
Abstract
Osteoprotegerin (OPG), a member of the tumor necrosis factor (TNF) receptor superfamily, contributes determinatively to the bone remodeling as well as to the pathogenetic mechanism of bone malignancies and disorders of mineral metabolism. There is additional evidence that OPG can promote cell survival by inhibiting TNF-related apoptosis-inducing ligand (TRAIL)-induced apoptosis. A number of recent in vitro, in vivo and clinical studies have defined the role of the RANK/RANKL/OPG pathway in skeletal and vascular diseases. These works were the milestone of the deep understanding of the mechanism of OPG. This review provides an overview of the potential innovative therapeutic strategies of OPG in metastatic breast and prostate carcinoma, multiple myeloma, postmenopausal osteoporosis, glucocorticoid-induced osteoporosis and rheumatoid arthritis. Special reference is given to the increasing evidence that RANKL and OPG may link the skeletal with the vascular system.
Similar content being viewed by others


Introduction
The key factor and dominant mediator of osteoclast differentiation, activation and survival is the Receptor Activator of NFkB Ligand (RANKL), which binds to its receptor (RANK) on osteoclast precursor cells and controls osteoclastogenesis and bone resorption. Osteoprotegerin (OPG), a secreted TNF receptor member that functions as a decoy of RANKL, regulates negatively this interaction [1–3]. Thereby inhibits osteoclast development and activation [1–4]. Understanding these molecular mechanisms of bone metabolism is crucial for developing novel drugs for treating such diseases [5]. Several studies in a variety of animal models have proved not only the dominance of RANKL blockade with OPG in inhibiting osteoclastogenesis and bone resorption, but also the role of deregulation of the RANK/RANKL/OPG system in the pathophysiology of multiple bone remodeling disorders such as osteoporosis, glucocorticoid-induced bone loss, erosive arthritis including rheumatoid arthritis, hypercalcemia of malignancy, Paget's disease of the bone and bone metastasis [6]. Moreover, clinical studies in post-menopausal women have shown that OPG can reduce the bone turnover [2, 3]. These different experimental and clinical studies shed light on the fact that designing novel drugs that target RANKL-RANK and their signalling pathways in osteoclasts could potentially revolutionize the treatment of many diseases [6].
Except from its ability to inhibit osteoclastic activity, OPG seems also to play a key role on cell survival, via its interaction with TNF-related apoptosis-inducing ligand (TRAIL) [7, 8]. TRAIL is a member of TNF superfamily. It is secreted by monocytes and seems to transduce apoptotic signals. More specifically, TRAIL binds to at least two membrane receptors (DR4 and DR5) and activates the death signaling pathway common to the TNF- family [7, 8].
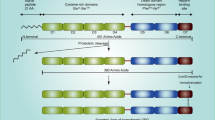
To date data show that OPG is a soluble receptor for TRAIL. Emery et al in their study have long demonstrated that OPG can bind to TRAIL in vitro[9]. Perhaps this interaction inhibits the binding of TRAIL to its death-activating receptors and therefore provides cells that produce OPG with a survival advantage [1, 9]. More recent studies have also shown that in vitro OPG (through the inhibition of TRAIL- induced apoptosis) could be a survival factor for prostate cancer cells [10], breast cancer cells [11] and multiple myeloma cells [12] (figure 1).

The OPG/RANK/RANKL system and its interaction with TRAIL. RANKL binds to RANK on osteoprogenitor cells and controls osteoclastogenesis and bone resorption. OPG regulates negatively this interaction and therefore inhibits osteoclast development and activation. TRAIL is being secreted by monocytes and is responsible for their ability to induce the apoptotic mechanisms of tumor cells. OPG facilitates the survival of prostate cancer cells due to its ability to bind to and inhibit the TRAIL death-activating receptors.
Despite the fact that till now there are not many data about OPG-TRAIL interaction in vivo, the exact regulating of the concentration, timing and location of expression of OPG and TRAIL in tissue microenvironment might determine the effects of their interactions in vivo [8]. OPG represents therefore a novel therapeutic option based on the remarkable advances occurring in the fields of cellular and molecular biology as applied to research in molecular medicine [13]. In the present review we want therefore to present the therapeutic potential of OPG for different diseases based on its molecular mode of action.
Bone malignancies
In patients with cancer, metastasis represents a major cause of morbidity and mortality. Bone is nowadays among the most frequent sites of hematogenous tumor metastasis in human body, representing around 20-40% of all sites of metastasis. Clinically, bone metastases cause severe complications (such as bone pain and fractures) and usually indicate a worse prognosis for the patient. Histologically, bone metastases are traditionally divided in osteolytic (where bone resorption is stimulated) and osteoblastic (where new bone formation is enhanced). However, today it is well known that in most osseous metastasis it's rather the balance between bone lysis and formation that is deregulated, leading to an unbalanced bone remodeling process and therefore to mixed lesions [1].
Bone pain, which represents one of the most severe complications of bone metastases, can be minimized by OPG. Honore et al. have proved that OPG blocks behaviors indicative of pain in mice with bone cancer [14]. Excessive tumor-induced bone destruction is involved in the generation of bone cancer pain and OPG by reducing bone resorption may provide an effective treatment for this common human condition [14].
