
Abstract
Background
Rootstocks can improve the chilling tolerance of grafted cucumbers, but their effectiveness varies. Rootstocks with strong de-blooming capacity may result in lower chilling tolerance of grafted cucumbers compared to those with weak de-blooming capacity, while also reducing the silicon absorption. However, it remains unclear whether this reduction in chilling tolerance is due to differences in rootstock genotypes or the reduction in silicon absorption.
Results
The chilling tolerance of cucumber seedlings was improved by using rootstocks and silicon nutrition. Rootstocks had a more significant effect than silicon nutrition, and the weak de-blooming rootstock ‘Yunnan figleaf gourd’ was superior to the strong de-blooming rootstock ‘Huangchenggen No. 2’. Compared to self-rooted cucumber, twelve miRNAs were regulated by two rootstocks, including seven identical miRNAs (novel-mir23, novel-mir26, novel-mir30, novel-mir37, novel-mir46, miR395a and miR398a-3p) and five different miRNAs (novel-mir32, novel-mir38, novel-mir65, novel-mir78 and miR397a). Notably, four of these miRNAs (novel-mir38, novel-mir65, novel-mir78 and miR397a) were only identified in ‘Yunnan figleaf gourd’-grafted cucumbers. Furthermore, six miRNAs (miR168a-5p, miR390a-5p, novel-mir26, novel-mir55, novel-mir67 and novel-mir70) were found to be responsive to exogenous silicon. Target gene prediction for 20 miRNAs resulted in 520 genes. Functional analysis of these target genes showed that ‘Yunnan figleaf gourd’ improves the chilling tolerance of cucumber by regulating laccase synthesis and sulfate metabolism, while ‘Huangchenggen No. 2’ and exogenous silicon reduced chilling stress damage to cucumber by regulating ROS scavenging and protein protection, respectively.
Conclusion
Among the identified miRNAs, novel-mir46 and miR398a-3p were found in cucumbers in response to chilling stress and two types of rootstocks. However, no identical miRNAs were identified in response to chilling stress and silicon. In addition, the differential expression of novel-mir38, novel-mir65, novel-mir78 and miR397a may be one of the important reasons for the differences in chilling tolerance of grafted cucumbers caused by two types of rootstocks.
Similar content being viewed by others

Background
Cucumber (Cucumis sativus L.) is an important vegetable crop grown in protected cultivation facilities. It is native to the tropics and is considered a cold-sensitive crop [1]. In some northern regions, low winter and spring temperatures can seriously affect the normal growth of cucumbers in protected cultivation facilities, leading to a reduction in both yield and quality.
Grafting is a significant agronomic technique that can improve the yield and stress tolerance of cucumber plants [2]. In the past, Cucurbita ficifolia Bouché has been the preferred rootstock due to its superior stress tolerance. However, the use of Cucurbita ficifolia Bouché as a rootstock leads to a thick layer of bloom on the surface of grafted cucumber fruit, which ultimately reduces the quality of the cucumber [3]. In recent years, Cucurbita moschata Duch has been gradually increased to improve the quality of cucumbers [4]. However, cucumbers grafted with Cucurbita moschata Duch have been found to have lower chilling tolerance compared to those grafted with Cucurbita ficifolia Bouché [5, 6].
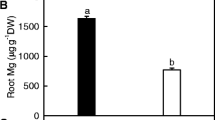
Several studies have shown that the amount of bloom on the fruit surface of cucumber is related to the ability of plants to absorb and metabolize silicon [7, 8]. Silicon is a beneficial element for plant development, as it increases cucumber yield and mitigates the negative impacts of abiotic stressors [9,10,11]. It has been speculated that the chilling tolerance of grafted cucumber may be related to its absorption and metabolism of silicon. However, it is unclear how rootstock genotypes and silicon nutrition differ in their respective roles and molecular mechanisms in improving the chilling tolerance of cucumber.
Plant microRNAs (miRNAs) are endogenous non-coding small RNAs of 21–24 nucleotides (nt) in length that are highly conserved among species and play an essential role in gene regulation [12]. miRNAs can target single or multiple genes to regulate plant responses to stressful environments [13]. Studies have shown that low temperatures can alter miRNAs expression in various plants, such as wheat [14], Arabidopsis [15], and tomato [16]. In addition, some studies have suggested that regulating miRNAs expressions through rootstocks and exogenous can improve plant stress tolerance [17,18,19,20]. These results have shown that miRNAs play an essential role in the response of plants to chilling stress.
The chilling tolerance of cucumber is influenced by rootstocks and silicon nutrition. However, determining whether the main cause of variation in the chilling tolerance of grafted cucumber is due to the rootstock genotype or silicon nutrition requires more systematic and in-depth studies. In this study, we used ‘Xintaimici’ cucumber together with two different rootstocks: the strong de-blooming rootstock ‘Huangchenggen No. 2’ (Cucurbita moschata D.) and the weak de-blooming rootstock ‘Yunnan figleaf gourd’ (Cucurbita ficifolia B.) as experimental materials. We employed high-throughput sequencing technology to identify miRNAs regulated by rootstocks and silicon under chilling stress. Our objective was to investigate the cause of altered chilling tolerance in cucumber from the perspective of miRNAs.
Results
Rootstock types and silicon nutrition on chilling tolerance of cucumber seedlings
The growth index and chilling injury index of cucumber seedlings in five treatments were measured to visually demonstrate the difference between the two rootstocks and silicon nutrition in reducing chilling injury (Table 1). In the TC/CC comparison pair, chilling stress inhibited the normal growth of cucumbers and caused severe chilling injury to the leaves. In the TS/TC, TH/TC and TY/TC comparison pairs, the application of two rootstocks and silicon nutrition reduced chilling injury, with rootstocks being more effective than silicon nutrition. Notably, the ‘Yunnan figleaf gourd’ showed the most significant reduction in chilling injury among the rootstocks.
Small RNA regulated by rootstock types and silicon nutrition under chilling stress
We constructed five RNA libraries (CC, TC, TS, TH and TY) to investigate the differential expression of small RNAs (sRNAs) in cucumber seedlings under chilling stress in response to rootstocks and silicon. The raw reads identified in these libraries were 13,472,603, 12,525,103, 12,975,897, 14,870,839, and 14,685,213, respectively (Table 2). After removing contaminating and low-quality sequences, 13,258,990 (98.41%), 12,315,436 (98.33%), 12,722,672 (98.05%), 14,607,618 (98.23%) and 14,377,447 (97.90%) clean reads were retained, respectively, and their lengths were calculated (Fig. 1). The results showed that the lengths of sRNAs in five libraries were mainly concentrated in 21 nt and 24 nt, accounting for 17.25% and 17.30% of the total reads, respectively, followed by sRNAs with the length of 23 nt, accounting for 14.22%.

