
Abstract
Background:
The rise of network pharmacology has led to the widespread use of network-based computational methods in predicting drug target interaction (DTI). However, existing DTI prediction models typically rely on a limited amount of data to extract drug and target features, potentially affecting the comprehensiveness and robustness of features. In addition, although multiple networks are used for DTI prediction, the integration of heterogeneous information often involves simplistic aggregation and attention mechanisms, which may impose certain limitations.
Results:
MSH-DTI, a deep learning model for predicting drug-target interactions, is proposed in this paper. The model uses self-supervised learning methods to obtain drug and target structure features. A Heterogeneous Interaction-enhanced Feature Fusion Module is designed for multi-graph construction, and the graph convolutional networks are used to extract node features. With the help of an attention mechanism, the model focuses on the important parts of different features for prediction. Experimental results show that the AUROC and AUPR of MSH-DTI are 0.9620 and 0.9605 respectively, outperforming other models on the DTINet dataset.
Conclusion:
The proposed MSH-DTI is a helpful tool to discover drug-target interactions, which is also validated through case studies in predicting new DTIs.
Similar content being viewed by others


Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Introduction
The development of new drugs has become increasingly challenging, from finding new candidate compounds, to hierarchical experiment approval, which often takes more than a decade and requires significant financial investment, exceeding $2.5 billion [1, 2]. In drug discovery and design, accurate prediction of drug-target Interactions (DTI) is of great significance for drug development. By utilizing the target information of drug molecules, we can discover their relevant pharmacological and pharmacodynamic properties with the development of machine learning method. In addition, the prediction of DTIs is also important in drug repositioning. Drug repositioning offers a promising alternative by discovering new uses for existing drugs, identifying new drug-disease relationships, which could skip the early stages of drug discovery, reducing time and cost while minimizing risk, and making the development process more efficient. However, due to the high cost of large-scale in vitro and in vivo experiments, with the development of machine learning method, computer-aided methods for predicting DTIs are gaining increasing attention [3, 4].
Existing DTI prediction methods can be divided into three categories: structure-based method [5], ligand-based method [6], and network-based method. The structure-based prediction method predicts DTI by analyzing the three-dimensional structural features of target proteins. Prava et al. [7] used proteomics and protein–protein interaction network data to predict new targets for a variety of leishmania strains. They developed 3D structural models of multiple hub proteins and docking experiments were done with some ligands. However, the structure-based method requires 3D structural information of both the drug and target, which is not always available since the 3D structure of some proteins has not been determined, thus limiting the applicability of this method [8]. The ligand-based prediction method predicts potential targets based on the similarity principle [9]. Woo et al. [10] developed a ligand structure similarity-based strategy to predict potential repurposed drugs that inhibit SARS-CoV-2 by comparing structure similarity and interaction similarity of drugs [11].
The relationships between drugs and targets are complex [12]. The emergence of network pharmacology [13]has activated the adoption of network-based methods for DTI prediction. IMCHGAN [14] uses a two-level neural attention mechanism to extract features of drugs and targets from the DTI heterogeneous network, which are fed into an inductive matrix completion (IMC) model to calculate the DTI scores. Cai et al. [15] used heterogeneous graph attention networks to enhance learning of the graph structure from both intra- and inter-layer perspectives, providing efficient connections between nodes, improving the effectiveness of message propagation from important nodes. HampDTI [16] extract features from drug molecular graphs and target sequences, and automatically learns meta-paths between drugs and targets using the heterogeneous graphs. The learned drug and target features are represented as nodes in the meta-path graphs, and graph convolution is applied for node embedding. The embeddings of all meta-path graphs are integrated for DTI prediction. MdDTI [17] predicts drug-target interactions (DTI) by constructing spatial feature representation based on Cartesian coordinates of heavy atoms (excluding hydrogen atoms) in drugs. These 3D spatial features are combined with 2D substructure features to provide a multidimensional representation of drug features. MINN-DTI [18] combines an Interacting-Transformer module (Interformer) with an improved Communicative Message Passing Neural Network (Inter-CMPNN). This hybrid approach aims to use molecular graphs of drugs and distance maps of targets to capture the bidirectional effects between drugs and targets. PGraphDTA [19] improves DTI prediction accuracy by integrating three-dimensional structural information of targets. However, existing network-based approaches focus on the relationship between drug and targets, and do not fully leverage the structural information of drug molecules and the sequence information of targets, which play an important role in DTI prediction [20].
In addition, to effectively utilize information from multiple networks [21, 22], GCHN-DTI [23]integrates information from drug-target interactions, drug–drug interactions, drug similarities, target–target interactions, and target similarities to generate heterogeneous networks. The heterogeneous network is then fed into graph convolution operation to obtain node embeddings for drugs and targets. EEG-DTI [24] constructs heterogeneous network including drugs, proteins, diseases, and side effects. During the embedding generation process, the features of neighboring nodes with different types are considered to derive the final features for drugs or proteins. However, for DTI prediction, the training model often struggles to balance the weights [25] between drugs and targets. Moreover, by introducing multiple networks, how to effectively and reasonably fuse the information of different sources is another issue to be solved [26].
