
Abstract
Background
Mutations in the Pleckstrin homology domain-containing, family G member 5 (PLEKHG5) gene has been reported in a family harboring an autosomal recessive lower motor neuron disease (LMND). However, the PLEKHG5 mutation has not been described to cause Charcot-Marie-Tooth disease (CMT).
Methods
To identify the causative mutation in an autosomal recessive intermediate CMT (RI-CMT) family with childhood onset, whole exome sequencing (WES), histopathology, and lower leg MRIs were performed. Expression and activity of each mutant protein were analyzed.
Results
We identified novel compound heterozygous (p.Thr663Met and p.Gly820Arg) mutations in the PLEKHG5 gene in the present family. The patient revealed clinical manifestations of sensory neuropathy. Fatty replacements in the distal lower leg muscles were more severe than in the thigh muscles. Although the symptoms and signs of this patient harboring slow nerve conduction velocities suggested the possibility of demyelinating neuropathy, a distal sural nerve biopsy was compatible with axonal neuropathy. Immunohistochemical analysis revealed that the patient has a low level of PLEKHG5 in the distal sural nerve and an in vitro assay suggested that the mutant proteins have a defect in activating the NF-κB signaling pathway.
Conclusions
This study identifies compound heterozygous PLEKHG5 mutations as the cause of RI-CMT. We suggest that PLEKHG5 might play a role in the peripheral motor and sensory nervous system. This study expands the phenotypic spectrum of PLEKHG5 mutations.
Similar content being viewed by others


Background
Mutation in Pleckstrin homology domain-containing, family G member 5 (PLEKHG5) has been reported only in a family with lower motor neuron disease (LMND) with childhood onset [1]. LMND, which is a large group of clinically and genetically heterogeneous disorders, is usually diagnosed by electrophysiological or histological evidence of muscle denervation without sensory neuropathy [2, 3]. Except for PLEKHG5, many other causative genes have been reported in LMNDs: SMN1 and DYNC1H1 in each autosomal recessive (AR) and dominant spinal muscular atrophy (SMA: MIM 253300 and 158600), HSPB1,HSPB8, and HSPB3 in distal hereditary motor neuronopathy type 2 (dHMN2: MIM 608634, 158590 and 613376), GARS, BSCL2 and REEP1 in dHMN5 (MIM 600794 and 614751), IGHMBP2 in dHMN6 (MIM 604320), DCTN1 and SLC5A7 in dHMN7 (MIM 607641 and 158580), ATP7A in X-linked dHMN and SMA (MIM 300489), and SETX in dHMN with pyramidal sign (MIM 602433) [2–11]. The affected individuals harboring a PLEKHG5 mutation presented with severe generalized distal and proximal muscle weakness and atrophy, but normal sensation; therefore, such mutations are often classified as a cause of dHMN [12] or generalized SMA [1]. However, there has been no report of a PLEKHG5 mutation relevant to Charcot-Marie-Tooth disease (CMT), which is also called hereditary motor and sensory neuropathy (HMSN).
CMT is a clinically and genetically heterogeneous disorder of the peripheral motor and sensory nervous system [13]. CMT has been reported to be associated with more than 50 causative genes or loci [14]. Based on nerve biopsy and neurophysiology, CMT falls into two main subtypes, a demyelinating form (CMT1), and an axonal form (CMT2) [15]. An intermediate group exists with nerve conduction velocities, which overlaps the two main groups, and is subdivided into dominant intermediate (DI-CMT) and recessive intermediate CMT (RI-CMT) by its pattern of inheritance [16]. Intermediate forms of CMT may affect both axons and Schwann cells [17, 18].
PLEKHG5 is predominantly expressed in the peripheral nervous system, and the protein contains a DH (Dbl homology)- PH (Pleckstrin homology) motif, which is known as the minimal unit for the nucleotide exchange-promoting function of guanine nucleotide exchange factors (GEFs) [19]. PLEKHG5 protein has been suggested to have a role in the activation of the RhoA exchange factor and NF-κB signaling pathway [20], and it thus appears to be involved in neuronal cell differentiation. It is suggested that both loss of function in the NF-κB transduction pathway and aggregate formation of mutant PLEKHG5 might contribute to neurotoxicity [1, 2].
In this study, we investigated a Korean RI-CMT family with childhood onset, and identified novel compound heterozygous mutations of PLEKHG5. To our knowledge, this is the first report of a PLEKHG5 mutation with motor and sensory neuropathy.