Length distribution of sRNA in CC, TC, TS, TH and TY libraries. Y-axis represents percentages of sRNAs identified in the study; X-axis represents the length of sRNAs. Five libraries are shown in different colours
All clean reads from the five libraries were then aligned with genome databases to classify the remaining sRNAs (Table 3). The results showed that 9,547,918 (53.4%), 8,366,100 (64.1%), 10,000,041 (67.62%), 10,628,208 (72.76%) and 9,993,536 (69.51%) of the reads in five libraries mapped to the cucumber genome database, respectively. Among them, 5,047,596, 4,339,259, 5,855,676, 5,093,037, and 4,231,038 reads in five libraries were annotated as tRNA, rRNA and other non-coding RNAs, respectively. In addition, 277,069, 232,908, 239,674, 333,648 and 321,294 clean reads matched exons or repetitive regions in the five libraries, respectively. Finally, among the remaining clean reads in each library, 548,451, 452,785, 762,301, 735,048 and 679,968 reads could be matched to known miRNAs in miRBase. The remaining 3,275,890, 2,839,006, 2,712,306, 3,915,178 and 4,217,851 reads in each library reached the non-coding region of the cucumber genome and might contain novel miRNAs.
Differentially expressed miRNAs regulated by rootstocks and silicon under chilling stress
We identified 64 known miRNAs belonging to 26 miRNA families in five sRNA libraries (Additional file 1: Table S1). After removing low-expressed sequences with less than 10 reads in each library for subsequent data analysis, 44 known miRNAs from 22 miRNA families were retained (Additional file 2: Table S2). Among these miRNA families, miR156/157, miR159, miR165/miR166, miR168, miR171, miR319, miR390, miR396 and miR398 had more than two members. The miR171 family had the highest number of members with five. In contrast, miRNA families such as miR160, miR162, and miR172 had only one member each. In addition, the expression of miRNA could be inferred from their reads. For example, miR159a and miR166a-3p had more than 100,000 reads in each library, indicating that they were highly expressed. However, the reads of miR390b-3p, miR858a, miR858b and miR2111a-5p were all less than 30 in all five libraries, indicating that their expression levels were significantly low. Finally, the classification of known miRNAs according to sequence length revealed that 21 nt was the major length of known miRNAs, accounting for 79%.
To identify potential novel miRNAs, we aligned the remaining reading fragments from the five libraries to the cucumber genome by using miREvo and mirdeep2 identifying 63 novel miRNAs after removing those with less than 10 reads in each library (Additional file 3: Table S3). Analysis of the length analysis of these novel miRNA revealed that 21 nt and 24 nt were the major lengths across the five libraries, accounting for 36.5% and 44.4%, respectively.

Heat map of differentially expressed miRNAs in four comparison pairs. a Differentially expressed miRNAs in TC/CC comparison pair. b Differentially expressed miRNAs in TS/TC comparison pair. c Differentially expressed miRNAs in TH/TC comparison pair. d Differentially expressed miRNAs in TY/TC comparison pair. Data are presented as heatmaps of Log2-transformed fold change. Red indicates high expression, white indicates intermediate expression, and blue indicates low expression
To investigate the effect of rootstock type and silicon nutrition under chilling stress on miRNA regulation, five libraries were divided into four comparison pairs (TC/CC, TS/TC, TH/TC, and TY/TC). The expression of miRNAs in each comparison pair was compared to obtain four groups of differentially expressed miRNAs (Fig. 2). In the TC/CC comparison pair, 6 miRNAs were identified in response to chilling stress, including 4 known miRNAs (miR408-3p, miR398a-3p, miR397a and miR157d) and 2 novel miRNAs (novel-mir21 and novel-mir46) (Fig. 2A). The TH/TC and TY/TC comparison pairs revealed 8 and 11 differentially expressed miRNAs, respectively (Fig. 2C and D). Among the miRNAs regulated by ‘Huangchenggen No. 2’, including 2 known miRNAs (miR395a and miR398a-3p) and 6 novel miRNAs (novel-mir23, novel-mir26, novel-mir30, novel-mir32, novel-mir37 and novel-mir46). There were 11 miRNAs regulated by ‘Yunnan figleaf gourd’, including 3 known miRNAs (miR395a, miR397a and miR398a-3p) and 8 novel miRNAs (novel-mir23, novel-mir26, novel-mir30, novel-mir38, novel-mir37, novel-mir46, novel-mir65, and novel-mir78). Finally, 6 miRNAs were identified from the TS/TC comparison pair, induced by exogenous silicon under chilling stress, including 2 known miRNAs (miR168a-5p and miR390a-5p) and 4 novel miRNAs (novel-mir26, novel-mir55, novel-mir67 and novel-mir70) (Fig. 2B).
The Venn diagram displayed the number of identical and differential miRNAs in four comparison pairs (Fig. 3). The results showed that 7 identical miRNAs (novel-mir23, novel-mir26, novel-mir30, novel-mir37, novel-mir46, miR395a and miR398a-3p) and 5 different miRNAs (novel-mir32, novel-mir38, novel-mir65, novel-mir78 and miR397a) were regulated by two rootstocks. Among the different miRNAs, novel-mir32 was solely identified in ‘Huangchenggen No. 2’-grafted cucumber, while novel-mir38, novel-mir65, novel-mir78 and miR397a were solely identified in ‘Yunnan figleaf gourd’-grafted cucumber. The expression of miR397a was regulated by both ‘Yunnan figleaf gourd’ and chilling stress, but the expression patterns were different. Six miRNAs (novel-mir26, novel-mir55, novel-mir67, novel-mir70, miR168a-5p and miR390a-5p) were regulated by silicon in the TS/TC comparison pair. Among them, novel-mir26 showed the same expression pattern in TY/TC and TH/TC comparison pairs. No miRNAs were identified as responsive to both low temperature and silicon in TC/CC and TS/TC comparison pairs.

Venn diagrams for analysis of miRNAs differentially expressed in TC/CC, TS/TC, TH/TC and TY/TC comparison pairs from cucumber seedlings
Target genes prediction and functional analysis of differentially expressed miRNAs
In our study, we identified 20 differentially expressed miRNAs in four comparison pairs, including 7 known miRNAs and 13 novel miRNAs. These miRNAs targeted a total of 520 genes, with the number of target genes per miRNA ranging from 0 to 120. Notably, miR157d had the highest number of target genes among all miRNAs (Additional file 4: Table S4).
These genes were subjected to GO (Gene Ontology) annotation classification analysis. In TC/CC, TS/TC, TH/TC and TY/TC comparison pairs, 140, 82, 219 and 257 target genes were assigned to GO terms, respectively. (Fig. 4). Notably, the ‘CCAAT-binding factor complex’ in Cell Component and ‘sulfate transporter activity’ in Molecular Function were commonly enriched in two grafted cucumbers. Some unique GO terms were identified in ‘Yunnan figleaf gourd’ compared to ‘Huangchenggen No. 2’, such as ‘preribosome’, ‘nucleolus’, ‘nuclear protein-containing complex’ and ‘ribonucleoprotein complex’ in Cell Component, and ‘ribosome biogenesis’ and ‘ribonucleoprotein complex biogenesis’ in Biological Process. The functional enrichment process of miRNAs regulated by silicon nutrition was significantly different from other treatments. In TS/TC comparison pair, the GO terms were mainly concentrated in ‘translation initiation factor activity’ in Molecular Function and ‘autophagy functions’ in Biological Process were significantly enriched.