Inspired by recommender-based systems [27], a MSH-DTI framework is proposed in this paper for DTI prediction. First, self-supervised learning methods [28, 29] are utilized to extract features of drugs and targets. Multiple graphs are then constructed based on the dataset to capture complex associations between drugs and targets. A Heterogeneous Interaction-enhanced Feature Fusion Module(HIFFM) is proposed to enriches the feature of drugs and targets. The graph convolutional network is applied to different graphs to aggregate multi-layer information for a better understanding of the DTI. Finally, an attention mechanism is introduced to obtain more accurate prediction results by weighting the feature of drugs and targets. The comparative experiments demonstrate that the MSH-DTI outperforms other baseline models in DTI prediction.
In summary, the main contributions of this paper are listed as below:
-
(1)
Introducing self-supervised learning methods into the heterogeneous graph to obtain high-level feature of drugs and target.
-
(2)
Constructing multiple types of graphs and introducing the HIFFM to fuse the information from multiple graphs.
-
(3)
Multiple attention mechanisms are constructed to aggregate drug or target feature in different graphs by assigning higher weights to important part of features.
Methods
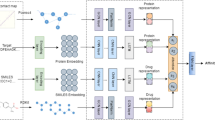
The overall framework of MSH-DTI is shown in Fig. 1. Firstly, the initial features of drug and target are obtained using the pre-trained self-supervised learning models, InfoGraph [28] and CPCPro [29], respectively. Next, three graphs including protein–protein interaction (PPI), drug–drug interaction (DDI) and drug–drug similarity (DDS) are constructed based on the drug and target initial features. In addition, another heterogeneous graph, protein-drug interaction (PDI), is constructed using multi-source data by a HIFFM, which could extract more comprehensive features in DTI. Next, multilayer graph convolutions are applied on four graphs to obtain the corresponding target features \(p^e\), \(p^c\) and drug feature \(d^e\), \(d^c\) and \(d^s\). Final target and drug feature are fused by the attention mechanism independently, which are multiplied together for DTI prediction.

Overall framework. a Feature extraction of drug and target using self-supervised learning method. b Construction of multiple graphs with HIFFM. c Multilayer graph convolution module. d Feature aggregation and result prediction with attention mechanism
Datasets
The DTINet dataset [30] is used in MSH-DTI from model training and test. The dataset consists of 708 drugs and 1512 targets, totaling 1923 drug-target associations, 10036 drug–drug associations, 7363 target–target associations. Among them, they extracted drug nodes, DTIs and drug–drug interactions from Drugbank 3.0 [31], and extracted target nodes and protein–protein interactions from HPRD [32]. The SMILES representations of drug are identified using DrugBank IDs, while the amino acid sequence are acquired according to the UniProt IDs of each target.
In addition, the ratio of negative to positive samples in the DTINet dataset is remarkable. The percentage of positive sample is 0.18%, and the percentage of negative samples is 99.82%.
Self-supervised feature extraction module
To fully utilize the structural information of drugs and targets, two self-supervised learning methods including Infograph and CPCProt are introduced to obtain more comprehensive representation for drug and target respectively.
However, it is important to note that self-supervised learning methods typically have higher computational complexity than traditional feature extraction approaches. Traditional methods often rely on hand-crafted feature extractors, which have relatively low computational costs, but may not capture the comprehensive intricate relationships within the data. Self-supervised learning methods, on the other hand, require more computational resources, more model training time and large-scale training data, which can involve more iterations and complex optimization processes.
10K molecules are used drug feature pre-training, which take about 0.07 h for each epoch, and 14 h in total. The protein feature pre-training process also need 1.5 h for each epoch on 5k protein sequences. However, through the pre-trained model, it only takes about 5 s to extract features of each drug and protein in the down-stream task.
Feature extraction of drug
The InfoGraph is an self-supervised learning model that utilizes graph neural networks to learn node and graph representations from graph data. By maximizing the mutual information between the graph representation and the patch representation, the InfoGraph model obtains an effective graph representation.
The SMILES strings of all drugs in the dataset are converted into molecular graphs using the RDKit [33], where each atom serves as a node and each bond serves as an edge. The generated drug molecular graph can be fed into the InfoGraph model which is pretrained on QM9 dataset [34]to extract structural features of the drug.
Feature extraction of target
To extract the structural features of the target, the CPCProt model is used, which is a self-supervised learning method by maximizing mutual information between both local and global information of protein sequence to obtain representation of the protein. The CPCProt model first divides the target sequence into fixed-size fragments and distinguishes between subsequent fragments from the same protein and fragments from random proteins using autoregressive modeling. Each fragment is then processed by the encoder to generate a feature. All features are concatenated to form the feature of protein.
In our model, all target sequences in the dataset are fed into the CPCProt which are already pre-trained on Pfam dataset [35] to extract the structural feature for each target.
To unify the dimensions of the target and drug features, both initial features of drug and target are transformed into the 128 dimension and fed into the proposed model. The target feature is denoted as p, while the drug feature is denoted as d.
Multiple graphs construction with heterogeneous interaction-enhanced feature fusion module
Multiple graphs construction
Once the self-supervised features have been extracted, they can be used to construct multiple graphs. For each graph, the self-supervised features of protein or drug are used as the feature each node in the graph. The relationship between each node according to the types of graphs. To capture the correlations between drug and target, multiple graphs are introduced into the model which capture different interaction information in each view. The first one is the drug–drug interaction graph (DDI), where the edges between drugs indicate their interaction relationships. The second one is the drug–drug similarity graph (DDS), where the edges between drugs indicate their similarity relationships. The similarity scores are Tanimoto coefficient calculated by extracting Morgan fingerprint [36] for each drug molecule. The other two graphs are protein–protein interaction graph (PPI) and protein-drug interaction graph (PDI) generated from the dataset.
