological features in hereditary and early onset colorectal cancer. Eur J Cancer. 1999, 35: 289-95. 10.1016/S0959-8049(98)00317-7.
Gryfe R, Kim H, Hsieh ET, Aronson MD, Holowaty EJ, Bull SB, Redston M, Gallinger S: Tumor microsatellite instability and clinical outcome in young patients with colorectal cancer. N Engl J Med. 2000, 342: 69-77. 10.1056/NEJM200001133420201.
Naidoo R, Tarin M, Chetty R, Phil D: A comparative microsatellite analysis of colorectal cancer in patients <35 years and >50 years of age. Am J Gastroeneterol. 2000, 95: 3266-75. 10.1016/S0002-9270(00)02001-3.
Su JH, Lim SD, Kim JC, Hong SH, Kang GH: Comparison of clinicopathologic characteristics and genetic alterations between microsatellite instability-positive and microsatellite instability-negative sporadic colorectal carcinomas in patients younger than 40 years old. Dis Colon Rectum. 2002, 45: 219-228.
Lengauer C, Kinzler KW, Vogelstein B: Genetic instability in colorectal cancers. Nature. 1997, 386: 623-627. 10.1038/386623a0.
Young J, Simms LA, Biden KG, Wynter C, Whitehall V, Karamatic R, George J, Goldblatt J, Walpole I, Robin S-A, et al: Features of colorectal cancers with high-level microsatellite instability occurring in familial and sporadic settings: parallel pathways of tumorigenesis. Am J Pathol. 2001, 159: 2107-2116.
Thibodeau SN, French AJ, Cunningham JM, Tester D, Burgart LJ, Roche PC, McDonnell SK, Schaid DJ, Walsh Vockley C, Michels VV, et al: Microsatellite instability in colorectal cancer: different mutator phenotypes and the principal involvement of hMLH1. Cancer Res. 1998, 58: 1713-18.
Lothe R, Peltomäki P, Meling GI, Aaltonen LA, Nyström-Lathi M, Pylkkanen L, Heimdal K, Andersen TI, Möller P, Rognum TO, et al: Genomic instability in colorectal cancer: relationship to clinicopathological variables and family history. Cancer Res. 1993, 53: 5849-52.
Soong R, Grieu F, Robbins P, Dix B, Chen D, Parsons R, House A, Iacopetta B: p53 alterations are associated with improved prognosis in distal colonic carcinomas. Clin Cancer Res. 1997, 3: 1405-11.
Nilbert M, Planck M, Fernebro E, Borg Å, Johnson A: Microsatellite instability is rare in rectal carcinomas and signifies hereditary cancer. Eur J Cancer. 1999, 35: 942-945. 10.1016/S0959-8049(99)00045-3.
Bleeker WA, Hayes VM, Karrenbeld A, Hofstra RM, Hermans J, Buys CC, Plukk JT: Impact of KRAS and TP53 mutations on survival in patients with left- and right-sided Dukes' C colon cancer. Am J Gastroenterol. 2000, 95: 2953-7. 10.1016/S0002-9270(00)01119-9.
Kapiteijn E, Liefers GJ, Los LC, Kranenbarg EK, Hermans J, Tollenaar RAEM, Moriya Y, van de Velde CJH, van Krieken JHJM: Mechanisms of oncogenesis in colon versus rectal cancer. J Pathol. 2001, 195: 171-178. 10.1002/path.918.
Boland CR, Thibodeau SN, Hamilton SR, Sidransky D, Eshleman JR, Burt RW, Meltzer SJ, Rodriguez-Bigas MA, Fodde R, Ranzani GN, et al: A national cancer institute workshop on microsatellite instability for cancer detection and familial predisposition: development of international criteria for the determination of microsatellite instability in colorectal cancer. Cancer Res. 1998, 58: 5248-5257.