Subjects and methods
Patients
This study enrolled 248 individuals in 160 unrelated families with variable types of CMT neuropathy. They were referred by their primary physicians or neurologists and selected negative for PMP22 duplication and major CMT gene mutations during the period 2003–2012. This study also included 300 healthy controls. Informed consent was obtained from all participants according to the protocol approved by the Institutional Review Board for EwhaWomans University, Mokdong Hospital (ECT 11-58-37).
DNA preparation and prescreening for CMT genes
DNA was purified from blood using a QIAamp blood DNA purification kit (Qiagen, Hilden, Germany). Patient samples were prescreened for 17p12 duplication, which is a major genetic cause of demyelinating CMT, by using hexaplex microsatellite PCR [21]. Sequencing was also determined for the coding exons of major CMT-relevant genes, including MPZ, PMP22, GJB1, and MFN2.
Exome sequencing and identification of causative mutations
Exome sequencing was performed for the proband of FC307 family according to Lee et al. [22]. Functionally significant variants (missense, nonsense, exonicindel and splicing site variants) were first selected from WES data, and then novel or uncommon variants (MAF ≤0.01) registered in the dbSNP137 [23] and 1000 Genomes database [24] were chosen. Homozygous or compound heterozygous mutations were finally selected. Candidate variants were confirmed by Sanger’s sequencing method using an ABI3130XL automatic genetic analyzer (Applied Biosystems, Foster City, CA). The underlying cause was determined by the presence of mutation only in the patient within the family and the absence in 300 controls. Conservation analysis of protein sequences was performed using MEGA5 version 5.05 software [25].
Clinical and electrophysiological assessments
The proband (Figure 1A, III-1), her parents (Figure 1A, II-3, and −7), and grandparents (Figure 1A, I-1, -2, -3, and −4) were examined for motor and sensory impairments, deep tendon reflexes, and muscle atrophy. The strength of flexor and extensor muscles was assessed manually using the medical research council (MRC) scale. Physical disabilities were assessed using a CMT neuropathy score (CMTNS) [26]. Sensory impairments were assessed in terms of the level and severity of pain, temperature, vibration, and position. Age at onset was determined by asking patients for the age when symptoms, i.e., distal muscle weakness, foot deformity, or sensory change, first appeared.

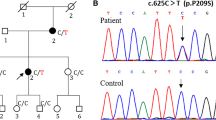
Pedigree, sequencing chromatograms and conservation in the FC307 family with compound heterozygous PLEKHG5 mutations. (A) Pedigree. Open symbols indicate unaffected individuals and the filled symbol indicates the affected individual. Half-filled symbols indicate carriers possessing one mutant allele. The arrow indicates the proband. Genotypes of both PLEKHG5 mutations are indicated at below each examined individual. (B) Sequencing chromatograms. Vertical arrows indicate the mutation site. (C) Conservation analysis of amino acid sequences. The analysis was conducted using MEGA5 version 5.05 software. Both mutation sites were well conserved across species.
Nerve conduction studies were performed with a surface electrode. Motor nerve conduction velocities (MNCVs) of the median and ulnar nerves were determined by providing stimulation at the elbow and wrist while recording compound muscle action potentials (CMAPs) over the abductor pollicis brevis and adductor digiti quinti, respectively. In the same manner, the MNCVs and CMAPs of the peroneal and tibial nerves were determined. Sensory nerve conduction velocities (SNCVs) and action potentials (SNAPs) were obtained from the median, ulnar, and sural nerves. Needle electromyography (EMG) was performed for bilateral proximal and distal limb muscles. The patient and her parents underwent tests for visually evoked potentials and brainstem auditory evoked potentials.
MRIs of the brain, hip, thigh and lower leg
The proband was studied by examining MRIs of the brain, hip, thigh, and lower leg, using a 1.5-T system (Siemens, Erlangen, Germany). Lower limb imaging was obtained in axial [field of view (FOV) 24–32 cm, slice thickness 6 mm, and slice gap 0.5–1.0 mm] and coronal planes (FOV 38–40 cm, slice thickness 4–5 mm, slice gap 0.5–1.0 mm). The following protocol was used: T1-weighted spin-echo (SE) (TR/TE 570–650/14–20, 512 matrices), T2-weighted SE (TR/TE 2800–4000/96–99, 512 matrices), and fat-suppressed T2-weighted SE (TR/TE 3090–4900/85–99, 512 matrices).