Heterogeneous interaction-enhanced feature fusion module
The feature matrix of DDI, DDS and PPI are constructed by initial feature of drug or target. In PDI, the types drug node and target node are different. When aggregating node features from its neighborhood, it is helpful to incorporate information from other relationship into the feature aggregation process. Therefore, a Heterogeneous Interaction-enhanced Feature Fusion Module (HIFFM) is proposed to updated node feature in PDI through the other three graphs. The specific process is shown in Fig. 2.

Firstly, the homogeneous features of each drug and target are aggregated through the initial features of the 1-hop neighbors in the PPI, DDI and DDS respectively, then the HA features of drug and target are aggregated by their 1-hop heterogeneous neighbors respectively, and the final fusion feature is obtained by the initial features and HA features of the drug and target respectively
The nodes encoding of the three homogeneous networks, PPI, DDI, and DDS, are initially obtained based on features from their 1-hop neighborhood nodes through Eqs. 1 and 2.
where \(N_{PPI}(m)\),\(N_{DDI}(i)\) and \(N_{DDS}(i)\) are the set of neighbors of node in corresponding graph, and \(p_m^*\) and \(d_i^*\) are the homogeneous feature of the target and drug respectively.
Next, features of each node in PDI aggregate their 1-hop heterogeneous neighboring node as the Heterogeneous Aggregation (HA) feature to enrich the target and drug representation, as Eqs. 3 and 4.
where \(N_{PDI-H}(m)\) and \(N_{PDI-H}(i)\) is set of 1-hop heterogeneous neighbor nodes of m and i, \(p_m^{**}\) and \(d_i^{**}\) are the HA features of the target and the drug.
Considering the different contributions of the initial features and HA features, the final fusion features of the target and the drug are calculated by combining with initial features and HA features by assigning different weights through Eq. 5. The feature \(p_m^{**}\) processed by the HIFFM module is added to the original feature \(p_m\). The original feature \(p_m\) retains a large part of the original information, while the feature \(p_m^{**}\) contains more interaction information. Combining the two allows the model to obtain more comprehensive and enriched information.
where \(\alpha\) and \(\beta\) are initialized weights, and \(\alpha\) + \(\beta\) = 1.
Multilayer graph convolution module
After obtaining the feature of each node in the four graphs, the graph convolutional neural networks (GCN) is used to capture the relationships between nodes. Due to the complex and highly correlated structure of the interaction network, traditional machine learning methods often struggle to capture the intricate patterns and correlations within it. However, GCN uses the connectivity between nodes to propagate information throughout the network and aggregate features from neighboring nodes. This approach preserves the global structure while capturing local features and relationships. Therefore, GCN is appropriate for updating node features. The core idea of GCN is to aggregate and update node features by exploiting the connectivity between nodes. Through iterative convolution operations and feature aggregation, each node can obtain more comprehensive information, improving the learning ability of graph data.
Take the target-centered PDI as an example, the initial features p of the targets and the fusion features \(d^f\) of the processed drugs from the previous section are fed into the GCN. Through multi-layer graph convolution operations, higher-order node information can be gradually passed and integrated. To effectively utilize the feature representations from each layer and improve the comprehensiveness of each node, features obtained from different layers are aggregated by mean pooling to obtain the final feature of each node.
For PDI, two GCNs are implemented to obtain the features of target and drug respectively. The first GCN uses the initial feature of drug and fusion feature of target as input to extract the target-centered PDI feature \(p^e\). The second GCN use the initial feature of target and fusion feature of drug as input to extract the drug-centered PDI feature \(d^e\).
For PPI, DDI, and DDS, only the initial features of the drug and target are used as inputs. The target PPI feature \(p^c\) and drug DDI feature \(d^c\) and DDS feature \(d^s\) are obtained.
Feature aggregation and result prediction
After obtaining multiple features, a simple concatenation operation of them as the final feature for prediction is not sufficient to adequately express the various relationships between drug and target. To better capture the association information and improve the accuracy of the model, inspired by Neural Attentive Item Similarity model(NAIS) [27],an attention-based weighted summation mechanism is introduced for feature representation of drugs and targets, The core principle of attention mechanism is to dynamically adjust the weights of each feature based on its importance to better capture associative information. The attention mechanism allows the model to be more flexible and accurate in handling feature representations of drugs and targets. Through computation, the attention weights for each feature are automatically adjusted according to its task relevance. As a result, during the feature weighting and summation process, the model is better equipped to capture the intricate associative information within the network, which is described as Eq. 6.
where \(z_{\alpha }\), \(z_{\beta }\), \(w_{\alpha }\), \(w_{\beta }\), \(b_{\alpha }\) and \(b_{\beta }\) are trainable weight parameters, and RELU denotes the activation function. After obtaining \(\alpha _p\) and \(\beta _p\), the softmax activation function is used for normalization to calculate the final target feature \(p'\):
Similarly, based on the three features \(d^e\), \(d^c\) and \(d^s\) of the drug, the final drug feature \(d'\) is calculated in the same way.