In treating of metastatic bone disease, the primary goals are the preventing a bone lesion from developing and limiting the progression of an established bone metastasis. However, the currently available therapies for bone metastasis such as bisphosphonates, radiotherapy, radiopharmaceuticals and surgery focus only on symptomatic management [15, 16]. However, biophosphates - for example - block not only bone resorption but also tumor-cell mitosis and stimulate tumor-cell apoptosis. Bisphosphonates also alleviate the above mentioned bone pain [17, 18]. Additionally, OPG- like peptidomimetics are also effective in suppressing bone resorption in patients with tumor-induced bone disease [19, 20]. Anti-RANKL antibodies, such as denosumad, represent a new class of RANKL inhibitors that are characterized by high specificity for RANKL and so they prevent RANKL from binding to its receptor and stimulating osteoclasts resulting in the reduction in bone resorption [21, 22].
Prostate cancer
Prostate cancer is among the malignancies that have great avidity to bone [1, 5]. OPG has been shown to be secreted by prostate cancer cells [10]. It can bind to RANKL and prevent osteoclastogenesis [23, 24]. It is also estimated that in vitro OPG can protect the tumor cells from apoptosis, via its ability to inhibit TRAIL (TNF- related Apoptosis Inducing Ligand) and the apoptotic mechanisms it activates [11, 10, 25]. On the contrary in vivo OPG was shown to inhibit the survival of prostate cancer cells in bone [23, 26]. Recent in vivo studies also show that OPG can inhibit RANKL-induced cancer cell migration to the bone. A possible mechanism is that RANKL can induce prostate cancer cell migration by binding to RANK which is expressed on the tumor cells [27, 28]. OPG binds to RANKL and blocks its interaction with RANK inhibiting RANKL-mediated cell migration to the bone [28]. This effect is not observed in other tissues, except from the bone. This tissue-specific inhibition of prostate cancer cell migration might be additionally partly due to other factors related to the local bone microenvironment [27, 28]. However, it is not yet clear whether this ability of OPG to inhibit the development of osseous lesions could have any clinical implications in the treatment of patients with tumor-related bone disease [26].
In clinical level, several studies have shown that serum OPG concentrations are significantly higher in patients with prostate cancer [29], and even higher in those with advanced disease [30], suggesting that prostate cancer cells over-secrete OPG. Such data indicate a potential role of serum OPG as a surrogate marker of the disease progression or early relapse in patients with prostate cancer.
Breast cancer
Concerning the role of OPG in breast cancer metastases to bone, it has been shown that breast cancer cells can produce OPG in vitro [11]. This seems to be very important, as OPG can protect breast cancer cells from undergoing TRAIL-induced apoptosis [11, 21, 31]. On the other hand, OPG treatment in animal models of breast cancer inhibited the development of osseous lesions [32, 33] and the growth of cancer cells in the bone [32]. This was a result of the treatment with high dose of OPG (up to 25 mg/kg) as controlled after 4 weeks with a Faxitron X-ray system and histological analysis [32]. Clinical studies do not yet exist. But these experimental data indicate that OPG could perhaps be a potential therapeutic option for the treatment of bone lysis observed in patients with metastatic breast cancer, but further studies are essential to establish this role.
Multiple myeloma
Multiple myeloma is a malignant clonal proliferation of B-lymphocyte- derived plasma cells, characterized by its ability to develop osteolytic bone lesions, via the induction of osteoclastic bone resorption [34].
Recent studies have revealed that myeloma cells do not express OPG, but via intracellular interactions with osteoblasts and Bone marrow stem cells (BMSCs) can down- regulate the OPG released from these cells [35]. Moreover, they can bind, internalize and degrade OPG in vitro [34]. This process leads to decreased OPG concentrations in bone marrow micro-environment and therefore to increased osteoclast activation via the RANKL pathway [34]. In the 5TMM murine model of multiple myeloma, treatment with OPG protected the mice from myeloma- induced bone disease as measured after 4 weeks by the radiographic Faxitron x-ray system [35, 36]. Recent experimental data also suggest that multiple myeloma cells enhance the expression of RANKL and reduce that of OPG in their microenvironment [37]. This seems to be a result of interaction between multiple myeloma cells and BMSCs indicating that multiple myeloma cells can cause an imbalance of OPG/RANKL axis and that this dysregylation is involved in the pathogenesis of multiple myeloma disease [37]. On the other hand, OPG is shown to prevent TRAIL-induced apoptosis in human myeloma cell lines [9]. It is therefore intriguing to investigate why the down-regulation of an anti-apoptotic factor may take part in the development of myeloma bone disease.
Clinical studies have shown that the serum concentrations of OPG are lower in patients with multiple myeloma, and even lower in those with evidence of bone lesions suggesting that OPG plasma concentrations may be related with the degree of osteolysis [38]. In a phase I clinical trial, the treatment of patients with a single-dose of OPG was shown to inhibit significantly bone lysis, as 56 days after the treatment the levels of bone resorption markers (and especially of urinary N- telopeptide/creatine) were decreased [10, 39].