Gene Ontology annotation analysis of genes targeted by miRNAs in four comparison pairs. The vertical axis represents enriched GO terms, the horizontal axis represents the enriched p-value of each term, and the numbers in bars indicate the number of genes enriched in each terms. Different colours are used to distinguish cellular components, biological processes and molecular functions
The target genes were then subjected to KEGG (Kyoto Encyclopedia of Genes and Genome) metabolic pathway analysis. The number of enriched pathways varied among the pairs, with the highest number being 36 in TY/TC comparison pair (Fig. 5). The pathways of TH/TC and TY/TC were found to be similar, such as ‘Stilbenoid, diarylheptanoid and gingerol biosynthesis’ and ‘Flavonoid biosynthesis’ were the common pathways in both types of grafted cucumber. In addition, ‘Flavonoid biosynthesis’ was identified as a common pathway in TC/CC, TH/TC and TY/TC pairs, indicating its potential fundamental role in the response to chilling stress in different types of grafted cucumber.

KEGG pathways of miRNAs target genes were significantly enriched in four comparison groups. The vertical axis represents the gene enrichment pathway, and the horizontal axis represents the rich factor. The size of the dot indicates the number of candidate target genes in the pathway, and the colour of the dot corresponds to different p-value ranges
qRT-PCR verification of differentially expressed miRNAs and their target genes
Three known miRNAs and three novel miRNAs were selected randomly for quantitative verification by qRT-PCR (Fig. 6). The results showed that the expression trends of the miRNAs detected by qRT-PCR were consistent with the sequencing results. For example, the expression of miR397a was down-regulated at low temperatures. Under chilling stress, ‘Yunnan figleaf gourd’ up-regulated its expression, while ‘Huangchenggen No. 2’ and silicon nutrition had no significant effect. The expression of novel-mir46 was inhibited by chilling stress and two types of rootstocks, but exogenous silicon up-regulated its expression. In addition, we quantified six target genes of differentially expressed miRNAs by qRT-PCR to explore the regulatory relationship between miRNAs and target genes (Fig. 7). The results showed that miRNAs regulated the expression of target genes through negative regulation. For example, low temperature down-regulated miR397a and up-regulated the expression of its target genes. The expression patterns of miRNAs identified by qRT-PCR and high-throughput sequencing were consistent, indicating the reliability of the sequencing results.

qRT-PCR expression profiling of six randomly selected miRNAs.The U6 snRNA was used as an internal control. Error bars represented the ± SD calculated from three replicates. Significant differences by Student`s t-test were indicated by different letters (p-value ≤ 0.05, n = 3)

qRT-PCR expression profiling of six randomly selected target genes. The actin gene was used as an internal control. Error bars represented the ± SD calculated from three replicates. Significant differences by Student`s t-test were indicated by different letters (p-value ≤ 0.05, n = 3)
Discussion
It is well known that plants can regulate the expression of genes related to chilling tolerance by mediating miRNAs in response to chilling injury [14,15,16]. The effects of rootstocks with different de-blooming capacities and exogenous silicon on the chilling tolerance of cucumber seedlings were significantly different (Table 1). In this study, we attempted to identify miRNAs that are regulated by two rootstocks and silicon nutrition under low temperatures, and to analyze their roles in regulating the chilling tolerance of cucumber seedlings.
miRNAs in response to chilling stress in cucumber seedlings
The study identified 6 miRNAs (miR157d, miR397a, miR398a-3p, miR408-3p, novel-mir21 and novel-mir46) that respond to chilling stress in cucumber seedlings. Some studies have shown that miR157d is a crucial miRNA related to cotton development and up-regulated miR157 can inhibit tissue cell proliferation and cell expansion [19]. In this study, chilling stress up-regulated miR157d, which was predicted to target various genes, including SQUAMOSA-promoter binding protein gene family (SP13B, SPL6, SPB1, SPL7 and SPL17), leucine-rich repeat receptor-like serine/threonine-protein kinase BAM1, cationic amino acid transporter 8 (CAT8), short-chain dehydrogenase TIC32 and serine/arginine-rich splicing factor RSZ2. BAM1 and SPL are essential transcription factors associated with floral organ development [20, 21]. CAT8 plays an essential role in maintaining intracellular amino acid homeostasis by regulating the amino acid content inside and outside the vacuole [22]. RSZ22 is homologous gene of human SRSF7 factor and may be involved in pre-mRNA splicing [23]. TIC32 is an integral part of chloroplast biogenesis [24]. These results indicated that cucumber seedlings might alter the expression of miR157d to control the growth of leaf cells in response to chilling stress.
miR397 can regulate the expression of laccase, POD and L-ascorbate oxidase, or directly target inducer of CBF expression 1 (ICE1) to regulate CBF genes, thereby participating in plant response to chilling stress [14, 25]. However, the expression patterns of miR397 in response to chilling stress vary among species. For example, the expression of miR397 decreased in wheat but increased in Arabidopsis under chilling stress [14, 15]. In this study, we observed that chilling stress suppressed the expression of miR397a in cucumber seedlings. LAC4, LAC11, LAC17 and aborted microspores (AMS) were predicted to be the primary target genes of miR397a. Some studies have shown that inhibiting the expression of LAC4 and LAC17 can significantly reduce the lignin content, resulting in a significant decrease in plant tolerance to chilling stress [26].
miR398 is a highly conserved miRNA that plays a crucial role in maintaining the balance of intracellular ROS content and mitigating stress damage in plants by regulating the expression of copper/zinc superoxide dismutase (CSD1 and CSD2) in plants [27, 28]. In this study, miR398a-3p was inhibited by chilling stress and was predicted to target CSD. Some studies have shown that CSD can enhance the ROS scavenging system and promote the plant response to chilling stress [29].
miR408 can affect plant tolerance to chilling stress by regulating ROS content, lignin synthesis and intracellular copper transport [30, 31]. Its expression pattern varies depending on the type of stress, such as salt stress, chilling stress and oxidative stress inducing its up-regulation, whereas drought and osmotic stress inhibit its expression [30]. In this study, the expression of miR408 was decreased after chilling stress, and miR408 was predicted to target Basic blue protein (BABL) and Copper-transporting ATPase PAA2. BABL, a structural protein of anthocyanins, positively affects plant tolerance to chilling stress [32]. Similarly, PAA2 plays an essential role in maintaining normal chloroplast function by transporting copper ions into the chloroplast [33].
