
Abstract
Background
Atopic dermatitis (AD) is a chronic inflammatory skin disorder caused by complex interaction of genetic and environmental factors. Besides mutations in the filaggrin gene, leading to impaired skin barrier function, variation in genes encoding additional skin proteins has been suggested to contribute to disease risk. Laminin 5, playing an important role in skin integrity, is composed of three subunits encoded by the LAMA3, LAMB3 and LAMC2 genes in which biallelic mutations cause epidermolysis bullosa junctionalis. We aimed at evaluating the role of variation in the LAMA3, LAMB3 and LAMC2 genes for AD pathogenesis.
Methods
29 single nucleotide polymorphisms (SNPs) were genotyped in the three genes in a German AD case–control cohort comprising 470 unrelated AD patients and 320 non-atopic controls by means of restriction enzyme digestion. Allele, genotype and haplotype frequencies were compared between cases and controls using chi-square testing and the Haploview software.
Results
Several SNPs in the LAMA3 gene showed significant association with AD in our cohort (p <0.01), while we did not detect association with variations in the LAMB3 and LAMC2 genes. Haplotype analysis additionally revealed several significantly associated haplotypes in the LAMA3 gene. Due to extensive linkage disequilibrium, though, we were not able to further differentiate the specific disease causing variation(s) in this region.
Conclusions
We established the LAMA3 gene as novel potential susceptibility gene for AD. Additional studies in independent cohorts are needed to replicate these results.
Similar content being viewed by others


Background
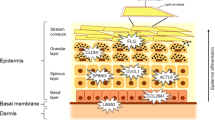
Atopic dermatitis (AD) is a chronic inflammatory skin disorder caused by complex interaction between genetic and environmental factors [1]. Besides dysregulated immune mechanisms that have long been suspected to play a major role for AD pathogenesis, the impact of an intact skin barrier in the protection against AD has gained increasing attention over the recent years [2]. In particular, the role of filaggrin, a major structural protein in the stratum corneum of the epidermis, has been extensively studied. Mutations in the FLG gene, located in the epidermal differentiation complex (EDC) on chromosome 1q21 [3], have consistently been associated with early-onset persistent AD [4]. It could be demonstrated that FLG mutations constitute the most significant known risk factor for AD development so far [5]. However, impaired skin barrier function has also been shown in AD patients without FLG mutations [6], suggesting that variation in genes encoding additional skin proteins may play a role in AD pathogenesis.
Laminin 5 is another protein that plays an important role for skin integrity [7]. It is comprised of three different subunits, built by the LAMA3 (alpha-3), LAMB3 (beta-3) and LAMC2 (gamma-2) polypeptide chains. Laminin 5 (also called laminin-332) is involved in connecting dermis and epidermis and induces adhesion, spreading and migration of human keratinocytes [8]. The three polypeptide chains are encoded by the LAMA3, LAMB3 and LAMC2 genes on chromosomes 18q11.2, 1q32.2 and 1q25.3, resp. [9]. Biallelic mutations in each of these three genes are known to cause epidermolysis bullosa junctionalis, a severe (type Herlitz) or less severe (type Non-Herlitz) skin disorder characterized by blisters and erosions of the skin [10]. Furthermore, laminin 5 synthesis is elevated during acute wound healing in healthy individuals [11].
Given the important role of mutations in skin barrier proteins in AD pathogenesis, we hypothesized that variation in one or more of the laminin 5 subunits may also confer risk for AD. Therefore we evaluated 29 single nucleotide polymorphisms (SNPs) in the three genes in a German AD case–control cohort and present first evidence that the LAMA3 gene may be a novel susceptibility gene for AD.
Methods
Subjects
470 unrelated patients with AD with a mean age of 19 ± 15 years (median 11 years) were recruited by a consultant specialist for AD (Q.P., Gladbeck, Germany) as described before [12]. AD diagnosis was established based on the criteria by Hanifin and Rajka [13]. Since the risk remains very high for primarily asymptomatic children to develop an allergic disease during childhood or even adulthood [14, 15], we chose to use non-allergic adults as a control group. Therefore, 320 individuals of at least 40 years (mean age 62 ± 11 years, median 63 years) that had neither self-reported allergies or allergic symptoms nor first degree relatives with allergic diseases were recruited in the same private practice as the patients. The controls further underwent clinical examination in order to exclude symptoms of AD, asthma or allergic rhinitis (see [12]). All participants were Germans of European ancestry and gave informed consent prior to enrolment. The Declaration of Helsinki protocols were followed and the study was approved by the Ethics Committee of the Ruhr-University Bochum.