These clinical and also experimental data strongly suggest that deregulation of the OPG expression is involved in multiple myeloma. OPG could be not only a possible surrogate marker for multiple myeloma in the management of patients with, but also a potential therapeutic target for the treatment of the multiple myeloma [34, 40].
It should also be mentioned that during the last years there has also been increasing interest in the role of Dickkopf 1 (DKK-1) in multiple myeloma. DKK-1 is a soluble inhibitor of Wnt signaling pathway, which prevents osteoblast differentiation. DKK-1 can also inhibit the expression of OPG in osteoblasts [41]. Therefore, DKK-1 both directly and indirectly enhances the osteoclastic activity in bone microenvironment. DKK-1 is secreted by myeloma cells and is over-expressed in patients with advanced myeloma bone disease, as shown by measurement of DKK-1 levels in both bone microenvironment and serum [42]. Overall, these data suggest that DKK-1 might partly be responsible for bone lysis observed in multiple myeloma disease [41, 42].
Hypercalcemia of malignancy
By binding and neutralizing RANKL, OPG diminishes osteoclast formation and activity resulting in blocking bone resorption in animal models of hypercalcemia of malignancy [43–45]. Studies have indicated that OPG, given either at the onset of hypercalcemia or after it had occurred (hypercalcemia over 3-5 days), blocks tumor-induced bone resorption and hypercalcemia and therefore leads to normalization of blood calcium within 48 hours [43–45] suggesting an effective method for curing this paraneoplasmatic syndrome. OPG can effectively reverse hypercalcemia in less than 3-5 days in most patients [44].
Disorders of mineral metabolism
As it has been already mentioned, OPG regulates negatively the interaction between RANKL and its receptor RANK and therefore inhibits osteoclast development and activation. This is the main pathway that explains that the RANK/RANKL/OPG system contributes significantly to the pathophysiological mechanism of several bone disorders by regulating osteoclastogenesis [2] and may represent the key factor for the discovery of the therapeutic properties of OPG for these diseases.
Postmenopausal osteoporosis
The discovery of the determinative contribution of the RANK/RANKL/OPG system in the osteoclastogenesis indicates its role in the pathogenesis of postmenopausal osteoporosis [2, 46]. Recent studies have shown that the up-regulation of RANKL on bone marrow cells induced by estrogen deficiency in postmenopausal women is responsible for the bone resorption [47]. By blocking the interaction of RANKL with RANK in vivo, OPG reduces the osteoresorptive results of this signaling pathway [48]. Furthermore, several investigations using a mouse ovariectomy model of estrogen deficiency-induced osteoporosis concluded to the fact that OPG not only prevents bone loss but also increases bone mineral density leading to osteopetrosis when over-expressed [48–50]. It has been also shown that a single injected dose of OPG in post-menopausal women causes rapidly and profoundly reducing bone turnover after 12 hours as measured by the decreasement of the biochemical markers, urinary N-telopeptide (NTX) and deoxypyridinoline (DPD), which are stable collagen degradation products [51].
Low-dosage intermittent parathyroid hormone (PTH) treatment has an anabolic effect on the skeleton and this ability makes PTH a promising therapeutic treatment for osteoporosis. However, intermittent administered PTH also stimulates bone resorption and therefore can cause hypercalcemia, which can be inhibited by co-administration of PTH and antiresorptives [48, 52]. A significantly greater and more rapid increase in bone mineral density is proved by OPG-PTH co-treatment rather than administering the agents separately [49]. Therefore the combination of OPG and PTH represents an effective therapeutic approach to treating or reversing severe osteoporosis in humans. A further advantage of PTH combined with OPG is the inhibition of the hypercalcemic effects of PTH treatment [48, 52].
Glucocorticoid-induced bone loss
Osteoporosis is a well known side-effect of long-term glucocorticoid treatment, but the precise underlying mechanism remains unclear. Recently, it has been proved that glucocorticoids affect the RANK/RANKL/OPG pathway by stimulating RANKL expression by osteoblasts and inhibiting OPG synthesis and thereby resulting in osteoclast differentiation, activation and survival [53–55]. These findings explain that glucocorticoid-induced bone loss develops rapidly because of the combination of increscent of bone resorption and suppression of bone formation [2, 53–55], suggesting that the reversal of this bone loss is possible by treating with OPG. These findings may open new therapeutic options to effectively treat the glycocorticoid-induced osteoporosis.
Rheumatoid arthritis
Rheumatoid arthritis is a chronic disease which is characterized by progressive synovial inflammation and joint destruction. Recently, it has been demonstrated that the RANK/RANKL/OPG system participates in the pathophysiological mechanism of focal and generalized bone loss in rheumatoid arthritis as it is described below [2, 4]. Osteoblastic-stromal cells, synovial fibroblasts and activated T-cells are important inducers of RANKL expression leading to loss of the RANKL/OPG-balance, resulting in the osteoclastogenic proceeds in erosive arthritis [4, 56]. Interestingly, studies have shown that higher serum levels of OPG and RANKL are present in patients with rheumatoid arthritis than in healthy people, but the ratio of OPG/RANKL is similar [40, 41, 42]. These data conclude to the fact that anti-TNF therapy normalizes the OPG/RANKL balance stimulating the bone erosion in arthritis [56–58].