Histopathological examination
The distal sural nerve of patient (III-1) was biopsied at 19 years of age. The density of myelinated fibers (MFs), axonal diameter, and myelin thickness were determined from the semi-thin transverse sections using a computer-assisted image analyzer (AnalySIS; Soft Imaging System, Münster, Germany). Ultrathin cut samples (60–65 nm) were contrasted with uranyl acetate and lead citrate for electron microscopy (H-7650, Hitachi, Japan). An immunohistochemical study was performed with anti-PLEKHG5 antibody (N-15: sc-130100, 1:50 dilution, Santa Cruz Biotech, Santa Cruz, CA) and the findings were compared with a control case (female, 39 years old).
Cloning, expression and reporter assay for PLEKHG5
PLEKHG5 (BC015231), which is fused with a c-myc epitope, was obtained from Capital Biosciences (Rockville, MD). Site-directed mutagenesis was performed to generate mutant PLEKHG5 (p.Thr663Met and p.Gly820Arg) using a QuickChange Site-directed mutagenesis kit (Stratagene, La Jolla, CA). DNA transfections into HEK293 cells were performed using each DNA and Lipofectamine 2000 (Invitrogen, Carlsbad, CA). Protein expression was analyzed by immunocytochemistry using anti-Myc antibody (Abcam, Cambridge, UK) after transfection of each vector. Reporter assay for NF-κB was achieved using a reporter vector, pNF-κB-Luc-reporter (from Dr. Kim HT, Sungkyunkwan University, Korea), and a luciferase assay system (Promega, Madison, WI).
Results
Identification of compound heterozygous mutations in PLEKHG5
To identify the disease-associated genetic defect of our patient, we performed exome sequencing of the affected individual (Table 1). We identified novel compound heterozygous mutations, c.1988C > T (p.Thr663Met, paternal origin) and c.2458G > C (p.Gly820Arg, maternal origin) in the PLEKHG5 gene in a Korean CMT family (family ID: FC307) (Figure 1A and 1B). The mutations of her parents were inherited from their mothers, respectively (Figure 1A). Neither mutation has been reported in dbSNP137 or the 1000 Genomes database. In the 300 Korean controls, the p.Thr663Met mutation was not found, while the p.Gly820Arg was found in three control samples. Both mutation sites were well conserved across species (Figure 1C). In silico analysis using PolyPhen 2 software [27] predicted that both mutations may affect protein function with scores of 0.993 (p.Thr663Met) and 1.000 (p.Gly820Arg). In addition to the compound heterozygous mutations in PLEKHG5, several other variants were identified in the more than 50 CMT-associated genes (Table 2). However, no variant was considered to be causative, because all the variants were polymorphic; moreover, they did not fit the recessive inheritance characteristic.
Clinical manifestations
The clinical manifestations of the present patient and the previously described patients with PLEKHG5 mutations are shown in Table 3. The proband, a 19-year-old woman, was the first child of healthy nonconsanguineous Korean parents. On neurological examination and after electrophysiological studies, the parents did not show any abnormalities. The proband was born at full term and the perinatal history was unremarkable. Early motor milestones were not delayed, and one year after her birth, she was able to walk. At age 8 years, she experienced frequent falling and noticed muscle weakness of the distal lower limbs. She started to walk with short leg braces at 16 years of age. Neurological examination at 19 years of age revealed muscle weakness and atrophy of bilateral distal muscles, predominantly at lower limbs rather than upper limbs. Bilateral pes cavus, steppage gait, and atrophic changes of intrinsic foot and calf muscles were noted, but scoliosis was not observed. Sensitivity to pinprick, touch, position, and vibration decreased. Vibration and position senses were more severely disturbed than pain and touch senses. Knee and ankle jerks were absent. No pyramidal or cerebellar signs were detected. CMTNS was 15. Elevated serum creatine kinase levels were revealed (246 μmol/L, reference value: <185 μmol/L).
Sensory and motor neuropathies
Data from the nerve conduction study are summarized in Table 4. Median MNCVs ranged from 24.7 m/s to 29.3 m/s. The results also revealed prolonged motor latencies, and no motor responses were elicited by stimulation of peroneal and tibial nerves at 19 years of age. SNAPs on bilateral sural nerves were absent on all electrophysiological studies (at 14, 15, 16 and 19 years old). In the upper limb, SNCVs and SNAPs were decreased on bilateral median and ulnar nerves. Needle EMG showed a neurogenic pattern of muscle degeneration. Visually evoked potentials and brainstem auditory-evoked potentials were normal.