In addition, novel-mir21 and novel-mir46 were predicted to respond to low temperatures. Novel-mir21 was predicted to target TCP2, TCP4, MYB33 and A20/AN1 zinc finger domain-containing stress-associated protein 1 (SAP1). The expression of SPA1 is induced by chilling stress, and the overexpression of SAP1 can improve the tolerance of tobacco to chilling stress [34]. TCP transcription factors can affect leaf development by regulating the cell cycle, and down-regulation of TCP2 expression leads to leaf deformation [35]. The novel-mir46 was predicted to target pectinesterase 3 (PME3). PME3 is capable of catalyzing the demethylation of pectin, resulting in the release of free carboxyl groups, methyl groups, and protons, which can improve cell wall stiffness and plant tolerance to chilling stress [36].
miRNAs associated with chilling tolerance affected by two rootstocks
Grafting is an important agronomic technique for improving the chilling tolerance of cucumbers. Compared with self-rooted cucumber, differential analysis of miRNAs in ‘Huangchenggen No. 2’-grafted cucumber and ‘Yunnan figleaf gorund’-grafted cucumber identified 8 and 11 different miRNAs, respectively (Fig. 2A). miR395a and miR398a-3p were identical miRNAs found in two types of grafted cucumber.
Previous studies have shown that miR395 plays a crucial role in the response of plants to salt and drought stress by regulating sulfate metabolism [37]. Recent studies have shown that miR395 expression can be altered in plants under chilling stress [16, 38]. In this study, rootstocks promoted the expression of miR395a in grafted cucumber seedlings under chilling stress. miR395a was predicted to target sulfate transport related proteins low affinity sulfate transporter 3 (SUT3), chloroplastic ATP sulfurylase 1 (APS1) and sulfate transporter 2.1 (SUT21) to participate in sulfate transport in plants [37]. In addition, mir395a was predicted to target gibberellin-regulatory protein 1 (GASA1), which is involved in the plant response to abiotic stress by regulating gibberellin signaling [39].
miR397 can influence plant tolerance to chilling stress by regulating CBF expression [40]. In addition, miR397 overexpression enhances the growth of banana plants while maintaining their salt stress resistance [41]. In this study, ‘Yunnan figleaf gourd’-grafted cucumber seedlings up-regulated miR397a expression, which was predicted to target several chilling stress-related genes, including LAC4, LAC11, LAC17 and Thaumatin-like protein 1 (TLP1). TLP1 is a high polyphenol oxidase (PPO) protein that can act as an antifreeze protein to improve chilling tolerance in plants [42,43,44]. Some studies have shown that miR398 is down-regulated by chilling stress, and it regulates intracellular ROS content by modulating CSD1 and CSD2 [45, 46]. In this study, rootstocks down-regulated the miR398a-3p expression, which was predicted to regulate ROS content by targeting superoxide dismutase 9 (Cu-Zn) [47].
In addition, we predicted 6 and 8 novel miRNAs from TH/TC and TY/TC comparison pairs, respectively. Among these novel miRNAs, there were 5 identical novel miRNAs (novel-mir23, novel-mir26, novel-mir30, novel-mir37, and novel-mir46) in two comparison pairs. Novel-mir23 was predicted to target deoxycytidine deaminase (DCTD), which can reduce the miscoding of intracellular genetic material by participating in the metabolism of intracellular cytosine nucleoside [48]. Novel-mir26 was predicted to target late embryogenesis abundant protein D-34 (LEA34) and cysteine-rich receptor-like protein kinase 10 (CRK10), both of which play a role in intracellular ROS scavenging, protein protection and ABA signaling [49,50,51]. Novel-mir30 was predicted to target cytoplasmic acetyl-CoA acetyltransferase 1 (AACT1) and Telomere repeat-binding protein 5 (TRP5), with the function of AACT1 remaining unclear and no significant difference in expression and metabolism between AACT1 mutants [52]. TRP5 specifically binds to plant double-stranded telomere repeats (TTTAGGG)n to reduce DNA damage and maintain normal cell development [53]. Novel-mir37 was predicted to target gibberellin acid-insensitive 1 (GAI1), a member of the DELLAs subfamily of the GRAS family and negatively regulates GA signaling. In addition, GAI1 can also bind to the ABF factor and is involved in plant ABA signal transduction in plants [54]. Novel-mir46 was up-regulated by two types of rootstocks and predicted to target PME3, which may be an important factor contributing to the promotion of cucumber seedling growth under chilling stress by rootstocks.
In response to chilling stress, novel-mir32 was up-regulated by ‘Huangchenggen No. 2’ and it was predicted to target PME3 and NAC. NACs regulate plant chilling tolerance by participating in the ICE-CBF signaling pathway [55]. The expressions of novel-mir38, novel-mir65 and novel-mir78 were regulated by ‘Yunnan figleaf gourd’. Among them, novel-mir38 was down-regulated and was predicted to target cysteine-rich receptor-like protein kinase 29 (CRK29). CRKs are involved in plant responses to various abiotic stresses by transmitting and sensing ROS/redox signals [56, 57]. Novel-mir65 was up-regulated by ‘Yunnan figleaf gourd’ and was predicted to target SUT3, SUT21 and APS1, which are all involved in sulfate transport in plants [58]. Novel-mir78 was down-regulated by ‘Yunnan figleaf gourd’ and was predicted to target putative pentatricopeptide repeat-containing protein and probable WRKY transcription factor 75 [59, 60].
miRNAs associated with chilling tolerance affected by silicon nutrition in seedlings
We identified 6 significantly differentially expressed miRNAs in response to silicon from TC/TS comparison pairs. These including 2 known miRNAs (miR168a-5p and miR390a-5p) and 4 novel miRNAs (novel-mir26, novel-mir55, novel-mir67, novel-mir70). miR168 is induced by abiotic stresses such as drought and low temperature, which indirectly regulates the function of other miRNAs by targeting argonaute1 (AGO1) [14, 61]. Up-regulation of miR168a has been reported to improve plant tolerance to drought stress, and the promoter region of miR168a contains ABA cis-acting elements that may be involved in the ABA signaling pathway [62]. In this study, silicon up-regulated miR168a-5p and was predicted to target AGO1. AGO1 associates with miRNAs to form an RNA-induced silencing complex (RISC), which is involved in target RNA cleavage and mediates post-transcriptional gene silencing [60].
miR390 plays a crucial role in regulating plant growth and abiotic stress response through the auxin signaling pathway by regulating the auxin response factor (ARF) [63, 64]. In this study, we observed that silicon increased the expression of miR390, which was predicted to target leucine-rich repeat receptor-like tyrosine-protein kinase PXC3 and E3 ubiquitin-protein ligase MARCH3. PXC3 is a member of the receptor-like protein kinase (RLK) subfamily, which binds to extracellular signaling molecules and activates intracellular kinase domains, thereby completing transmembrane signal transduction [65]. RLKs have been shown to be involved in plant responses to abiotic stresses, such as low temperature and drought, by regulating mitogen-activated protein kinase (MAPK) cascade, ABA and ROS signaling [66, 67]. MARCH3 is a crucial component of the ubiquitin-26S proteasome system (UPS) and plays an essential role in plant response to abiotic stress [68, 69]. For example, E3 ubiquitin-protein ligase is up-regulated by low temperature, which attenuates the damage caused by chilling stress in rice [70].