In a T-cell-dependent model of rat adjuvant arthritis characterized by severe joint inflammation, bone and cartilage destruction and crippling, blocking of RANKL through treatment with OPG at the onset of disease prevents bone and cartilage destruction but not inflammation [59]. These results show that both systemic and local T-cell activation leads to RANKL production and therefore bone loss. This study of Kong et al. demonstrates nicely that bone remodelling and bone loss are controlled by a balance between RANKL and OPG [59].
Fractures
Although the effects of OPG on fracture healing are not so clear, it has been proved that expression of OPG and RANKL in callus tissue is involved in the mechanism of endochondral resorption and remodeling [4]. The clinical study of Vinther et al. including 80 patients has demonstrated that although OPG treatment impairs remodeling of healing fractures and decreases the mechanical quality of the callus tissue, OPG does not influence the strength of the fractures in total [4].
Renal osteodystrophy
In patients with uremia either increased or suppressed bone turnover can be observed [2]. However, secondary hyperparathyroidism is the most common type of renal osteodystrophy and is characterized by PTH-related bone loss. OPG serum levels are increased in patients with chronic kidney disease [2] and it might inhibit osteoclastogenesis and bone resorption induced by PTH in patients with uremia [60]. Therefore, OPG serum concentrations could possibly be used as surrogate markers of the progression of bone disease in patients with uremia, but further studies are needed to establish this role [2, 61].
Vascular Diseases
Several epidemiological studies have shown that osteoporosis and vascular calcification could be associated [2, 62, 63] and share many common risk factors [62]. Although the mechanisms responsible for this phenomenon are not clear yet [64], OPG is today believed to be an important mediator of it. More specifically, it is estimated that a deregulation of calcium allocation results in its movement from the bone to the endothelium of the vessels through mechanisms that involve OPG [62].
It is also interesting that endothelial cells (ECs) and vascular smooth muscle cells (VSMCs) produce and express all members of the OPG/RANK/RANKL-axis [62]. RANKL produced by ECs and VSMCs interacts with RANK- receptors on the surface of ECs and might participate in the process of atherosclerosis via mechanisms that are today under investigation. In a recent in vitro study, RANKL caused a dose-dependent increase of calcification in VSMCs, which was inhibited by OPG. The researchers hypothesize that RANKL binds to RANK and through the activation of an intracellular pathway leads to an increased production of bone morphogenetic protein 4 (BMP4) in VSMCs, which induces vascular calcification [65].
OPG is believed to antagonize RANKL actions in vessels as well [62]. However its role on vascular tissue is much more complicated, as its production can either be induced or down-regulated in response to various inflammatory mediators, and can therefore affect the process of vascular calcification in multiple ways [62].
Vascular calcification
The actions of OPG have been studied in a number of animal models [62, 66–68]. In vivo studies in OPG knockout mice indicate calcification of the aorta and renal arteries [62, 66]. These are both sites where OPG is normally expressed [67]. These findings suggest that OPG produced in these arteries might protect them from early calcification and that its lack or deficiency might take part in pathological artery calcification [67].
Such a vascular protective role is also suggested by a study in which treatment by OPG could inhibit the vascular calcification induced by warfarin and vitamin D [68]. Warfarin causes calcification by inhibiting the vitamin K- dependent carboxilation of matrix Cla protein (MGP). Injections of OPG in rats treated previously with warfarin inhibited arterial calcification [68].
On the second animal model of this study, treatment with high doses of vitamin D caused extended calcification of the arteries in rats, which was also inhibited by injections of OPG [68]. The researchers suppose that the vascular-protective role of OPG in their animal models is a result of two possible mechanisms; on the one hand limited bone resorption and on the other hand direct promotion of the expression of calcification inhibitors (e.g. MGP) in vascular cells [68].
On the contrary, clinical studies have revealed that serum OPG concentrations are increased in patients with cardiovascular disease and may serve as an independent risk factor of cardiovascular events [63, 69, 70]. Moreover, its key- role in the regulation of the inflammation, proliferation and apoptosis of various cell types in tissues where it is expressed (e.g. the bone) indicate that OPG may also participate in such procedures in the vascular tissue as well [63]. Therefore, further researches and essential in order to establish a possible role of OPG not only as a marker, but also as a therapeutic target of cardiovascular disease.
Conclusion
The RANK/RANKL/OPG system plays a determinative role in the pathophysiology of different bone and vascular diseases and increases our understanding of the contribution of OPG to the treatment of those disorders. However, greater insights into the healing mechanism of OPG concerning bone diseases would lead to new therapeutic approaches that limit or prevent such disorders. Therefore, further researches are indispensable, since questions related to the therapeutic role of OPG have to be answered, but OPG remains a breakthrough in the treatment of bone disorders.