After obtaining the final feature representations of the target and drug, the inner product operation is used to predict the drug-target interaction through Eq. 9. The inner product of the drug feature \(d_i'\) and target feature \(p_m'\) could be represented as the relationship between drug and target, since the inner product of two vectors is defined to be the amount that a vector is pointing in the same direction as the other vector. A larger inner product indicates that they are more similar or correlated.
\(y_{im}'\) denotes the label predicted by the model. Finally, the loss function is used to optimize the model:
\(\mu\) is the weight parameter, \(N_d\) is the number of drugs, \(N_p\) is the number of targets, \(y_{ij}\) is the true value of drug i and target j, \(y_{ij}'\) is the predicted value of drug i and target j. \(\odot\) denotes element-by-element multiplication, \(\parallel \parallel ^2\) is the squared Frobenius norm. Figure 3 shows the entire feature variation process.

The workflow of feature processing
Results
In order to address the issue of data imbalance, a 1:1 ratio of positive to negative samples were chosen. Specifically, to match the number of positive samples, equal number of negative samples were randomly selected to construct the training set, which ensures that the model utilizes equal number of positive and negative samples during training, thereby mitigating bias towards either class. The known drug target pairs in the dataset are considered as positive samples, while the rest of the unknown drug target pairs are considered as negative samples. Specifically, 10% of the positive samples and the same number of negative samples were used as the test set and the remaining samples are used as the training set. Moreover, 10-fold cross-validation is utilized to assess the model’s performance. This method partitions the dataset into ten equal subsets, with nine subsets used for training during each iteration and the remaining subset used for validation.
The data imbalance is a big issue due to significantly fewer positive samples than negative samples in the dataset. To better evaluate the performance of model on the imbalance dataset, AUROC (Area Under the Receiver Operating Characteristic Curve) and AUPR (Area Under the Precision versus Recall Curve) are chosen as evaluation metrics, which are widely used for evaluating binary classifiers. AUROC and AUPR are two commonly used evaluation metrics to assess the performance of classification models in predicting drug-target interactions. AUROC reflects the ability of the model to correctly identify drug-target interactions by considering both the true positive rate and the false positive rate.
To better demonstrate the superiority of the MSH-DTI, several baselines are used for performance comparison, including DTINet [30], IMCHGAN [14], HAS-DTI [37], EEG-DTI [24], and MultiDti [22]. DTINet [30] integrates a heterogeneous network to predict novel drug-target interactions. IMCHGAN [14] uses a two-level neural attention mechanism on the DTI heterogeneous network to extract drug and target features to calculate DTI scores. HAS-DTI [37] create multi-view graphs by sampling diverse neighbors and ensures consistency through graph contrastive optimization, integrating information from drug-side-effect networks, drug structures, drug-disease network and target-related networks for prediction. EEG-DTI [24] constructs a heterogeneous network by integrating features from diverse neighboring nodes to generate drug and protein embeddings. MultiDTI [22] integrates heterogeneous network interactions with drug and target sequence data to predict interactions for new chemical entities based on their structures and the learned representation of the network.
Performance comparison between MSH-DTI and other baseline methods
The evaluation results of MSH-DTI in comparison with baselines are shown in Table 1. The AUROC and AUPR of MSH-DTI are 0.9620 and 0.9605 respectively. MSH-DTI outperforms other baseline methods on AUROC, and obtains the second best result on AUPR. EEG-DTI outperformed MSH-DTI in terms of AUPR by 0.4%, probably due to the introduction of additional data. Unlike MSH-DTI, EEG-DTI incorporates additional information such as drug-side effect associations and drug-disease interactions, which are connected with the drug-target interaction networks. These additional sources of data are likely to provide EEG-DTI with a more comprehensive understanding of the DTI. However, the proposed MSH-DTI solely on drug-target interaction networks could also achieve the similar AUPR value and the best AUROC value, which also demonstrate its scalability and practicality, making it applicable to a wider range of scenarios. The introduction of self-supervised pre-trained models is helpful to extract more comprehensive information from drug and target. In addition, MSH-DTI can construct and utilize multiple types of graphs to capture drug-target interactions from different perspectives, thereby improving the ability of the model to capture relevant features and patterns. For example, drug-drug interaction netwokrs could extract complex information of drug [38, 39]. With the help of the attention mechanisms, the model can effectively capture key information related to drug-target interactions.
Hyperparameter experiments
Hyperparameter configurations are crucial for model performance and generalization. A series of experiments were implemented to find the optimal hyperparameters, including the dimensions of hidden layers of feature, and the number of convolutional layers of GCN. The hyperparameters in this study is given in Table 2.
Firstly, the dimension of hidden layers ranging from 64 to 512 are tested to analyze the performance of model, as shown in Fig. 4a. The AUROC value exhibit an increasing trend until reaching the peak at 128 dimensions, after which it declines with dimension increase. The lower dimension of hidden layers may struggle to capture the complexity of node. Conversely, higher dimension of hidden layers may result in the overfitting problem. Finally, the number of convolutional layers are evaluated as Fig. 4b. The best prediction results are achieved with 6 convolutional layers. More or less layers may result in information loss or over smoothing of the model.