Genotyping
DNA of AD patients and controls was extracted as described before [16]. We selected 29 SNPs in the three genes (17 in the LAMA3 gene, 8 in LAMB3, and 4 in LAMC2) that represented the haplotype block structures according to HapMap [17]. Genotyping was performed by polymerase chain reaction (PCR) followed by restriction enzyme digestion. PCR was done in a total volume of 10 μl, containing 40 ng DNA, 200 mmol of each dNTP, 1.5-3 mmol MgCl2, 5 pmol of each primer, and 0.5 U Taq-DNA-polymerase (Genecraft, Münster, Germany) on the RoboCycler or Biometra T cycler (Stratagene, Heidelberg, Germany and Biometra GmbH, Göttingen, Germany, respectively). After two initial cycles at 6° C and 3°C above the annealing temperature, 28–32 cycles of 95°C (30 sec), annealing temperature (30 sec) and 72°C (30 sec) were run. PCR products were subsequently digested with the respective restriction enzyme, the fragments separated on 2.5%-3.5% agarose gels in 1xTBE buffer (30–60 min, 200 V) and visualized with ethidium bromide (0.5% [w/v]). Additional information about primer sequences, PCR conditions and restriction enzymes is summarized in Additional file 1.
Statistics
Genotype and allele frequencies were compared between AD patients and controls according to the χ2 method; the significance threshold was set at p < 0.05. We evaluated every SNP for deviations from Hardy-Weinberg equilibrium (HWE) using the deFinetti program [18]. The program Haploview 4.0 [19] was used to estimate haplotype frequencies and test for haplotypic association. We applied Bonferroni correction for multiple tests; however, since this approach has been controversially discussed for genetic case–control studies [20], especially if several tightly linked SNPs within one gene are analyzed as in the present study, we also present the uncorrected p-values and discuss them as hypothesis-generating.
Results
All 29 SNPs showed genotypic distributions according to HWE. Of the 17 SNPs chosen for the LAMA3 gene, ten showed significant associations (p < 0.01 in the uncorrected analysis) with AD in the present cohort. The most significant results that also survived Bonferroni correction were obtained for rs8083184 and rs1711450, both located in the promoter region of LAMA3 (p = 0.0003, pcorr = 0.0087, Table 1; full genotype data is presented in Additional file 2). A high degree of linkage disequilibrium (LD) was observed for the SNPs in the LAMA3 gene region (Figure 1). Significant association extended into the 5′ region of LAMA3, including rs1613739 which is located in the neighbouring ANKRD29 gene. Two other SNPs in ANKRD29 (rs7238623 and rs8096061), however, did not show significant association results. Haplotype analyses revealed the existence of two haploblocks (Figure 1) with a common haplotype in block 1 that was highly significantly associated with AD (61.9% in cases vs. 51.1% in controls, p = 2.94×10-5, Table 2).

Haploblock structure of the LAMA3 region as revealed by Haploview 4.0 [19]. *SNPs associated with AD at p < 0.01 (uncorrected values) **SNPs associated with AD at p < 0.001 (uncorrected values).
In the LAMB3 gene, neither single SNP nor haplotype analyses revealed significant association with AD in the present cohort (Table 1; haplotype data not shown). In LAMC2, no single SNP showed association with AD; however, haplotype analyses revealed the existence of a rare protective haplotype (4% in cases vs. 6.9% in controls, p = 0.01; Table 3).
Discussion
To our knowledge, this is the first report of an association of AD with variation in the LAMA3 gene, encoding the alpha-chain of laminin 5. In a well-characterized German case–control cohort, we found significant association of both allelic and haplotypic frequencies in this gene with AD, suggesting that it may constitute a novel susceptibility gene for this frequent skin disease.