References
Fili S, Karalaki M, Schaller B: Mechanisms of bone metastasis: The role of osteoprotegerin and of the host-tissue microenvironment-related survival factors. Cancer Lett. 2009, 283: 10-9. 10.1016/j.canlet.2009.01.011.
Vega D, Maalouf NM, Sakhaee K: The role of receptor activator of nuclear factor-kB (RANK)/RANK ligand/osteoprotegerin: clinical implications. J Clinical Endocrinol Metab. 2007, 92: 4514-21. 10.1210/jc.2007-0646.
Ulrich-Vinther M, Soeballe K, Andreasen TT: Effects of osteoprotegerin treatment on healing fractures. Bone. 2005, 37: 751-8. 10.1016/j.bone.2005.07.021.
Romas E, Sims NA, Hards DK, Lindsay M, Quinn JWM, Ryan PFJ, Dunstan CR, Martin TJ, Gillespie MT: Osteoprotegerin reduces osteoclast numbers and prevents bone erosion in collagen-induced arthritis. Am J Pathol. 2002, 161: 1419-1427.
Schaller B, Merlo A, Kirsch E, Lehmann K, Huber PR, Lyrer P, Steck AJ, Gratzl O: Prostate-specific antigen in the cerebrospinal fluid leads to diagnosis of solitary cauda equina metastasis: A unique case report and review of the literature. Br J Cancer. 1998, 77: 2386-9.
Wada T, Nakashima T, Hiroshi N, Penninger JM: RANKL-RANK signaling in osteoclastogenesis and bone disease. Trends Mol Med. 2006, 12: 17-25. 10.1016/j.molmed.2005.11.007.
Degli-Esposti Mariapia: To die of not to die- the quest of the TRAIL receptors. Journal of leukocyte biology. 1999, 65: 535-542.
Holen I, Shipman CM: Role of osteoprotegerin (OPG) in cancer. Clinical Science. 2006, 110: 279-291. 10.1042/CS20050175.
Emery J, McDonnell P, Burke MB, Deen KC, Lyn S, Silverman C, Dul E, Appelbaum ER, Eichman C, DiPrinzio R, Dodds RA, James IE, Rosenberg M, Lee JC, Young PR: Osteoprotegerin is a receptor for the cytotoxic ligand TRAIL. J Biol Chem. 1998, 273: 14363-14367. 10.1074/jbc.273.23.14363.
Holen I, Croucher PI, Hamdy FC, Eaton CL: Osteoprotegerin (OPG) is a survival factor for human prostate cancer cells. Cancer Res. 2002, 62: 1619-23.
Holen I, Cross SS, Neville-Webbe HL, Cross NA, Balasubramanian SP, Croucher PI, Evans CA, Lippitt JM, Coleman RE, Eaton CL: Osteoprotegerin (OPG) expression by breast cancer cells in vitro and breast tumours in vivo--a role in tumour cell survival?. Breast Cancer Res Treat. 2005, 92: 207-215. 10.1007/s10549-005-2419-8.
Shipman CM, Croucher PI: Osteoprotegerin is a solube decoy receptor fot tumor necrosis factor- related apoptosis- inducing ligand/Apo2 ligand and can function as a paracrine survival factor for human myeloma cells. Cancer Res. 2003, 63: 912-916.
Schaller B, Cornelius JF, Sandu N: Molecular medicine successes in neuroscience. Mol Med. 2008, 14: 361-4. 10.2119/2008-00055.Schaller.
Honore P, Luger NM, Sabino MAC, Schwei MJ, Rogers SD, Mach DB, O'keefe PF, Ramnaraine ML, Clohisy DR, Mantyh PW: Osteoprotegerin blocks bone cancer-induced skeletal destruction, skeletal pain and pain-related neurochemical reorganization of the spinal cord. Nature Medicine. 2000, 6: 521-528. 10.1038/74999.
Roodman GD: Mechanisms of bone metastasis. N Engl J Med. 2004, 350: 1655-1664. 10.1056/NEJMra030831.
Coleman RE: Metastatic bone disease: clinical features, pathophysiology and treatment strategies. Cancer Treat Rev. 2001, 27: 165-176. 10.1053/ctrv.2000.0210.
Mundy GR: Metastasis to bone: causes, consequences and therapeutic opportunities. Nature. 2002, 2: 584-493.
Petrut B, Simmons C, Broom R, Trinkaus M, Clemons M: Pharmacotherapy of bone metastases in breast cancer patients. Expert Opin Pharmacother. 2008, 9: 937-45. 10.1517/14656566.9.6.937.
Schoppet M, Preissner KT, Hofbauer LC: RANK ligand and osteoprotegerin: paracrine regulators of bone metabolism and vascular function. Arterioscler Thromb Vasc Biol. 2002, 22: 549-553. 10.1161/01.ATV.0000012303.37971.DA.
Body JJ, Greipp P, Coleman RE, Facon T, Geurs F, Fermand JP, Harousseau JL, Lipton A, Mariette X, Williams CD: A phase I study of AMGN-0007 a recombinant osteoprotegerin construct, in patients with multiple myeloma or breast carcinoma related bone metastases. Cancer. 2003, 1: 887-892. 10.1002/cncr.11138.