Performance analysis of MSH-DTI model hyperparameter configurations
Ablation experiments
To further investigate the importance of individual components proposed in this paper, several variants are considered in this section for ablation experiments. The first step is to determine the contribution role of the initial features extraction method of drug and protein through self-supervised pre-training including InfoGraph and CPCProt. Three variants are used for comparison, the first one is ECFP+FOAA. Two hand-craft feature extraction method are used for drug and protein. The ECFP [36] is a Circular fingerprint for drug and FOAA [40] analyzing the frequency of occurrence of different amino acids in the protein sequence to generate the protein feature. The second one is GNN+LSTM. The GNN is used for extracting drug features through molecular graph and the LSTM is used for extracting protein features through sequence. The last one is Morgan+CPCProt, which uses the Morgan fingerprint method to extract drug features, and radius is set as 2 to consider the local environment of the molecule. At the same time, the CPCProt method is used to process protein sequences.
The results are shown in Fig. 5. Specifically, the AUROC is improved by 3.17% and the AUPR is improved by 1.87% of MSH-DTI compared to the Morgan+CPCP algorithm, while the results of ECFP+FOAA is close to that of GNN+LSTM. The results indicate that the feature representations obtained through self-supervised models play a positive role in improving model performance, which could be pre-trained with large-scale unlabeled data, and the model can learn a more robust and generalized representation based on large-scale data.

Comparison of different feature extraction methods
In addition, to evaluate the contribution of the HIFFM and the attention mechanism in the model, three variants are set up.
MSH-NS: The similarity network is removed in the model. MSH-NN: The HIFFM is removed in the model, which utilized multiple graphs to construct fusion features. MSH-NA: The attention mechanism is removed in the model.
The performance of the three variants of the model and the proposed model are shown in Fig. 6, all variants exhibit a slight reduction in performance compared to the proposed model, which suggests that the similarity network, HIFFM and the attention mechanism play important roles in enhancing feature transfer and aggregation between drugs and targets. Through the utilization of a similarity network, the model is encouraged to acquire more meaningful features, thereby improving its ability to comprehend the input data. Furthermore, the HIFFM provides an effective mechanism for guiding the model to learn relationships between nodes and facilitate feature propagation. The attention mechanism enables the model to focus on key aspects of the final features and assign varying weights to different output features, thereby better capturing the important features of the input data.

Comparison of the three variants with the proposed model
SHAP analysis
SHAP (SHapley Additive exPlanations) analysis is a method used to interpret the predictions of machine learning models, which provides explanations by calculating the contribution of each feature to the model predictions. It is an intuitive and interpretable way to understand the basis of model decisions. Figure 7 shows the results of the SHAP analysis and the top 15 features ranked by their contributions are selected, with red representing high value and blue representing low value.

The importance of features to prediction results
In Fig. 7, it can be observed that Feature103 has positive SHAP values (becoming increasingly red towards the right) when its value is higher, and negative SHAP values (becoming increasingly blue towards the left) when its value is lower. The red points of Features 103, 34, 231, 167, 203, 51 are clustered to the right of the x-axis respectively, indicating that higher values of these features have positive effects on the prediction results. Conversely, the red points of Features 150, 87 are clustered to the left of the x-axis respectively, indicating that at higher values of these features, the model tends to predict the absence of DTIs.
Case study
In this section, the ability of MSH-DTI applied in the real scenario is evaluated. We selected one drugs and one target from the DTINet dataset in order to predict their potential targets and drugs to validate the performance of the model, which are Cefazolin and Muscarinic acetylcholine receptor M2. Cefazolin, a broad-spectrum cephalosporin antibiotic, is predominantly indicated for the treatment of skin infections and moderate to severe infections affecting the respiratory system. On the other hand, Muscarinic acetylcholine receptor M2 is a receptor protein that can interact with neurotransmitters such as acetylcholine and plays an important role in the nervous system.
For testing drug, all known associations in the dataset were utilized to train the model, and unknown associations between all targets in the dataset with testing drug were predicted by the model. The top 10 candidate targets were selected according to their prediction scores, and relevant literature was consulted to verify the validity of the prediction, which are shown in Table 3. Similarly, the top 10 candidate drugs for Muscarinic acetylcholine receptor M2 were selected according to their prediction scores, which are shown as Table 4.
For Cefazolin, the Glucocorticoid receptor owns the highest prediction score, suggesting a potential interaction with Cefazolin, which is consistent with reports [41] that Glucocorticoid receptor interacts with Cefazolin by modulating the inflammatory response and immune function. The Fibroblast growth factor 2 is the second predicted drugs. It is reported that Cefazolin could potentially affect fibrinogen concentrations and basic fibroblast growth factor (bFGF) levels in vivo, which exhibits anti-angiogenic activities [42].
For Muscarinic acetylcholine receptor M2 (M2 receptor), Olanzapine and Lidocaine are shown to interact with M2 receptor. Among them, Olanzapine increases the release of acetylcholine by antagonising the M2 receptor, which could can affect the function of nervous system, including cognition and mood [45]. According to the literature [46], when used in combination with muscarinic receptor agonists (such as methylcholine), Lidocaine enhances the atrial natriuretic peptide (ANP) relaxation response in bovine tracheal smooth muscle by interacting with the M2 receptor.
In summary, over half of the top 10 interactions predicted by the MSH-DTI for the two entities have been well verified in the literature, which further demonstrated the generalization performance of MSH-DTI in the real scenario.