Of the 19 SNPs evaluated across the LAMA3 gene, ten showed genotypic or allelic association with AD at p < 0.01 (uncorrected values). Due to the extensive LD evident at the LAMA3 locus we were not able to further differentiate the specific disease causing variation(s) in this region. Significant association extended into the 5′ neighbouring gene ANKRD29, but additional SNPs in this gene did not show association with AD. The biological function of ANKRD29, encoding the ankyrin repeat domain-containing protein 29, is not yet known; however, for another protein of the same family, ANKRD17, a role in anti-bacterial innate immune pathways has been suggested [21]. For the gene located 3′ of LAMA3 (TTC39C), encoding the tetratricopeptide repeat protein 39C, the biological function is not yet clear either, and since the most significant results for LAMA3 were at the 5′ end of this large gene, we did not evaluate additional SNPs in the TTC39C gene. Taken together, even though we cannot exclude that the observed association with AD may be due to LD with a susceptibility variant in another gene in this region, our data highly points to LAMA3 as susceptibility gene for AD in the 18q11.2 region. SNPs in the genes encoding the beta- and gamma-chains of laminin 5, on the other hand, did not show convincing evidence for association with AD in this analysis. However, haplotype analysis suggested the existence of a rare protective haplotype in LAMC2.
The main role of laminin 5 in normal tissues is the maintenance of epithelial-mesenchymal cohesion in tissues exposed to external forces, such as skin and stratified squamous mucosa [22]. Genetic variation in laminin 5 components may thus contribute to reduced skin integrity and barrier function, as has been observed for FLG mutations [5], even though the exact underlying mechanisms still have to be elucidated. In each of the three genes analysed here, rare null mutations are known to cause epidermolysis bullosa junctionalis, a severe autosomal recessive skin disorder characterized by the development of blisters and skin erosions in response to minor injury or friction [10]. In contrast, we observed association of common variation in LAMA3 with the common complex disorder AD. This phenomenon is in line with findings for the SPINK5 gene, in which null mutations cause autosomal recessive Netherton syndrome while common variations have been associated with AD [23].
Our association results further contribute to the hypothesis that an intact skin barrier function plays a key part in AD pathogenesis. Additional to the established role of FLG mutations [24], associations with some other skin-related genes have been described recently. For example, mutations in the claudin-1 gene (CLDN1), encoding a major tight junction protein in the granular layer of the epidermis, were associated with AD in North American cohorts of both European and African American origin [25]. Further, polymorphisms near the OVOL1 and ACTL9 genes, both involved in epidermal proliferation and differentiation, showed genome-wide significant association with AD in a large meta-analysis of GWAS data, including 5,606 AD patients and 20,565 controls [26]. In a study cohort comprising AD patients from Germany, Poland and the Czech Republic, a 24-bp deletion in the gene encoding small proline-rich protein 3 (SPRR3), located within the EDC, was significantly associated with disease risk [27]. On the other hand, a deletion of the cornified envelope 3B and 3C genes within in the EDC was not associated with AD in a European cohort [28]. All of these results still await replication in independent cohorts. Altogether, though, evidence is accumulating that additional genes involved in epidermal differentiation and stability may be important for AD pathogenesis.
We are conscious of the fact that the cohort sizes of the present study are comparatively small so that the statistical power is only moderate. Furthermore, only for two SNPs in LAMA3 results remained significant after strict Bonferroni correction for multiple testing, leaving open the risk for false-positive results. However, the Bonferroni correction has been controversially discussed for genetic case–control studies [20], especially if several tightly linked SNPs within one gene are analyzed, as was the case in the present study. Therefore, we regard the results as hypothesis-generating and strongly encourage replication in additional cohorts.
Conclusions
We presented initial evidence for association of LAMA3 variation with AD, suggesting that variation in this gene may contribute to skin fragility and impaired barrier function underlying AD pathogenesis. Additional studies in independent populations as well as functional analyses of the associated variations appear warranted to replicate or extend these findings, since the genetic risk factors for AD might be increasingly included into prognostic and therapeutic strategies in the future. In more detail, an integrated approach including genotypic and phenotypic information as well as genetic and biological biomarkers has been proposed for example to identify patients who are prone to develop persistent AD and/ or additional asthma and start early intervention [29]. Further, therapeutic strategies targeting the skin barrier function are already under way [30]. Thus, elucidating the complex genetic background of AD is essential for paving the way towards a more individualized therapy in the future.
Abbreviations
- AD:
-
Atopic dermatitis
- EDC:
-
Epidermal differentiation complex
- HWE:
-
Hardy-Weinberg equilibrium
- LD:
-
Linkage disequilibrium
- SNP:
-
Single nucleotide polymorphism.