Neville-Webbe HL, Cross NA, Eaton CL, Nyambo R, Evans CA, Coleman RE, Holen I: Osteoprotegerin (opg) produced by bone marrow stromal cells protects breast cancer cells from trail-induced apoptosis. Breast Cancer Research and Treatment. 2004, 86: 269-279. 10.1023/B:BREA.0000036900.48763.b3.
Body JJ, Facon T, Coleman RE, Lipton A, Geurs F, Fan M, Holloway D, Peterson MC, Bekker PJ: A study of the biological receptor activator of nuclear factor-kappaB ligand inhibitor, denosumab, in patients with multiple myeloma or bone metastases from breast cancer. Clin Cancer Res. 2006, 12: 1221-1228. 10.1158/1078-0432.CCR-05-1933.
Zhang J, Dai J, Qi Y, Lin DL, Smith P, Strayhorn C, Mizokami A, Fu Z, Westman J, Keller ET: Osteoprotegerin inhibits prostate cancer induced osteoclastogenesis and prevents tumor growth in the bone. J Clin Invest. 2001, 107: 1235-1244. 10.1172/JCI11685.
Miller RE, Roudier M, Jones J, Armstrong A, Canon J, Dougall WC: RANK ligand inhibition plus docetaxel improves survival and reduces tumor burden in a murine model of prostate cancer bone metastasis. Mol Cancer Ther. 2008, 7: 2160-9. 10.1158/1535-7163.MCT-08-0046.
Nyambo R, Cross N, Lippitt J, Holen I, Bryden G, Hamdy FC, Eaton CL: Human bone marrow stromal cells protect prostate cancer cells from TRAIL- induced apoptosis. J Bone Miner Res. 2004, 19: 1712-1721. 10.1359/JBMR.040703.
Corey E, Brown LG, Kiefer JA, Quinn JE, Pitts TE, Blair JM, Vessella RL: Osteoprotegerin in prostate cancer bone metastases. Cancer Res. 2005, 65: 1710-1718. 10.1158/0008-5472.CAN-04-2033.
Ando K, Mori K, Rédini F, Heymann D: RANKL/RANK/OPG; key therapeutic target in bone oncology. Curr Drug Discov Technol. 2008, 5: 263-268. 10.2174/157016308785739857.
Mori K, Ando K, Heymann D, Rédini F: Receptor activator of nuclear factor- kappa B ligand (RANKL) stimulates bone- assosiated tumors through functional RANK expressed on bone- assosiated cancer cells. Histol Histopathol. 2009, 24: 235-242.
Brown JM, Corey E, Lee ZD, True LD, Yun TJ, Tondravi M, Vessella RL: Osteoprotegerin and rank ligand expression in prostate cancer. Urology. 2001, 57: 611-616. 10.1016/S0090-4295(00)01122-5.
Chen G, Sircar K, Aprikian A, Potti A, Goltzman D, Rabbani SA: Expression of RANK/RANKL/OPG in primary and metastatic human prostate cancer as markers of disease stage and functional regulation. Cancer. 2006, 107: 289-298. 10.1002/cncr.21978.
Park HR, Min SK, Cho HD, Kim DH, Shin HS, Park YE: Expression of osteoprotegerin and RANK ligand in breast cancer bone metastasis. J Korean Med Sci. 2003, 18: 541-546.
Morony S, Capparelli C, Sarosi I, Lacey DL, Dunstan CR, Kostenuik PJ: Osteoprotegerin inhibits osteolysis and decreases skeletal tumor burden in syngeneic and nude mouse models of experimental bone metastasis. Cancer Res. 2001, 61: 4432-6.
Canon JR, Roudier M, Bryant R, Morony S, Stolina M, Kostenuik PJ, Dougall WC: Inhibition of RANKL blocks skeletal tumor progression and improves survival in a mouse model of breast cancer bone metastasis. Clin Exp Metastasis. 2008, 25: 119-29. 10.1007/s10585-007-9127-1.
Terpos E, Dimopoulos MA: Myeloma bone disease: pathophysiology and management. Ann Oncol. 2005, 16: 1223-1231. 10.1093/annonc/mdi235.
Croucher PI, Shipman CM, Lippitt J, Perry M, Asosingh K, Hijzen A, Brabbs AC, van Beek EJ, Holen I, Skerry TM, Dunstan CR, Russell GR, Van Camp B, Vanderkerken K: Osteoprotegerin inhibits the development of osteolytic bone disease in multiple myeloma. Blood. 2001, 15: 3534-40. 10.1182/blood.V98.13.3534.
Croucher PI, Shipman CM, Van Camp B, Vanderkerken K: Bisphosphonates and osteoprotegerin as inhibitors of myeloma bone disease. Cancer. 2003, 97: 818-24. 10.1002/cncr.11125.
Guiliani N, Bataille R, Mancini C, Lazzaretti M, Barille S: Myeloma cells induce imbalance in the osteoprotegerin/osteoprotegerin ligand system in the human bone marrow environment. Blood. 2001, 98: 3527-3533. 10.1182/blood.V98.13.3527.