Cold-start experiment
In order to demonstrate the applicability of the proposed method in real-world scenario, a cold-start experiment which excluding repetitive drugs from the training set is conducted to evaluating the generalization ability of the model facing unseen data.
First, from all positive drug-target interaction samples, 10% of the drugs are selected as new drugs, along with an equal number of negative samples, to form the test set. Next, we remove all known associations between these drug candidates and targets from the drug-target interaction network. The remaining positive samples involving other drugs, along with an equal number of negative samples, are used as the training set to train the MSH-DTI. This method aims to evaluate the ability of the model to predict potential associations for new drugs, which is crucial for assessing the ability of the model to handle unknown data in practical applications.
As shown in Table 5, MSH-DTI showed a significant improvement over EEG-DTI in the cold start experiment. This improvement can be attributed to the superior feature representation capability of MSH-DTI, which uses advanced pre-trained models to capture deeper and more complex relationships within the data. In addition, the pre-trained features enable MSH-DTI to better generalize to new and unseen data, making it particularly effective in cold-start scenarios.
Discussion
In this paper, two self-supervised learning methods are utilized to represent drug molecules and target sequences, which are InfoGraph and CPCProt. The core idea of InfoGraph is to learn the representation of nodes by utilizing local and total information in graph data, while CPCProt is a self-supervised learning framework for protein sequences. With the help of self-supervised learning, effective representations can be learned from large-scale unlabeled molecular data and protein sequence data to improve the comprehensiveness and robustness of the features of molecule and protein, which can achieve better performance in property prediction and drug discovery.
Secondly, the introduction of multiple source information to the graph convolution process improves the model to extract useful information from graph data more efficiently. In addition, the HIFFM proposed in this paper could take advantage of connectivity in multiple homogeneous and heterogeneous graphs, and the feature of each node could be aggregated by multiple source data, thereby facilitating a better representation of the relationships between nodes in the graph.
Finally, it is observed that the performance the model is improved by applying the attention mechanism to DTI. This may be attributed to the fact that through the attention mechanism, the model could dynamically adjust the attention weights between the drug and the target, which improves the accuracy of the prediction.
Conclusion
The MSH-DTI, a deep learning framework for drug target interaction prediction is proposed in this paper. Two self-supervised learning methods are introduced to extract the initial feature of drugs and targets. By combining multiple graphs, the heterogeneous aggregation features are constructed to learn effective feature of node. The attention mechanism is then employed for feature aggregation, improving overall performance. Experimental results demonstrate that MSH-DTI outperforms other prediction methods. Moreover, MSH-DTI could be extended to the prediction of drug–drug and target–target interactions.
Availability of data and materials
The MSH-DTI code and data are provided at https://github.com/Beiyi0719/MSH-DTI. The DTINet dataset and the source code are acquired through https://github.com/luoyunan/DTINet.
Abbreviations
- DTI::
-
Drug-target interaction
- SMILES::
-
Simplified molecular-input line-entry system
- HIFFM::
-
Heterogeneous interaction-enhanced feature fusion module
- GCN::
-
Graph convolutional neural networks
- ECFP::
-
Extended-connectivity fingerprint
- FOAA::
-
Frequency Of amino acid occurrence
- GNN::
-
Graph neural networks
- LSTM::
-
Long short-term memory network
References
Deng J, Yang Z, Ojima I, Samaras D, Wang F. Artificial intelligence in drug discovery: applications and techniques. Brief Bioinform. 2021;23(1):430. https://doi.org/10.1093/bib/bbab430.
Li Z, Jiang M, Wang S, Zhang S. Deep learning methods for molecular representation and property prediction. Drug Discov Today. 2022;27: 103373.
Xia L, Xu L, Pan S, Niu D, Zhang B, Li Z. Drug–target binding affinity prediction using message passing neural network and self supervised learning. BMC Genomics. 2023;24(1):557.
Pan S, Xia L, Xu L, Li Z. SubMDTA: drug target affinity prediction based on substructure extraction and multi-scale features. BMC Bioinf. 2023;24(1):334.
Jarada TN, Rokne JG, Alhajj R. A review of computational drug repositioning: strategies, approaches, opportunities, challenges, and directions. J Cheminf. 2020;12(1):1–23.
Shim J, Hong Z-Y, Sohn I, Hwang C. Prediction of drug–target binding affinity using similarity-based convolutional neural network. Sci Rep. 2021;11(1):4416.
Prava J, Pan A. In silico analysis of leishmania proteomes and protein–protein interaction network: prioritizing therapeutic targets and drugs for repurposing to treat leishmaniasis. Acta Trop. 2022;229: 106337. https://doi.org/10.1016/j.actatropica.2022.106337.
Bai Q, Tan S, Xu T, Liu H, Huang J, Yao X. MolAICal: a soft tool for 3D drug design of protein targets by artificial intelligence and classical algorithm. Brief Bioinform. 2020;22(3):161. https://doi.org/10.1093/bib/bbaa161.
Fan J, Fu A, Zhang L. Progress in molecular docking. Quant Biol. 2019;7:83–9.
Jang WD, Jeon S, Kim S, Lee SY. Drugs repurposed for COVID-19 by virtual screening of 6218 drugs and cell-based assay. Proc Natl Acad Sci USA. 2021;118(30):2024302118.