References
Holloway JW, Yang IA, Holgate ST: Genetics of allergic disease. J Allergy Clin Immunol 2010, 125: S81-S94. 10.1016/j.jaci.2009.10.071
Boguniewicz M, Leung DY: Atopic dermatitis: a disease of altered skin barrier and immune dysregulation. Immunol Rev 2011, 242: 233–246. 10.1111/j.1600-065X.2011.01027.x
Kypriotou M, Huber M, Hohl D: The human epidermal differentiation complex: cornified envelope precursors, S100 proteins and the ‘fused genes’ family. Exp Dermatol 2012, 21: 643–649. 10.1111/j.1600-0625.2012.01472.x
Irvine AD, McLean WH, Leung DY: Filaggrin mutations associated with skin and allergic diseases. N Engl J Med 2011, 365: 1315–1327. 10.1056/NEJMra1011040
McAleer MA, Irvine AD: The multifunctional role of filaggrin in allergic skin disease. J Allergy Clin Immunol 2013, 131: 280–291. 10.1016/j.jaci.2012.12.668
Jakasa I, Koster ES, Calkoen F, McLean WH, Campbell LE, Bos JD, Verberk MM, Kezic S: Skin barrier function in healthy subjects and patients with atopic dermatitis in relation to filaggrin loss-of-function mutations. J Invest Dermatol 2011, 131: 540–542. 10.1038/jid.2010.307
Nishiyama T, Amano S, Tsunenaga M, Kadoya K, Takeda A, Adachi E, Burgeson RE: The importance of laminin 5 in the dermal-epidermal basement membrane. J Dermatol Sci 2000, 24(Suppl 1):S51-S59.
Schneider H, Muhle C, Pacho F: Biological function of laminin-5 and pathogenic impact of its deficiency. Eur J Cell Biol 2007, 86: 701–717. 10.1016/j.ejcb.2006.07.004
Akutsu N, Amano S, Nishiyama T: Quantitative analysis of laminin 5 gene expression in human keratinocytes. Exp Dermatol 2005, 14: 329–335. 10.1111/j.0906-6705.2005.00275.x
Kiritsi D, Has C, Bruckner-Tuderman L: Laminin 332 in junctional epidermolysis bullosa. Cell Adh Migr 2013, 7: 135–141. 10.4161/cam.22418
Amano S, Akutsu N, Ogura Y, Nishiyama T: Increase of laminin 5 synthesis in human keratinocytes by acute wound fluid, inflammatory cytokines and growth factors, and lysophospholipids. Br J Dermatol 2004, 151: 961–970. 10.1111/j.1365-2133.2004.06175.x
Macaluso F, Nothnagel M, Parwez Q, Petrasch-Parwez E, Bechara FG, Epplen JT, Hoffjan S: Polymorphisms in NACHT-LRR (NLR) genes in atopic dermatitis. Exp Dermatol 2007, 16: 692–698. 10.1111/j.1600-0625.2007.00589.x
Hanifin JM, Rajka G: Diagnostic features of atopic dermatitis. Acta Derm Venereol 1980, 92: 44–47.
Bel EH: Clinical phenotypes of asthma. Curr Opin Pulm Med 2004, 10: 44–50. 10.1097/00063198-200401000-00008
De Marco R, Locatelli F, Cerveri I, Bugiani M, Marinoni A, Giammanco G: Incidence and remission of asthma: a retrospective study on the natural history of asthma in Italy. J Allergy Clin Immunol 2002, 110: 228–235. 10.1067/mai.2002.125600
Miller SA, Dykes DD, Polesky HF: A simple salting out procedure for extracting DNA from human nucleated cells. Nucleic Acids Res 1988, 16: 1215. 10.1093/nar/16.3.1215
International HapMap Consortium: The International HapMap Project. Nature 2003, 426: 789–796. 10.1038/nature02168
DeFinetti test for Hardy-Weinberg equilibrium. http://ihg.gsf.de/cgi-bin/hw/hwa1.pl
Barrett JC: Haploview: Visualization and analysis of SNP genotype data. Cold Spring Harb Protoc 2009, pdb ip71.
Boehringer S, Epplen JT, Krawczak M: Genetic association studies of bronchial asthma–a need for Bonferroni correction? Hum Genet 2000, 107: 197. 10.1007/s004390000353
Menning M, Kufer TA: A role for the Ankyrin repeat containing protein Ankrd17 in Nod1- and Nod2-mediated inflammatory responses. FEBS Lett 2013, 587: 2137–2142. 10.1016/j.febslet.2013.05.037
Rousselle P, Beck K: Laminin 332 processing impacts cellular behavior. Cell Adh Migr 2013, 7: 122–134. 10.4161/cam.23132
Norgett EE, Kelsell DP: SPINK5: both rare and common skin disease. Trends Mol Med 2002, 8: 7.