Lipton A, Ali SM, Leitzel K, Chinchilli V, Witters L, Engle L, Holloway D, Bekker P, Dunstan CR: Serum osteoprotegerin levels in healthy controls and cancer patient. Clin Cancer Res. 2002, 8: 2306-10.
Body JJ, Greipp P, Coleman RE, Facon T, Geurs F, Fermand JP, Harousseau JL, Lipton A, Mariette X, Williams CD, Nakanishi A, Holloway D, Martin SW, Dunstan CR, Bekker PJ: A phase I study of AMGN- a recombinant osteoprotegerin construct, in patients with multiple myeloma or breast carcinoma related bone metastases. Cancer. 0007, 97: 887-892. 10.1002/cncr.11138.
Guiliani N, Colla S, Rizzoli V: Update on the pathogenesis of osteolysis in multiple myeloma patients. Acta Bio Medica Ateneo Parmense. 2004, 75: 143-152.
Oyajobi MO: Multiple myeloma/hypercalcemia. Arthritis Res ther. 2007, 9 (supp l): S4-10.1186/ar2168.
Gavriatopoulou M, Dimopoulos MA, Christoulas D, Migkou M, Iakovaki M, Gkotzamanidou M, Terpos E: Dickkopf- 1: a suitable target for managment of myeloma bone disease. Expert Opin Ther Targets. 2009, 13: 839-848. 10.1517/14728220903025770.
Goltzman D: Osteolysis and cancer. J Clin Invest. 2001, 107: 1219-1220. 10.1172/JCI13073.
Capparelli C, Kostenuik PJ, Morony S, Starnes C, Weimann B, Van G, Scully S, Qi M, Lacey DL, Dunstan CR: Osteoprotegerin prevents and reverses hypercalcemia in a murine model of humoral hypercalcemia of malignancy. Cancer Res. 2000, 60: 783-787.
Martin TJ: Hypercalcemia of malignancy epidemiology and pathogenesis. Clinical Review in Bone and Mineral Metabolism. 2002, 1: 51-64. 10.1385/BMM:1:1:51.
Hamdy NAT: Osteoprotegerin as a potential therapy for osteoporosis. Current Rheumatology Reports. 2006, 8: 50-54. 10.1007/s11926-006-0026-2.
Eghbali-Fatourechi G, Khosla S, Sanyal A, Boyle WJ, Lacey DL, Riggs BL: Role of RANK ligand in mediating increased bone resorption in early postmenopausal women. J Clin Invest. 2003, 111: 1221-30.
Kostenuik PJ, Capparelli C, Morony S, Adamu S, Shimamoto G, Shen V, Lacey DL, Dunstan CR: OPG and PTH-(1-34) have additive effects on bone density and mechanical strength in osteopenic ovariectomized rats. Endocrinology. 2001, 142: 4295-4304. 10.1210/en.142.10.4295.
Bolon B, Carter C, Daris M, Morony S, Campagnuolo G, Feige U, Sheng J: Osteoprotegerin (OPG) gene therapy in animal models of osteoarticular disease. Arthritis Res. 2001, 3: 5-10.1186/ar371.
Hofbauer LC, Khosla S, Dunstan CR, Lacey DL, Spelsberg TC, Riggs BL: Estrogen stimulates gene expression and protein production of osteoprotegerin in human osteoblastic cells. Endocrinology. 1999, 140: 4367-70. 10.1210/en.140.9.4367.
Bekker PJ, Holloway D, Nakanishi A, Arrighi M, Leese PT, Dunstan CR: The effect of a single dose of osteoprotegerin in postmenopausal women. J Bone Miner Res. 2001, 16: 348-360. 10.1359/jbmr.2001.16.2.348.
Coetzee M, Kruger MC: Osteoprotegerin-receptor activator of nuclear factor-[kappa]B ligand ratio: A new approach to osteoporosis treatment?. South Med J. 2004, 97: 506-11. 10.1097/00007611-200405000-00018.
Humphrey E, Williams J, Davie M, Marshall M: Effects of dissociated glucocorticoids on OPG and RANKL in osteoblastic cells. Bone. 2006, 38: 652-61. 10.1016/j.bone.2005.10.004.
Nobuhiro S, Eiji K, Yasuhiro A, Kazuki Y, Eisuke T: Glucocorticoid decreases circulating osteoprotegerin (OPG): Possible mechanism for glucocorticoid induced osteoporosis. Nephrol Dial Transplant n. 2001, 16 (3): 479-482. 10.1093/ndt/16.3.479.
Sasaki N, Kusano E, Ando Y, Nemoto J, Iimura O, Ito C, Takeda S, Yano K, Tsuda E, Asano Y: Changes in osteoprotegerin and markers of bone metabolism during glucocorticoid treatment in patients with chronic glomerulonephritis. Bone. 2002, 30: 853-8. 10.1016/S8756-3282(02)00742-1.