In addition, 4 novel miRNAs (novel-mir26, novel-mir55, novel-mir67 and novel-mir70) were predicted to respond to exogenous silicon. Notably, novel-mir26 was responsive to both silicon and two rootstocks and was predicted to target late embryogenesis abundant protein 34 (LEA34) and cysteine-rich receptor-like protein kinase 10 (CRK10). LEA34 and CRK10 may be involved in intracellular ROS scavenging, protein protection and ABA signaling [71]. Under chilling stress, silicon reduced the expression of novel-mir55 and novel-mir70, which were predicted to target expansin A23 (EXPA23). EXPA23 is a member of the expansin superfamily, which disrupts non-covalent bonds between cell wall polysaccharides and leads to stress-dependent cell expansion in response to various abiotic stresses [72, 73]. Overexpression of EXPA8-B and EXPA8-D has been shown to improve the chilling tolerance in Arabidopsis [74]. Under chilling stress, novel-mir70 was up-regulated by exogenous silicon and was predicted to target the transcription factor GTE12. Although twelve GTE proteins have been found in Arabidopsis thaliana, their functions are not well understood.
Conclusion
In this study, we used high-throughput sequencing technology to identify miRNAs that respond to low temperature, exogenous silicon and two rootstocks in cucumber seedlings. Our results showed that novel-mir46 and miR398a-3p were observed in TC/CC, TH/TC and TY/TC comparison pairs, indicating that these miRNAs respond not only to chilling stress but also to the rootstocks. However, we did not identify the same miRNAs in TC/CC and TS/TC comparison pairs. Furthermore, the differential expression of novel-mir38, novel-mir65, novel-mir78 and miR397a could be an important factor contributing to the differences in chilling tolerance of grafted cucumbers caused by two rootstocks. The identification of these miRNAs helps us to understand the differences in molecular mechanisms of chilling stress mitigation between rootstocks with different de-blooming capacities, which is essential for the subsequent research to improve the chilling tolerance of grafted cucumber.
Methods
Plant materials and treatments
The scion was ‘Xintaimici’ cucumber (Cucumis sativus L.), and the rootstocks were ‘Huangchenggen No. 2’ (Cucurbita moschata D.) with strong de-blooming capacity and ‘Yunnan figleaf gourd’ (Cucurbita ficifolia B.) with weak de-blooming capacity. The experiment was conducted from June 2021 to April 2022 in an artificial climate chamber at Shandong Agricultural University. Cucumber seeds and rootstock seeds were immersed in water at 30°C for 4 h and 6 h, respectively, and placed in an incubator (HZQ-F160a, Yiheng, China) to stimulate germination for 24 h and 48 h at 28 ± 1°C under humid (relative humidity between 85% and 100%) and dark conditions, respectively. The germinated seeds were sown into available seedling substrate plugs (peat: vermiculite: perlite = 1:1:1 (v:v:v)). When the rootstock seeds germinated, the scion seeds were sown into the plugs. When the cotyledon of the scion and the first true leaf of the stock had fully expanded, the seedlings were grafted by using ‘hole insertion’ method. To increase the survival rate of the grafted cucumber seedlings, the temperature was maintained at 28°C /24°C (day/night) and the relative humidity was kept between 85% and 100% for about 7 days. When the grafted seedlings survived, they were transferred to an environment with a temperature of 28°C/18°C, relative humidity of 75%~85% and light intensity of 400 µmol·m− 2·s− 1 for further cultivation. When the first true leaves of self-rooted cucumber seedlings and grafted cucumber seedlings spread out, 18 healthy seedlings with similar growth were selected and placed in 3 incubators, each containing 6 L of Yamazaki cucumber nutrient solution. The solution was replaced every 3 days.
The experiment consisted of five treatments: (1) CC: Self-rooted cucumber seedlings were grown in nutrient solution and maintained at 28 ± 1 °C/18 ± 1 °C. (2) TC: Self-rooted cucumber seedlings were grown in nutrient solution and maintained at 10 ± 1 °C/5 ± 1 °C. (3) TS: Self-rooted cucumber seedlings were grown in nutrient solution containing 0.5mmol·L− 1 Na2SiO3·9H2O and maintained at 10 ± 1 °C/5 ± 1 °C. (4) TH: ‘Huangchenggen No. 2’-grafted cucumber seedlings were grown in nutrient solution and maintained at 10 ± 1 °C/5 ± 1 °C. (5) TY: ‘Yunnan figleaf gourd’-grafted cucumber seedlings were cultured in nutrient solution and maintained at 10 ± 1 °C/5 ± 1 °C. TC served as the controlled experiment among the five treatments. After 12 and 24 h of treatment, respectively, 8 seedlings were randomly selected for sampling. The sampling methods were as follows: the third leaf was taken, the veins were removed, mixed well, and cut into pieces. Samples were frozen in liquid nitrogen and immediately stored at -80 °C.
Determination of growth index
The plants were exposed to the low temperature of 10 ± 1 °C/5 ± 1 °C (day/night) for 2 days, and 8 plants were randomly selected from each treatment group to measure the growth index of cucumber seedlings. The length of cucumber cotyledon to growing point was measured with a ruler as plant height, and the diameter of cucumber cotyledon node position was measured with a vernier caliper as stem diameter. After measuring the plant height and stem diameter, the seedlings were divided into roots and shoots with scissors, washed and blotted with deionized water, and the fresh weight of the branches was determined. The third functional leaf above the cotyledon of cucumber seedlings was taken. The leaf area was measured using LA-S root scanner (WSeen, China) and WinRHIZO image analysis software. The shoots were then placed in an oven and kept at 105 °C for 30 min, and then the temperature was lowered to 75 °C until they maintained a constant weight, and the dry weight of shoots was recorded.
Determination of chilling injury index
After 24 h of chilling stress, the chilling injury index of cucumber seedlings was evaluated according to the chilling injury index classification established in a previous report [75]. The chilling injury index was calculated as follows: Grade 0: No injury symptoms. Grade 1: The edge of the first leaf is yellow or slightly dehydrated. Grade 2: Dehydration spots appear on a small portion of the first leaf; other leaves are slightly dehydrated. Grade 3: Dehydration spots appear on the first half of the leaves; other leaves are slightly dehydrated. Grade 4: Desiccated spots appear on most of the leaf area; the other half of the leaves are dehydrated. Grade 5: Almost all leaves are severely dehydrated and wilted. Chilling injury index = Σ (plants of different grades × grade) / (total plants × maximum grade).