McLean WH, Irvine AD: Heritable filaggrin disorders: the paradigm of atopic dermatitis. J Invest Dermatol 2012, 132: E20-E21.
De Benedetto A, Rafaels NM, McGirt LY, Ivanov AI, Georas SN, Cheadle C, Berger AE, Zhang K, Vidyasagar S, Yoshida T, Boguniewicz M, Hata T, Schneider LC, Hanifin JM, Gallo RL, Novak N, Weidinger S, Beaty TH, Leung DY, Barnes KC, Beck LA: Tight junction defects in patients with atopic dermatitis. J Allergy Clin Immunol 2010, 127: 773–786.
Paternoster L, Standl M, Chen CM, Ramasamy A, Bonnelykke K, Duijts L, Ferreira MA, Alves AC, Thyssen JP, Albrecht E, Baurecht H, Feenstra B, Sleiman PM, Hysi P, Warrington NM, Curjuric I, Myhre R, Curtin JA, Groen-Blokhuis MM, Kerkhof M, Sääf A, Franke A, Ellinghaus D, Fölster-Holst R, Dermitzakis E, Montgomery SB, Prokisch H, Heim K, Hartikainen AL, Pouta A, et al.: Meta-analysis of genome-wide association studies identifies three new risk loci for atopic dermatitis. Nat Genet 2012, 44: 187–192.
Marenholz I, Rivera VA, Esparza-Gordillo J, Bauerfeind A, Lee-Kirsch MA, Ciechanowicz A, Kurek M, Piskackova T, Macek M, Lee YA: Association screening in the Epidermal Differentiation Complex (EDC) identifies an SPRR3 repeat number variant as a risk factor for eczema. J Invest Dermatol 2011, 131: 1644–1649. 10.1038/jid.2011.90
Bergboer JG, Zeeuwen PL, Irvine AD, Weidinger S, Giardina E, Novelli G, Den Heijer M, Rodriguez E, Illig T, Riveira-Munoz E, Campbell LE, Tyson J, Dannhauser EN, O'Regan GM, Galli E, Klopp N, Koppelman GH, Novak N, Estivill X, McLean WH, Postma DS, Armour JA, Schalkwijk J: Deletion of Late Cornified Envelope 3B and 3C genes is not associated with atopic dermatitis. J Invest Dermatol 2010, 130: 2057–2061. 10.1038/jid.2010.88
Bieber T, Cork M, Reitamo S: Atopic dermatitis: a candidate for disease-modifying strategy. Allergy 2012, 67: 969–975. 10.1111/j.1398-9995.2012.02845.x
Heimall J, Spergel JM: Filaggrin mutations and atopy: consequences for future therapeutics. Expert Rev Clin Immunol 2012, 8: 189–197. 10.1586/eci.11.100
Pre-publication history
The pre-publication history for this paper can be accessed here:http://www.biomedcentral.com/1471-5945/14/17/prepub
Acknowledgments
We thank Maike Kallenbach, Natascha Wirkus, Monika Harazin and Katharina Batzke for technical assistance and the patients for their cooperation in this study.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
SS was in charge of study design and statistical analysis and drafted the manuscript. QP and EPP were involved in patient recruitment and helped to draft the manuscript. JTE and SH conceived of the study, participated in its design and coordination and helped to draft the manuscript. All authors read and approved the final manuscript.
Electronic supplementary material
12895_2014_171_MOESM1_ESM.docx
Additional file 1: Primers, PCR conditions and restriction enzymes used for genotyping of polymorphisms in the LAMA3, LAMB3 and LAMC2 genes. (DOCX 17 KB)
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
This article is published under an open access license. Please check the 'Copyright Information' section either on this page or in the PDF for details of this license and what re-use is permitted. If your intended use exceeds what is permitted by the license or if you are unable to locate the licence and re-use information, please contact the Rights and Permissions team.
About this article

Cite this article
Stemmler, S., Parwez, Q., Petrasch-Parwez, E. et al. Association of variation in the LAMA3 gene, encoding the alpha-chain of laminin 5, with atopic dermatitis in a German case–control cohort. BMC Dermatol 14, 17 (2014). https://doi.org/10.1186/1471-5945-14-17
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1471-5945-14-17