Marcus VA, Madlensky L, Gryfe R, Kim H, Millar A, Temple LKF, Hsieh E, Hiruki T, Narod S, Bapat BV, et al: Immunohistochemistry for hMLH1 and hMSH2: a practical test for DNA mismatch repair-deficient tumors. Am J Surg Pathol. 1999, 23: 1248-55. 10.1097/00000478-199910000-00010.
Lindor NM, Burgart LJ, Leontovich O, Goldberg RM, Cunningham JM, Sargent DJ, Walsh-Vockley C, Petersen GM, Walsh MD, Leggett BA, et al: Immunohistochemistry versus microsatellite instability testing in phenotyping colorectal tumors. J Clin Oncol. 2002, 20: 1043-1048. 10.1200/JCO.20.4.1043.
Mutter GL, Wada H, Faquin WC, Enomoto T: K-ras mutations appear in the premalignant phase of both microsatellite stable and unstable endometrial carcinogenesis. Mol Pathol. 1999, 52: 257-62.
Samowitz WS, Holden JA, Curtin K, Edwards SL, Walker AR, Lin HA, Robertson MA, Nichols MF, Gruenthal KM, Lynch BJ, et al: Inverse relationship between microsatellite instability and K-ras and p53 gene alterations in colon cancer. Am J Pathol. 2001, 158: 1517-21.
Andreyev HJN, Norman AR, Cunningham D, Oates J, Dix BR, Iacopetta BJ, Young J, Walsh T, Ward R, Hawkins N, et al: Kirsten ras mutations in patients with colorectal cancer: the "RASCAL II" study. Br J Cancer. 2001, 85: 692-96. 10.1054/bjoc.2001.1964.
Hugh TJ, Dillon SA, O'Dowd G, Getty B, Pignatelli M, Poston GJ, Kinsella AR: B-catenin expression in primary and metastatic colorectal carcinoma. Int J Cancer. 1999, 82: 504-511. 10.1002/(SICI)1097-0215(19990812)82:4<504::AID-IJC6>3.0.CO;2-6.
Sparks AB, Morin PJ, Vogelstein B, Kinzler KW: Mutational analysis of the APC/B-catenin/Tcf pathway in colorectal cancer. Cancer Res. 1998, 58: 1130-34.
Miyaki M, Iijma T, Kimura J, Yasuno M, Mori T, Hayasi Y, Koike M, Shitara N, Iwama T, Kuroki T: Frequent mutation of B-catenin and APC genes in primary colorectal tumors from patients with hereditary nonpolyposis colorectal cancer. Cancer Res. 1999, 59: 4506-9.
Pre-publication history
The pre-publication history for this paper can be accessed here:http://www.biomedcentral.com/1471-2407/2/25/prepub
Acknowledgments
We would like to acknowledge Eva Rambech and Annette Persson for technical assistance performing the immunohistochemistry. Financial support was granted from the Swedish Cancer Society, the G. Nilsson Cancer Fund, the King Gustaf V Jubilee Foundation and the Mrs. B. Kamprad Cancer Fund.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
None declared.
Authors' contributions
Author 1 was responsible for the design of the study, obtained tumor material, evaluated the immunohistochemistry, performed clinical correlations and summarized the results. Author 2 reviewed the histopathological material. Author 3 performed the flow cytometric DNA analyses. Author 4 conceived of the study, evaluated the immunohistochemistry, the MSI and the mutation analyses, and coordinated the results. Authors 1 and 4 both contributed to the writing of the paper.
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
This article is published under an open access license. Please check the 'Copyright Information' section either on this page or in the PDF for details of this license and what re-use is permitted. If your intended use exceeds what is permitted by the license or if you are unable to locate the licence and re-use information, please contact the Rights and Permissions team.
About this article
Cite this article
Fernebro, E., Halvarsson, B., Baldetorp, B. et al. Predominance of CIN versus MSI in the development of rectal cancer at young age. BMC Cancer 2, 25 (2002). https://doi.org/10.1186/1471-2407-2-25
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1471-2407-2-25