Wang Z, Liang L, Yin Z, Lin J. Improving chemical similarity ensemble approach in target prediction. J Cheminf. 2016;8:1–10.
Li M, Cai X, Xu S, Ji H. Metapath-aggregated heterogeneous graph neural network for drug–target interaction prediction. Brief Bioinform. 2023;24(1):578. https://doi.org/10.1093/bib/bbac578.
Thafar MA, Olayan RS, Ashoor H, Albaradei S, Bajic VB, Gao X, Gojobori T, Essack M. DTiGEMS+: drug–target interaction prediction using graph embedding, graph mining, and similarity-based techniques. J Cheminf. 2020;12(1):1–17.
Li J, Wang J, Lv H, Zhang Z, Wang Z. IMCHGAN: inductive matrix completion with heterogeneous graph attention networks for drug–target interactions prediction. IEEE/ACM Trans Comput Biol Bioinf. 2021;19(2):655–65.
Li M, Cai X, Li L, Xu S, Ji H. Heterogeneous graph attention network for drug-target interaction prediction. In: Proceedings of the 31st ACM international conference on information and knowledge management 2022. pp. 1166–1176.
Wang H, Huang F, Zhang W. HampDTI: a heterogeneous graph automatic meta-path learning method for drug-target interaction prediction 2021. arXiv:2112.08567.
Li H, Wang S, Zheng W, Yu L. Multi-dimensional search for drug–target interaction prediction by preserving the consistency of attention distribution. Comput Biol Chem. 2023;107: 107968. https://doi.org/10.1016/j.compbiolchem.2023.107968.
Li F, Zhang Z, Guan J, Zhou S. Effective drug–target interaction prediction with mutual interaction neural network. Bioinformatics. 2022;38(14):3582–9. https://doi.org/10.1093/bioinformatics/btac377.
Bal R, Xiao Y, Wang W. Pgraphdta: improving drug target interaction prediction using protein language models and contact maps 2023. arXiv:2310.04017.
Yu Z, Huang F, Zhao X, Xiao W, Zhang W. Predicting drug-disease associations through layer attention graph convolutional network. Brief Bioinform. 2021;22(4):243.
Zhao T, Hu Y, Valsdottir LR, Zang T, Peng J. Identifying drug–target interactions based on graph convolutional network and deep neural network. Brief Bioinform. 2020;22(2):2141–50. https://doi.org/10.1093/bib/bbaa044.
Zhou D, Xu Z, Li W, Xie X, Peng S. MultiDTI: drug–target interaction prediction based on multi-modal representation learning to bridge the gap between new chemical entities and known heterogeneous network. Bioinformatics. 2021;37(23):4485–92. https://doi.org/10.1093/bioinformatics/btab473.
Wang W, Liang S, Yu M, Liu D, Zhang H, Wang X, Zhou Y. GCHN-DTI: predicting drug–target interactions by graph convolution on heterogeneous networks. Methods. 2022;206:101–7.
Peng J, Wang Y, Guan J, Li J, Han R, Hao J, Wei Z, Shang X. An end-to-end heterogeneous graph representation learning-based framework for drug-target interaction prediction. Brief Bioinform. 2021;22(5):bbaa430. https://doi.org/10.1093/bib/bbaa430.
Cheng Z, Yan C, Wu F-X, Wang J. Drug–target interaction prediction using multi-head self-attention and graph attention network. IEEE/ACM Trans Comput Biol Bioinf. 2022;19(4):2208–18. https://doi.org/10.1109/TCBB.2021.3077905.
Shao K, Zhang Y, Wen Y, Zhang Z, He S, Bo X. DTI-HETA: prediction of drug–target interactions based on GCN and GAT on heterogeneous graph. Brief Bioinform. 2022;23(3):109. https://doi.org/10.1093/bib/bbac109.
He X, He Z, Song J, Liu Z, Jiang Y-G, Chua T-S. NAIS: neural attentive item similarity model for recommendation. IEEE Trans Knowl Data Eng. 2018;30(12):2354–66. https://doi.org/10.1109/TKDE.2018.2831682.
Sun F, Hoffmann J, Tang J. Infograph: Unsupervised and semi-supervised graph-level representation learning via mutual information maximization. In: CoRR; 2019. arXiv: 1908.01000.
Lu AX, Zhang H, Ghassemi M, Moses A. Self-supervised contrastive learning of protein representations by mutual information maximization. bioRxiv 2020. https://doi.org/10.1101/2020.09.04.283929
Luo Y, Zhao X, Zhou J, Yang J, Zhang Y, Kuang W, Peng J, Chen L, Zeng J. A network integration approach for drug–target interaction prediction and computational drug repositioning from heterogeneous information. Nat Commun. 2017;8(1):573.
Knox C, Law V, Jewison T, Liu P, Ly S, Frolkis A, Pon A, Banco K, Mak C, Neveu V, et al. Drugbank 3.0: a comprehensive resource for ‘omics’ research on drugs. Nucleic Acids Res. 2010;39(1):1035–41.
Keshava-Prasad T, Goel R, Kandasamy K, Keerthikumar S, Kumar S, Mathivanan S, Telikicherla D, Raju R, Shafreen B, Venugopal A, et al. Human protein reference database-2009 update. Nucleic Acids Res. 2009;37(1):767–72.