Fatty replacement of lower limb muscles
Brain and hip MRIs were normal. However, thigh and lower leg MRIs of the patient demonstrated hyperintense signal abnormalities. T1-weighted images showed severe muscle atrophy and fatty replacement in the lower leg muscles compared with the thigh, which is compatible with a hypothesis of length-dependent axonal degeneration (Figure 2A and 2B). At the thigh level, fatty replacement of the vastus lateralis and semimembranous muscles was observed, but more muscle remained than fat. Sartorius, gracilis, biceps femoris, and rectus femoris muscles contained some fatty streaks (Figure 2C). We could observe a distinct pattern of muscle involvement; lower leg MRI showed selective and predominant involvement of anterior and lateral compartment muscles; however, superficial and deep posterior compartment muscles revealed mild manifestations (Figure 2D and 2E).

T1-weighted MRI of the proband (III-1). (A, B) Coronal images of thigh (A) and lower leg (B). T1-weighted coronal leg MRIs of lower extremities demonstrated more severe fatty replacement and muscle atrophies in the lower leg than did those of the thigh. (C) Axial image of the middle thigh. Vastus lateralis, semimembranous muscles showed moderate to severe fatty replacements, but they revealed more muscles than fat. However, sartorius, gracilis, biceps femoris, and rectus femoris muscles revealed fatty streaks. (D, E) Axial images at the upper third (D) and lower third calf (E). T1-weighted images revealed predominantly severe involvement of anterior (black arrowhead; anterior tibialis, extensor hallucis longus and digitorum longus) and lateral (white arrowhead; peroneus longus and brevis) compartment muscles, and superficial and deep posterior compartment (arrow; soleus and tibialis posterior) muscles showed mild involvement.
Histopathological findings
Semi-thin transverse sections showed loss of large- and medium-sized MFs, with small MFs remaining (Figure 3A). The number of MFs was reduced (297/mm2) compared with a control (20-year-old female, 9,800/mm2) (Figure 3B). The range (1.2-4.6 μm) and average (2.4 μm) MF diameter were also reduced compared with the control (2–12.5 μm and 4.5 μm, respectively). The distribution pattern of MF diameter was unimodal and composed of 75% of <3 μm MFs. In addition, MF% area and g-ratio (axonal diameter/MF diameter) were also reduced. The MF% area in this case was 0.2% (normal distal sural nerve in 20-year-old female: 25.2%). The range and average of g-ratio (axonal diameter/MF diameter) were 0.38-0.73 and 0.56 ± 0.09, respectively (mean g-ratio at age 21–50 years old: 0.66). The g-ratio >0.7 (abnormally thin myelin sheath) consisted of 10% of MFs and g-ratio <0.4 (abnormally thick myelin sheath) consisted of 1.7%. Electron microscopic examination revealed that the remaining small MFs showed occasional focal folding of myelin with very rare evidence of regeneration (clusters of regenerating fibers) (Figure 3C and 3D). The unmyelinated axons showed suggestive clustering (reinnervated Büngner bands with unmyelinated fibers) and atrophy. Endoneurial fibroblast proliferation and large amounts of collagen deposition were well documented.

Histopathological findings of the distal sural nerve of the patient. (A) Toluidine blue-stained semi-thin transverse section revealed loss of large MFs and remaining medium- and small-sized MFs with rarely noted regenerating axonal clusters and unmyelinated axons with suggestive clustering and atrophy. (B) Histogram showing unimodal distribution pattern of myelinated fibers. (C and D) Electron micrographs showed axonal atrophy, rare axonal clustering, occasional focal folding of myelin, and increased collagen deposition in the endoneurium. (E) Immunohistochemical study with anti-PLEKHG5 antibody showed a focal positive reaction in the patient’s Schwann cell nuclei. (F) Comparison with diffuse strong positive reaction in axons and Schwann cell nuclei in the control case (original magnification: A, ×400; C, ×3000, D, ×8000, E and F, ×400).
Expression of mutant proteins
The immunohistochemical study with anti-PLEKHG5 antibody showed a focal positive reaction in Schwann cell nuclei in this patient (Figure 3E) compared with a diffuse positive reaction in axons and Schwann cell nuclei in the control case (Figure 3F). To further investigate the impact of the mutant proteins in cellular function and localization, we generated mutants using a BC015231 clone as a template. Cellular localization of the proteins was analyzed after transfection of the genes into HEK293 cells. Overall, the proteins were localized in the cytoplasm and there was no distinctive aggregation of any of the three proteins (Figure 4).