Lubberts E, Bersselaar van den L, Oppers-Walgreen B, Schwarzenberger P, Coenen-de Roo CJ, Kolls JK, Joosten LA, Berg van den WB: IL-17 promotes bone erosion in murine collagen-induced arthritis through loss of the receptor activator of NF-kappa B ligand/osteoprotegerin balance. J Immunol. 2003, 170: 2655-62.
Ziolkowska M, Kurowska M, Radzikowska A, Luszczykiewicz G, Wiland P, Dziewczopolski W, Filipowicz-Sosnowska A, Pazdur J, Szechinski J, Kowalczewski J, Rell-Bakalarska M, Maslinski W: High levels of osteoprotegerin and soluble receptor activator of nuclear factor kappa B ligand in serum of rheumatoid arthritis patients and their normalization after anti-tumor necrosis factor alpha treatment. Arthritis Rheum. 2002, 46: 1744-53. 10.1002/art.10388.
Catrina AI, Af Klint E, Ernestam S, Catrina S, Makrygiannakis D, Botusan Ir, Klareskog L, Ulfgren AK: Anti-tumor necrosis factor therapy increases synovial osteoprotegerin expression in rheumatoid arthritis. Arthritis Rheum. 2006, 54: 76-81. 10.1002/art.21528.
Kong YY, Feige U, Sarosi I, Bolon B, Tafuri A, Morony S, Capparelli C, Li J, Elliott R, McCabe S, Wong T, Campagnuolo G, Moran E, Bogoch ER, Van G, Nguyen LT, Ohashi PS, Lacey DL, Fish E, Boyle WJ, Penninger JM: Activated T cells regulate bone loss and joint destruction in adjuvant arthritis through osteoprotegerin ligand. Nature. 1999, 402: 304-9. 10.1038/46303.
Coen G, Ballanti P, Balducci A, Calabria S, Fischer MS, Jankovic L, Manni M, Morosetti M, Moscaritolo E, Sardella D, Bonucci E: Serum osteoprotegerin and renal osteodystriphy. Nephrol Dial Transplant. 2002, 17: 233-238. 10.1093/ndt/17.2.233.
Fukagawa M, Kurokawa K: Renal osteodystrophy and secondary hyperparathyroidism. Nephrol Dial Transplant. 2002, 17: 2-5. 10.1093/ndt/17.9.1553.
Collin-Osbody P: Regulation of vascular calcification by osteoclast regulatory factors RANKL and osteoprotegerin. Circ Res. 2004, 95: 1046-1057. 10.1161/01.RES.0000149165.99974.12.
Van Campenhout A, Golledge J: Osteoprotegerin, vascular calcification and atherosclerosis. Atherosclerosis. 2009, 204: 321-329. 10.1016/j.atherosclerosis.2008.09.033.
Schaller B: Reply to "dissection of the internal carotid artery and hemicraniectomy. Am J Med. 2007, 120: e17-10.1016/j.amjmed.2005.12.033.
Panizo S, Cardus A, Encinas M, Parisi E, Valcheva P, López-Ongil S, Coll B, Fernandez E, Valdivielso JM: RANKL increases vascular smooth muscle cell calcification through a RANKL-BMP4- dependent pathway. Circ Res. 2009, 104: 1041-1048. 10.1161/CIRCRESAHA.108.189001.
Tintut Y, Demer L: Role of osteoprotegerin and its ligands and competing receptors in atherosclerotic calcification. J Investig Med. 2006, 54: 395-401. 10.2310/6650.2006.06019.
Bucay N, Sarosi I, Dunstan CR, Morony S, Tarpley J, Capparelli C, Scully S, Tan HL, Xu W, Lacey DL, Boyle WJ, Simonet WS: Osteoprotegerin- deficient mice develop early onset osteoporosis and arterial calcification. Genes Dev. 1998, 12: 1260-1268. 10.1101/gad.12.9.1260.
Price PA, June HH, Buckley JR, Williamson MK: Osteoprotegerin inhibits artery calcification induced by warfarin and vitamin D. Arterioscler Thromb Vasc Biol. 2001, 21: 1610-1616. 10.1161/hq1001.097102.
Nybo M, Rasmussen LM: The capability of plasma osteoprotegerin as a predictor of cardiovascular disease: a systematic literature review. Eur J Endocrinol. 2008, 159: 603-608. 10.1530/EJE-08-0554.
Shargorodsky M, Boaz M, Luckish A, Matas Z, Gavish D, Mashavi M: Osteoprotegerin as an independent marker of subclinical atherosclerosis in osteoporotic postmenopausal women. Atherosclerosis. 2009, 204: 608-611. 10.1016/j.atherosclerosis.2008.10.024.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors' contributions
FS and KM have equally contributed to the elaboration of the review. SB was the senior author. All authors read and approved the final manuscript.
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
This article is published under license to BioMed Central Ltd. This is an Open Access article distributed under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Fili, S., Karalaki, M. & Schaller, B. Therapeutic implications of osteoprotegerin. Cancer Cell Int 9, 26 (2009). https://doi.org/10.1186/1475-2867-9-26
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1475-2867-9-26