Small RNA libraries construction and sequencing
Total RNA was extracted from cucumber leaves of 8 plants in 3 replicates per treatment using Trizol reagent (Vazyme, Nanjing, China). The degree of RNA degradation and contamination was analyzed by agarose gel electrophoresis, and then the purity of RNA samples was detected by NanoDrop (D260 nm/D280 nm ≥ 1.8 × OD260, OD230 ≥ 1.0). The Qubit detection method was used to accurately quantify the concentration of RNA samples (total RNA concentration ≥ 250 ng/µL); Agilent 2100 was used to accurately detect the integrity of RNA. Finally, the sRNA library was produced by using samples with RNA integrity ≥ 8. Agilent 2100 and other methods were used to accurately detect the integrity of RNA. Finally, high-throughput sequencing was performed on the HiSeq2000 instrument (Illumina, USA), and each treatment was sequenced three times.
Raw data pre-processing
The raw sequencing data contained low-quality reads, which were filtered for subsequent analysis [76]. Data processing methods are as follows: (1) Remove the low-quality reads (the quality value sQ ≤ 20 accounts for more than 30% of the total reads). (2) Remove the reads with a proportion of N was greater than 10. ‘N’ indicates that the base cannot be determined. (3) Remove the reads with 5` primer contamination. (4) Remove the reads with poly A/T/G/C. (5) Remove the reads shorter than 18 nt. The clean reads with 18 nt to 30 nt were obtained, and the unique reads and total reads were calculated. Finally, the length distribution of clean reads in each library is calculated.
Identification of known miRNAs and novel miRNAs in cucumber leaves
The 18 nt to 30 nt length sRNAs were mapped to Cucumber (Chinese Long) v3 genome [77] by Bowtie (no mismatch), then their expression and distribution on reference sequence were analyzed. To remove tags from protein-coding genes, repetitive sequences, rRNA, tRNA, snRNA and snoRNA, small RNA tags were mapped to Repeat Masker (http://repeatmasker.org/cgi-bin/WEBRepeatMasker), Rfam database (http://rfam.janelia.org/) and GenBank database (http://ftp.ncbi.nlm.nih.gov/genbank/). The remaining unique sequences were matched to miRbase 22.1 database (https://www.mirbase.org/sea-rch.shtml) using BLAST to identify conservative and known miRNAs in cucumber (no mismatch). Perfect match between sRNA and precursor sequences. However, reads with at least 16 nt overlap consistent with mature miRNAs in miRBase were allowed to offset. Count the number of miRNAs matching the conditions and analyze the base deviations at each position of all identified miRNAs.
To predict novel miRNAs, we utilized miREvo software to align reads with the reference genome and predict miRNA precursors [78]. Briefly, we performed a 70 nt sliding window based on the positions of reads mapped to reference genome to determine the suitable location, from which we extracted the upstream 20 nt and downstream 70 nt to predict the secondary structure of the miRNA precursor. Next, we analyzed the predicted precursor sequences using the miRDeep2 software, which was accessed through the miREvo interface [79]. After obtaining the precursor sequences, we applied a series of selection criteria to filter out unsuitable candidates, including (1) no branched secondary structures upon folding; (2) categorization of precursor sequences into mature, loop, or star strands based on mapping results; (3) at least 60% of the mature strand sequence being covered by reads. Using the predicted mature regions of the precursor sequences, we identified novel sRNA sequences. We used miREvo_v1.1 with the following primary parameters: -i, -r, -M, -m, -k, -p 10, -g 50,000; and miRDeep2_0_0_5 with the following primary parameters: quantifier.pl, -p, -m, -r, -y, -g 0, -T 10.
Analysis of differentially expressed miRNA
The expression of miRNAs in the sample was counted, and the expression level was normalized using the TPM algorithm (actual miRNA count / total count of clean reads × 1,000,000). Then, we used the DESeq2 software package (3.0.3) to analyze the differential expression between two groups. The p-value ≤ 0.05 and | log2 (foldchange) | ≥ 1 were set as the threshold for screening differentially expressed genes, and the p-value was calculated according to Benjamini & Hochberg method. The results of four comparison pairs were obtained by difference analysis. The differentially expressed miRNAs in TC/CC comparison pair might be related to low-temperature response, and the differentially expressed miRNAs in TS/TC comparison pair might be related to the response to exogenous silicon under chilling stress. The differentially expressed miRNAs in TH/TC comparison pair might be related to the induction of the ‘Huangchenggen No. 2’ under chilling stress. The differentially expressed miRNAs in TY/TC comparison pair might be related to the induction of ‘Yunnan figleaf gourd’ under chilling stress.
Prediction and analysis of miRNAs target genes
The miRNA sequence was compared with the genomic EST sequence of cucumber, and TargetFinder predicted the target genes. To further understand the biological function of the target genes, we mapped all candidate genes to GO terms in the database (http://www.geneontology.org/) and performed quantitative statistics. Compared with the internal reference gene background, a hypergeometric test was used to find the significantly enriched GO terms in candidate target genes. The GO term with p-value ≤ 0.05 is defined as significantly enriched in candidate target genes. Similarly, all candidate genes were mapped to KEGG [80, 81] pathways in database and determined the significantly enriched KEGG metabolic pathways in target gene candidates were determined by hypergeometric test (p-value ≤ 0.05).
Identification of miRNAs and target gene expression by qRT-PCR
Eight plants were randomly selected from each treatment group, and the materials were mixed and subjected to qRT-PCR analysis three times for each treatment group. The differentially expressed miRNAs were verified by qRT-PCR using the miRNA 1st Strand cDNA Synthesis Kit (by STEM-Loop) detection Kit (Vazyme, Nanjing, China). The volume of each reaction was 20 µL, including 10 µL of 2 × miRNA Universal SYBR qPCR Master Mix, 0.4 µL of Specific Primer (10 µM), 0.4 µL of universal reverse primer (10 µM) and 2 µL of Template cDNA. The PCR program was as follows: The pre-denaturation was performed at 95 °C for 5 min, cycling was performed at 95 °C for 40 cycles (5 s per cycle), and the melting curve was generated at 60 °C for 30 s. Finally, the data were calculated using the 2−ΔΔCt method (U6 snRNA as the reference gene).
The expression level of mRNA was verified by qRT-PCR. The HiScript II One Step RT-PCR kit (Vazyme Biotech, Nanjing, China) was used to prepare the first strand cDNA according to the instructions. qRT-PCR was performed on the ABI 75,000 Real-Time PCR machine (Applied Biosystems, Foster City, CA, USA). The reaction volume of qPCR was 20 µL, including 10µL of SYBR Premix EX TaqII, 0.4 µL of forward primers (10 µM), 0.4 µL of reverse primers (10 µM), and Template cDNA of 2 uL. The PCR programme was as follows: The predenaturation was performed at 95 °C for 30 s, cycling was performed at 95 °C for 40 cycles (5 s per cycle), and the melting curve was generated at 60 °C for 30 s. Finally, the 2−ΔΔCt method was used to calculate the data (actin as the reference gene). Additional file 5: Table S5 lists the sequences of the primers.
Data Availability
The datasets generated and analyzed during the current study are available in the NCBI Sequence Read Archive (SRA) repository, at.
https://www.ncbi.nlm.nih.gov/bioproject/938179 and accession #: PRJNA938179.