Cellular localization of wild-type and mutant PLEKHG5 proteins. Immunocytochemical analysis reveals that wild-type and mutant PLEKHG5 proteins were localized in the cytoplasm and there was no distinctive aggregation of any of the proteins. HEK293 cells transfected with wild-type or mutant PLEKHG5-expressing vectors were stained with DAPI (nucleus) and c-Myc antibody.
The proteins were examined for ability to activate the NF-κB pathway using a luciferase assay. Transfection of wild-type PLEKHG5 significantly induced luciferase activity. However, elevated levels of the activity were reduced in mutant protein-expressing cells (Figure 5).

Activities of wild-type and mutant PLEKHG5. Compared with wild-type, mutant PLEKHG5 proteins have defect in the activation of NF-κB pathway. Luciferase activity measured in HEK293 cells, which were treated with pNF-κB-Luc-reporter and PLEKHG5-expressing vectors. Statistical significance was calculated using Student’s t test: *, p < .05.
Discussion
We have identified a pair of novel compound heterozygous mutations of PLEKHG5 as the underlying cause of the disease in the CMT family with autosomal recessive inheritance. Although the sample size in this family was small, the mutations were in good agreement with affected or carrier members in the pedigree. These identified mutations have not been reported at either the CMT or LMND mutation database or in related papers. We believe that the identified PLEKHG5 mutations are responsible for the CMT for the following reasons: (1) the patient harbors paternal p.Thr663Met and maternal p.Gly820Arg mutations, but no other family members or controls had both heterozygous mutations simultaneously, (2) amino acids at the mutation sites among different species are well conserved, and (3) exome sequencing revealed that no causative mutation of the many known CMT genes was found in the present family.
Sensory neuropathy is a useful discriminating factor between CMT and LMND [1, 13]. LMND is characterized by motor nerve degeneration in the anterior horn cell of the spinal cord or the brainstem, and the diagnosis is confirmed by the evidence of muscle degeneration with abnormal motor conduction velocities without sensory neuropathy [1, 3]. However, CMT involves not only motor, but also sensory neuropathies. The present patient showed clinical, electrophysiological, and pathological evidence of sensory nerve involvement; therefore, she was diagnosed as having CMT.
It is interesting that the symptoms and signs of this patient such as sensory loss and slow nerve conduction velocities suggested the possibility of demyelinating neuropathy; however, the distal sural nerve biopsy revealed similarities with axonal neuropathy harboring absence of medium- and large-sized MFs. Absence of large MFs could explain the slow nerve conduction velocities and action potentials in this patient. The intermediate form of CMT has been observed in cases with median MNCVs between 25 and 45 m/s, and in our patient, median MNCVs were at the lower extreme of the range [16–18, 28]. By neurophysiological and neuropathological criteria, the current case was consistent with the RI-CMT.
Currently, information on the molecular function and disease-associated mechanism of PLEKHG5 is limited: only one homozygous mutation, p.F647S, was reported as a causative for autosomal recessive LMND. The site locates within the PH domain and the mutation was suggested to deteriorate the function on activation of NF-κB signaling pathway. Thr663 lies within the PH domain as does the previously reported Phe647, while Gly820 does not belong to any specific domain. Our in vitro data also suggest that these patients might have a defect in activation of the NF-κB signaling pathway. According to immunohistochemical data from the patient and immunocytochemical study using cloned mutants, the expression levels of the mutant proteins are lower than that of wild-type protein. By contrast with the p.F647S mutation, however, there was no indication of intracellular aggregation of current mutants. Therefore, these data imply that the low level of expression and NF-κB activation might induce peripheral neuropathy in the patient.
The PH domain of GEFs for Rho-related GTPase plays a critical role in the spatial organization of major signaling pathways, through their interaction with lipids (especially with phosphoinositides) and proteins [29]. NF-κB signaling could be included in those pathways in conjunction with the function of RhoA in triggering the translocation of NF-κB into the nucleus, because PLEKHG5 is known as an activator of RhoA exchange factor. As well as its well-known function in immune and inflammatory responses, the NF-κB signaling pathway is also involved in the development and activity of the nervous system. Particularly in the peripheral nervous system, NF-κB stimulation has been shown to have essential roles in the differentiation and myelinating programs as well as Schwann cell regeneration [30]. However, several questions regarding the pathological mechanism in our patient remain to be answeredabout the PH domain defect of the PLEKHG5 protein and congenital dysfunction in peripheral nervous system via NF-κB signaling pathway. Other human neurodegenerative disease-related genes sharing a PH/RhoGEF domain, such as DNM2, SBF2, and ALS2, could provide important clues for these elusive questions [31–33].