Landrum G, et al. RDKit: a software suite for cheminformatics, computational chemistry, and predictive modeling. Greg Landrum. 2013;8:31.
Ramakrishnan R, Dral PO, Rupp M, Von Lilienfeld OA. Quantum chemistry structures and properties of 134 kilo molecules. Sci Data. 2014;1(1):1–7.
El-Gebali S, Mistry J, Bateman A, Eddy SR, Luciani A, Potter SC, Qureshi M, Richardson LJ, Salazar GA, Smart A, Sonnhammer EL, Hirsh L, Paladin L, Piovesan D, Tosatto SC, Finn RD. The Pfam protein families database in 2019. Nucleic Acids Res. 2018;47(D1):427–32. https://doi.org/10.1093/nar/gky995.
Rogers D, Hahn M. Extended-connectivity fingerprints. J Chem Inf Model. 2010;50(5):742–54.
Wang R, Zhang Z, Zhang Y, Jiang Z, Sun S, Zhang C. Sparse imbalanced drug-target interaction prediction via heterogeneous data augmentation and node similarity. In: Pacific-Asia conference on knowledge discovery and data mining. Springer; 2022. pp. 548–561.
Niu D, Xu L, Pan S, Xia L, Li Z. SRR-DDI: a drug–drug interaction prediction model with substructure refined representation learning based on self-attention mechanism. Knowl Based Syst. 2024;285: 111337. https://doi.org/10.1016/j.knosys.2023.111337.
Niu D, Zhang L, Zhang B, Zhang Q, Li Z. DAS-DDI: a dual-view framework with drug association and drug structure for drug–drug interaction prediction. J Biomed Inf. 2024;156: 104672. https://doi.org/10.1016/j.jbi.2024.104672.
Godoy-Ruiz R, Ariza F, Rodriguez-Larrea D, Perez-Jimenez R, Ibarra-Molero B, Sanchez-Ruiz JM. Natural selection for kinetic stability is a likely origin of correlations between mutational effects on protein energetics and frequencies of amino acid occurrences in sequence alignments. J Mol Biol. 2006;362(5):966–78. https://doi.org/10.1016/j.jmb.2006.07.065.
Golikov P. Change in the function of glucocorticoid receptors and translocation of glucocorticoid receptor complexes in hepatocyte nuclei exposed to nonsteroidal drugs. Patologicheskaia fiziologiia i eksperimental’naia terapiia. 1991;6:34–6.
Michalska M, Palatyńska-Ulatowska A, Palatyński A, Mirowski M, Kaplińska K, Nawrot-Modranka J, Lazarenkow A. Influence of antibiotic therapy on the level of selected angiogenic factors in patients with benign gynecologic tumors-preliminary report. Pharmazie. 2011;66(8):619–22.
Chung AH, Watson K. Cefazolin-induced hypoprothrombinemia. Am J Health-Syst Pharm. 2008;65(9):823–6. https://doi.org/10.2146/ajhp070243.
Aslan E, Adem S. Investigation of the effects of some drugs and phenolic compounds on human dihydrofolate reductase activity. J Biochem Mol Toxicol. 2015;29(3):135–9.
Johnson DE, Nedza FM, Spracklin DK, Ward KM, Schmidt AW, Iredale PA, Godek DM, Rollema H. The role of muscarinic receptor antagonism in antipsychotic-induced hippocampal acetylcholine release. Eur J Pharmacol. 2005;506(3):209–19.
Yunoki M, Nakahara T, Mitani A, Sakamoto K, Ishii K. Role of the M\(_2\) muscarinic receptor pathway in lidocaine-induced potentiation of the relaxant response to atrial natriuretic peptide in bovine tracheal smooth muscle. Naunyn-Schmiedeberg’s Arch Pharmacol. 2003;367:76–9.
Zhu S, Khalafi S, Chen Z, Poveda J, Peng D, Lu H, Soutto M, Que J, Garcia-Buitrago M, Zaika A, et al. Silencing of miR490–3p by H. pylori activates DARPP-32 and induces resistance to gefitinib. Cancer Lett. 2020;491:87–96.
Benavides-Haro DE, Sánchez-Chapula JA. Chloroquine blocks the background potassium current in guinea pig atrial myocytes. Naunyn-Schmiedeberg’s Arch Pharmacol. 2000;361:311–8.
Kume H, Nishiyama O, Isoya T, Higashimoto Y, Tohda Y, Noda Y. Involvement of allosteric effect and \(\text{ K}_{{\rm CA}}\) channels in crosstalk between \(\beta\)2-adrenergic and muscarinic \(\text{ M}_2\) receptors in airway smooth muscle. Int J Mol Sci. 2018;19(7):1999.
Acknowledgements
Not applicable.
Funding
This work has been supported by National Natural Science Foundation of China (12371491).
Author information
Authors and Affiliations
Contributions
B.Z. designed the method and wrote the manuscript. D.N. designed the experiments and collected the data. L.Z. and Q.Z. analyzed the results. Z.L. designed the method and revised the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Conflict of interest
The authors declare that they have no Conflict of interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Zhang, B., Niu, D., Zhang, L. et al. MSH-DTI: multi-graph convolution with self-supervised embedding and heterogeneous aggregation for drug-target interaction prediction. BMC Bioinformatics 25, 275 (2024). https://doi.org/10.1186/s12859-024-05904-5
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12859-024-05904-5