References
Cabrera R, Saltveit M, Owens K. Cucumber Cultivars Differ in their response to. Chilling temperatures. Journal of the American Society for Horticultural Science American Society for Horticultural Science. 1992;117.
Xu Y, Guo SR, Li H, Sun H, Na L, Shu S, Sun J. Resistance of cucumber grafting. Rootstock pumpkin cultivars to chilling and salinity stresses. Korean Journal of Horticultural Science & Technology. 2017;35:220–231.
Lee JM, Bang HJ, Ham HS. Quality of cucumber fruit as affected by rootstock. Acta Horticulturae. 1999;483:117–123.
Mitani N, Yamaji N, Ago Y, Iwasaki K, Ma JF. Isolation and functional characterization of an influx silicon transporter in two pumpkin cultivars contrasting in silicon accumulation. Plant Journal, 2011, 66(2):231–240.
Li H, Wang F, Chen XJ, Shi K, Xia XJ, Considine MJ, Yu JQ, Zhou YH. The sub/supra-optimal temperature-induced inhibition of photosynthesis and oxidative damage in cucumber leaves are alleviated by grafting onto figleaf gourd/luffa rootstocks. Physiologia Plantarum. 2014;152(3):571–584.
Guan WJ, Haseman D, Nowaskie D. Rootstock evaluation for grafted cucumbers grown in high tunnels: yield and plant growth. Hortscience. 2020;55(6):914–919.
Li Y, Tian XM, Wei M, Shi QH, Yang FJ, Wang XF. Mechanisms of tolerance. Differences in cucumber seedlings grafted on rootstocks with different tolerance to low temperature and weak light stresses. Turkish Journal of Botany. 2015;39:606–614.
Haynes RJ. A contemporary overview of silicon availability in agricultural soils. Journal of Plant Nutrition and Soil Science. 2014;177(6):831–844.
Hu W, Su Y, Zhou J, Zhu H, Guo J, Huo H, Gong H. Foliar application of silicon. And selenium improves the growth, yield and quality characteristics of cucumber in field conditions. Scientia Horticulturae. 2022;294:110776.
Bu R, Xie J, Yu J, Liao W, Xiao X, Lv J, Wang C, Ye J, Calderón-Urrea A. Autotoxicity in cucumber (Cucumis sativus L.) seedlings is alleviated by silicon through an increase in the activity of antioxidant enzymes and by mitigating lipid peroxidation. Journal of Plant Biology. 2016;59(3):247–259.
Gou T, Chen X, Han R, Liu J, Zhu Y, Gong H. Silicon can improve seed germination. And ameliorate oxidative damage of bud seedlings in cucumber under salt stress. Acta Physiologiae Plantarum. 2020;42(1):12.
Budak H, Akpinar BA. Plant miRNAs: biogenesis, organization and origins. Funct. Integr Genomics. 2015;15(5):523–531.
Banerjee S, Sirohi A, Ansari AA, Gill SS. Role of small RNAs in abiotic stress responses in plants. Plant Gene. 2017;11:180–189.
Gupta OP, Meena NL, Sharma I, Sharma P. Differential regulation of microRNAs in. Response to osmotic, salt and cold stresses in wheat. Mol Biol Rep. 2014;41(7):4623–4629.
Sunkar R, Zhu JK. Novel and stress-regulated microRNAs and other small RNAs from Arabidopsis. Plant Cell. 2004;16(8):2001–2019.
Liu WH, Cheng CZ, Chen FL, Ni SS, Lin YL, Lai ZX. High-throughput sequencing of small RNAs revealed the diversified cold-responsive pathways during cold stress in the wild banana (Musa itinerans). BMC Plant Biol. 2018;18.
Fang CX, Zhang PL, Jian X, Chen WS, Lin HM, Li YZ, Lin WX. Overexpression of Lsi1 in cold-sensitive rice mediates transcriptional regulatory networks and enhances resistance to chilling stress. Plant Science. 2017;262:115–126.
Li Z, Khan MU, Yan X, Mu D, Xie YB, Waqas M, Wu X, Letuma P, Fang CX, Lin WX. Deciphering the Molecular Mechanisms of Chilling Tolerance in Lsi1-Overexpressing Rice. International Journal of Molecular Sciences. 2022;23(9)
Liu N, Tu L, Wang L, Hu H, Xu J, Zhang X. MicroRNA 157-targeted SPL genes regulate floral organ size and ovule production in cotton. BMC Plant Biol. 2017;17(1):7.
Xing S, Salinas M, Höhmann S, Berndtgen R, Huijser P. miR156-targeted and. Nontargeted SBP-box transcription factors act in concert to secure male fertility in Arabidopsis. Plant Cell. 2010;22(12):3935–3950.
DeYoung BJ, Bickle KL, Schrage KJ, Muskett P, Patel K, Clark SE. The CLAVATA1-related BAM1, BAM2 and BAM3 receptor kinase-like proteins are required for meristem function in Arabidopsis. Plant J. 2006;45(1):1–16.
Arun-Chinnappa KS, McCurdy DW. De novo assembly of a genome-wide transcriptome map of Vicia faba (L.) for transfer cell research. Front Plant Sci. 2015;6:217.
Tillemans V, Dispa L, Remacle C, Collinge M, Motte P. Functional distribution and dynamics of Arabidopsis SR splicing factors in living plant cells. The Plant Journal. 2005;41(4):567–582.
Hörmann F, Küchler M, Sveshnikov D, Oppermann U, Li Y, Soll J. Tic32, an essential component in Chloroplast Biogenesis*. Journal of Biological Chemistry. 2004;279(33): 34756–34762.
Shen Y, Zhang Z, Lin H, Liu H, Chen J, Peng H, Cao M, Rong T, Pan G. Cytoplasmic male sterility-regulated novel microRNAs from maize. Funct Integr Genomics. 2011;11(1):179–191.
Berthet S, Demont-Caulet N, Pollet B, Bidzinski P, Cézard L, Le Bris P, Borrega N, Hervé J, Blondet E, Balzergue S, et al. Disruption of LACCASE4 and 17 results in tissue-specific alterations to lignification of Arabidopsis thaliana stems. The Plant Cell. 2011;23(3):1124–1137.
Sunkar R, Kapoor A, Zhu JK. Posttranscriptional induction of two Cu/Zn superoxide dismutase genes in Arabidopsis is mediated by downregulation of miR398 and important for oxidative stress tolerance. Plant Cell. 2006;18(8):2051–2065.
Wang B, Sun YF, Song N, Wei JP, Wang XJ, Feng H, Yin ZY, Kang ZS. MicroRNAs involving in cold, wounding and salt stresses in Triticum aestivum L. Plant Physiol Biochem. 2014;80:90–96.
Leng X, Wang P, Zhu X, Li X, Zheng T, Shangguan L, Fang J. Ectopic expression of CSD1 and CSD2 targeting genes of miR398 in grapevine is associated with oxidative stress tolerance. Funct Integr Genomics. 2017;17(6):697–710.