Conclusions
We identified novel compound heterozygous PLEKHG5 mutations as the underlying cause of RI-CMT. In the present case, nerve conduction studies showed slow motor and sensory nerve conduction, and the distal sural nerve revealed marked loss of myelinated fibers. Our findings significantly expand the phenotype of PLEKHG5 mutation; therefore, genetic screening of PLEKHG5 should be considered in not only LMND but also RI-CMT families. We suggest that PLEKHG5 plays an important role in both the peripheral motor and sensory nervous systems.
Abbreviations
- CMAP:
-
compound muscle action potential
- CMT:
-
Charcot-Marie-Tooth disease
- CMTNS:
-
CMT neuropathy score
- EMG:
-
electromyography
- dHMN:
-
distal hereditary motor neuronopathy
- HMSN:
-
hereditary motor and sensory neuropathy
- LMND:
-
lower motor neuron disease
- MNCV:
-
motor nerve conduction velocities
- PLEKHG5:
-
Pleckstrin homology domain-containing, family G member 5
- SMA:
-
spinal muscular atrophy
- SNAP:
-
sensory nerve action potentials
- SNCV:
-
sensory nerve conduction velocities
- WES:
-
whole exome sequencing.
References
Maystadt I, Zarhrate M, Leclair-Richard D, Estournet B, Barois A, Renault F, Routon MC, Durand MC, Lefebvre S, Munnich A, Verellen-Dumoulin C, Viollet L: A gene for an autosomal recessive lower motor neuron disease with childhood onset maps to 1p36. Neurology. 2006, 67: 120-124. 10.1212/01.wnl.0000223834.55225.2d.
Maystadt I, Rezsöhazy R, Barkats M, Duque S, Vannuffel P, Remacle S, Lambert B, Najimi M, Sokal E, Munnich A, Viollet L, Verellen-Dumoulin C: The nuclear factor κB-activator gene PLEKHG5 is mutated in a form of autosomal recessive lower motor neuron disease with childhood onset. Am J Hum Genet. 2007, 81: 67-76. 10.1086/518900.
Lefebvre S, Bürglen L, Reboullet S, Clermont O, Burlet P, Viollet L, Benichou B, Cruaud C, Millasseau P, Zeviani M: Identification and characterization of a spinal muscular atrophy-determining gene. Cell. 1995, 80: 155-165. 10.1016/0092-8674(95)90460-3.
Evgrafov OV, Mersiyanova I, Irobi J, Van Den Bosch L, Dierick I, Leung CL, Schagina O, Verpoorten N, Van Impe K, Fedotov V, Dadali E, Auer-Grumbach M, Windpassinger C, Wagner K, Mitrovic Z, Hilton-Jones D, Talbot K, Martin JJ, Vasserman N, Tverskaya S, Polyakov A, Liem RK, Gettemans J, Robberecht W, De Jonghe P, Timmerman V: Mutant small heat-shock protein 27 causes axonal Charcot-Marie-Tooth disease and distal hereditary motor neuropathy. Nat Genet. 2004, 36: 602-606. 10.1038/ng1354.
Windpassinger C, Auer-Grumbach M, Irobi J, Patel H, Petek E, Hörl G, Malli R, Reed JA, Dierick I, Verpoorten N, Warner TT, Proukakis C, Van den Bergh P, Verellen C, Van Maldergem L, Merlini L, De Jonghe P, Timmerman V, Crosby AH, Wagner K: Heterozygous missense mutations in BSCL2 are associated with distal hereditary motor neuropathy and Silver syndrome. Nat Genet. 2004, 36: 271-276. 10.1038/ng1313.
Harms MB, Ori-McKenney KM, Scoto M, Tuck EP, Bell S, Ma D, Masi S, Allred P, Al-Lozi M, Reilly MM, Miller LJ, Jani-Acsadi A, Pestronk A, Shy ME, Muntoni F, Vallee RB, Baloh RH: Mutations in the tail domain of DYNC1H1 cause dominant spinal muscular atrophy. Neurology. 2012, 78: 1714-1720. 10.1212/WNL.0b013e3182556c05.
Kolb SJ, Snyder PJ, Poi EJ, Renard EA, Bartlett A, Gu S, Sutton S, Arnold WD, Freimer ML, Lawson VH, Kissel JT, Prior TW: Mutant small heat shock protein B3 causes motor neuropathy: utility of a candidate gene approach. Neurology. 2010, 74: 502-506. 10.1212/WNL.0b013e3181cef84a.
Beetz C, Pieber TR, Hertel N, Schabhüttl M, Fischer C, Trajanoski S, Graf E, Keiner S, Kurth I, Wieland T, Varga RE, Timmerman V, Reilly MM, Strom TM, Auer-Grumbach M: Exome sequencing identifies a REEP1 mutation involved in distal hereditary motor neuropathy type V. Am J Hum Genet. 2012, 91: 139-145. 10.1016/j.ajhg.2012.05.007.
Barwick KES, Wright J, Al-Turki S, McEntagart MM, Nair A, Chioza B, Al-Memar A, Modarres H, Reilly MM, Dick KJ, Ruggiero AM, Blakely RD, Hurles ME, Crosby AH: Defective presynaptic choline transport underlies hereditary motor neuropathy. Am J Hum Genet. 2012, 91: 1103-1107. 10.1016/j.ajhg.2012.09.019.
Kennerson ML, Nicholson GA, Kaler SG, Kowalski B, Mercer JFB, Tang J, Llanos RM, Chu S, Takata RI, Speck-Martins CE, Baets J, Almeida-Souza L, Fischer D, Timmerman V, Taylor PE, Scherer SS, Ferguson TA, Bird TD, De Jonghe P, Feely SM, Shy ME, Garbern JY: Missense mutations in the copper transporter gene ATP7A cause X-linked distal hereditary motor neuropathy. Am J Hum Genet. 2010, 86: 343-352. 10.1016/j.ajhg.2010.01.027.
Chen Y-Z, Bennett CL, Huynh HM, Blair IP, Puls I, Irobi J, Dierick I, Abel A, Kennerson ML, Rabin BA, Nicholson GA, Auer-Grumbach M, Wagner K, De Jonghe P, Griffin JW, Fischbeck KH, Timmerman V, Cornblath DR, Chance PF: DNA/RNA helicase gene mutations in a form of juvenile amyotrophic lateral sclerosis (ALS4). Am J Hum Genet. 2004, 74: 1128-1135. 10.1086/421054.
Rossor AM, Kalmar B, Greensmith L, Reilly MM: The distal hereditary motor neuropathies. J Neurol Neurosurg Psychiatry. 2012, 83: 6-14. 10.1136/jnnp-2011-300952.
Shy ME, Garbern JY, Kamholz J: Hereditary motor and sensory neuropathies: a biological perspective. Lancet Neurol. 2002, 1: 110-118. 10.1016/S1474-4422(02)00042-X.
Inherited Peripheral Neuropathies Mutation Database. [http://www.molgen.ua.ac.be/CMTMutations/mutations]
Harding AE, Thomas PK: The clinical features of hereditary motor and sensory neuropathy types I and II. Brain. 1980, 103: 259-280. 10.1093/brain/103.2.259.
Davis CJ, Bradley WG, Madrid R: The peroneal muscular atrophy syndrome: clinical, genetic, electrophysiological and nerve biopsy studies. I. Clinical, genetic and electrophysiological findings and classification. J Genet Hum. 1978, 26: 311-349.
Nicholson G, Nash J: Intermediate nerve conduction velocities define X-linked Charcot-Marie-Tooth neuropathy families. Neurology. 1993, 43: 2264-2558.
Nicholson G, Myers S: Intermediate forms of Charcot-Marie-Tooth neuropathy: a review. Neuromolecular Med. 2006, 8: 123-130.
Chhatriwala MK, Betts L, Worthylake DK, Sondek J: The DH and PH domains of Trio coordinately engage Rho GTPases for their efficient activation. J Mol Biol. 2007, 368: 1307-1320. 10.1016/j.jmb.2007.02.060.
Matsuda A, Suzuki Y, Honda G, Muramatsu S, Matsuzaki O, Nagano Y, Doi T, Shimotohno K, Harada T, Nishida E, Hayashi H, Sugano S: Large-scale identification and characterization of human genes that activate NF-κB and MAPK signaling pathways. Oncogene. 2003, 22: 3307-3318. 10.1038/sj.onc.1206406.
Choi BO, Kim J, Lee KL, Yu JS, Hwang JH, Chung KW: Rapid diagnosis of CMT1A duplications and HNPP deletions by multiplex microsatellite PCR. Mol Cells. 2007, 23: 39-48.
Lee SS, Lee HJ, Park JM, Hong YB, Park KD, Yoo JH, Koo H, Jung SC, Park HS, Lee JH, Lee MG, Hyun YS, Nakhro K, Chung KW, Choi BO: Proximal dominant hereditary motor and sensory neuropathy with proximal dominance association with mutation in the TRK-fused gene. JAMA Neurol. 2013, 70: 607-615.
dbSNP137. [http://www.ncbi.nlm.nih.gov/snp/]
1000 Genomes database. [http://www.1000genomes.org/]
Tamura K, Peterson D, Peterson N, Stecher G, Nei M, Kumar S: MEGA5: molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol Biol Evol. 2011, 28: 2731-2739. 10.1093/molbev/msr121.
Shy ME, Blake J, Krajewski K, Fuerst DR, Laura M, Hahn AF, Li J, Lewis RA, Reilly M: Reliability and validity of the CMT neuropathy score as a measure of disability. Neurology. 2005, 64: 1209-1214. 10.1212/01.WNL.0000156517.00615.A3.
PolyPhen2. [http://genetics.bwh.harvard.edu/pph2/]
Rouger H, LeGuern E, Birouk N, Gouider R, Tardieu S, Plassart E, Gugenheim M, Vallat JM, Louboutin JP, Bouche P, Agid Y, Brice A: Charcot-Marie-Tooth disease with intermediate motor nerve conduction velocities: characterization of 14 Cx32 mutations in 35 families. Hum Mutat. 1997, 10: 443-452. 10.1002/(SICI)1098-1004(1997)10:6<443::AID-HUMU5>3.0.CO;2-E.
Viaud J, Gaits-Iacovoni F, Payrastre B: Regulation of the DH-PH tandem of guanine nucleotide exchange factor for Rho GTPases by phosphoinositides. Adv Biol Regul. 2012, 52: 303-314. 10.1016/j.jbior.2012.04.001.
Memet S: NF-κB functions in the nervous system: from development to disease. Biochem Pharmacol. 2006, 72: 1180-1195. 10.1016/j.bcp.2006.09.003.
Bitoun M, Stojkovic T, Prudhon B, Maurage CA, Latour P, Vermersch P, Guicheney P: A novel mutation in the dynamin 2 gene in a Charcot–Marie–Tooth type 2 patient: clinical and pathological findings. Neuromuscul Disord. 2008, 18: 334-338. 10.1016/j.nmd.2008.01.005.
Senderek J, Bergmann C, Weber S, Ketelsen UP, Schorie H, Rudnik-Schoneborn S, Buttner R, Buchheim E, Zerres K: Mutation of the SBF2 gene, encoding a novel member of the myotubularin family, in Charcot-Marie-Tooth neuropathy type 4B2/11p15. Hum Mol Genet. 2003, 12: 345-356.
Hadano S, Kunita R, Otomo A, Suzuki-Utsunomiya K, Ikeda JE: Molecular and cellular function of ALS2/alsin: implication of membrane dynamics in neuronal development and degeneration. Neurochem Int. 2007, 51: 74-84. 10.1016/j.neuint.2007.04.010.
Acknowledgements
This study was supported by the Korean Health Technology R&D Project, Ministry of Health & Welfare, Republic of Korea (A120182).
Author information
Authors and Affiliations
Corresponding authors
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
KHJ and CB-O: study concept and design; HYB, CYR, PM, KH, CKW and CB-O: acquisition of data; HYB, PJ-M, KYJ, YBR, and KH: analysis and interpretation of data; KHJ and PJ-M: drafting of the manuscript; KH, CKW, and CB-O: critical revision of the manuscript for important intellectual content; KYJ, YBR and PJ-M: administrative, technical, and material support; CKW, and CB-O: provision of funding and study supervision. All authors read and approved the final manuscript.
Hyeon Jin Kim, Young Bin Hong contributed equally to this work.
An erratum to this article is available at http://dx.doi.org/10.1186/1750-1172-8-165.
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
Open Access This article is published under license to BioMed Central Ltd. This is an Open Access article is distributed under the terms of the Creative Commons Attribution License ( https://creativecommons.org/licenses/by/2.0 ), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Kim, H.J., Hong, Y.B., Park, JM. et al. Mutations in the PLEKHG5 gene is relevant with autosomal recessive intermediate Charcot-Marie-Tooth disease. Orphanet J Rare Dis 8, 104 (2013). https://doi.org/10.1186/1750-1172-8-104
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1750-1172-8-104