Ma C, Burd S, Lers A. miR408 is involved in abiotic stress responses in Arabidopsis. Plant J. 2015;84(1):169–187.
Sun M, Yang J, Cai X, Shen Y, Cui N, Zhu Y, Jia B, Sun X. The opposite roles of OsmiR408 in cold and drought stress responses in Oryza sativa. Molecular Breeding. 2018;38(10):120.
Li SJ, Bai YC, Li CL, Yao HP, Chen H, Zhao HX, Wu Q. Anthocyanins accumulate in tartary buckwheat (Fagopyrum tataricum) sprout in response to cold stress. Acta Physiol Plant. 2015;37(8):159.
Tapken W, Kim J, Nishimura K, van Wijk KJ, Pilon M. The clp protease system is required for copper ion-dependent turnover of the PAA2/HMA8 copper transporter in chloroplasts. New Phytol. 2015;205(2):511–517.
Mukhopadhyay A, Vij S, Tyagi AK. Overexpression of a zinc-finger protein gene from rice confers tolerance to cold, dehydration, and salt stress in transgenic tobacco. Proceedings of the National Academy of Sciences. 2004;101(16):6309–6314.
Koyama T, Mitsuda N, Seki M, Shinozaki K, Ohme-Takagi M. TCP transcription factors regulate the activities of ASYMMETRIC LEAVES1 and miR164, as well as the auxin response, during differentiation of leaves in Arabidopsis. Plant Cell. 2010;22(11):3574–3588.
Solecka D, Żebrowski J, Kacperska A. Are pectins involved in Cold Acclimation and de-acclimation of Winter Oil-seed rape plants? Annals of Botany. 2008;101(4):521–530.
Frazier TP, Sun G, Burklew CE, Zhang B. Salt and Drought stresses induce the aberrant expression of microRNA genes in Tobacco. Molecular Biotechnology. 2011;49(2):159–165.
Jiang W, Shi W, Ma X, Zhao J, Wang S, Tan L, Sun C, Liu F. Identification of microRNAs responding to cold stress in Dongxiang common wild rice. Genome. 2019;62(9):635–642.
Martinelli F, Ibanez AM, Reagan RL, Davino S, Dandekar AM. Stress responses in citrus peel: comparative analysis of host responses to Huanglongbing disease and puffing disorder. Scientia Horticulturae. 2015;192:409–420.
Dong CH, Pei H. Over-expression of miR397 improves plant tolerance to cold stress in Arabidopsis thaliana. Journal of Plant Biology. 2014;57(4):209–217.
Patel P, Yadav K, Srivastava AK, Suprasanna P, Ganapathi TR. Overexpression of native Musa-miR397 enhances plant biomass without compromising abiotic stress tolerance in banana. Sci Rep. 2019;9(1):16434.
Ma Y, Luo M, Xu Y, Liu Y, Liu X, Bi X, Yuan Y, Su F, Yin X. Purification and characterization of a thaumatin-like protein-1 with polyphenol oxidase activity found in Prunus mume. RSC Adv. 2020;10(48):28746–28754.
Zhang M, Xu J, Liu G, Yang X. Antifungal properties of a thaumatin-like protein from watermelon. Acta Physiol Plant. 2018;40(11):186.
Gupta R, Deswal R. Low-temperature stress modulated secretome analysis and purification of antifreeze protein from Hippophae rhamnoides, a himalayan wonder plant. J Proteome Res. 2012;11(5):2684–2696.
Chen Y, Jiang J, Song A, Chen S, Shan H, Luo H, Gu C, Sun J, Zhu L, Fang W, et al. Ambient temperature enhanced freezing tolerance of Chrysanthemum dichrum CdICE1 Arabidopsis via miR398. BMC Biol. 2013;11:121.
Lu Q, Guo F, Xu Q, Cang J. LncRNA improves cold resistance of winter wheat by interacting with miR398. Functional Plant Biology. 2020;47(6):544.
Zhao H, Zhang R, Yan X, Fan K. Superoxide dismutase nanozymes: an emerging star for anti-oxidation. J Mater Chem B. 2021;9(35):6939–6957.
Xi Y, Shivarov V, Yaari G, Kleinstein S, Strout MP. DNA Demethylation by Activation-Induced Cytidine Deaminase in B Cell Lymphoma. Blood. 2014;124(21):3549–3549.
Tunnacliffe A, Hincha DK, Leprince O, Macherel D. LEA Proteins: versatility of form and function. In: Lubzens E, Cerda J, Clark M, editors. Dormancy and Resistance in Harsh environments. Berlin, Heidelberg: Springer Berlin Heidelberg; 2010. 91–108.
Tanaka H, Osakabe Y, Katsura S, Mizuno S, Maruyama K, Kusakabe K, Mizoi J, Shinozaki K, Yamaguchi-Shinozaki K. Abiotic stress-inducible receptor-like kinases negatively control ABA signaling in Arabidopsis. Plant J. 2012;70(4):599–613.
Mowla SB, Cuypers A, Driscoll SP, Kiddle G, Thomson J, Foyer CH, Theodoulou FL. Yeast complementation reveals a role for an Arabidopsis thaliana late embryogenesis abundant (LEA)-like protein in oxidative stress tolerance. The Plant Journal. 2006;48(5):743–756.
Ahumada I, Cairó A, Hemmerlin A, González V, Pateraki I, Bach TJ, Rodríguez-Concepción M, Campos N, Boronat A. Characterisation of the gene family encoding acetoacetyl-CoA thiolase in Arabidopsis. Funct Plant Biol. 2008;35(11):1100–1111.
Hong JP, Byun MY, Koo DH, An K, Bang JW, Chung IK, An G, Kim WT. Suppression of RICE TELOMERE BINDING PROTEIN 1 results in severe and gradual developmental defects accompanied by genome instability in rice. Plant Cell. 2007;19(6):1770–1781.
Wang Z, Liu L, Cheng C, Ren Z, Xu S, Li X. GAI functions in the plant response to dehydration stress in Arabidopsis thaliana. Int J Mol Sci. 2020;21(3).
Puranik S, Sahu PP, Srivastava PS, Prasad M. NAC proteins: regulation and role in stress tolerance. Trends Plant Sci. 2012;17(6):369–381.
Wrzaczek M, Brosché M, Salojärvi J, Kangasjärvi S, Idänheimo N, Mersmann S, Robatzek S, Karpiński S, Karpińska B, Kangasjärvi J. Transcriptional regulation of the CRK/DUF26 group of receptor-like protein kinases by ozone and plant hormones in Arabidopsis. BMC Plant Biol. 2010;10:95.
Bourdais G, Burdiak P, Gauthier A, Nitsch L, Salojärvi J, Rayapuram C, Idänheimo N, Hunter K, Kimura S, Merilo E et al. Large-scale Phenomics identifies primary and fine-tuning roles for CRKs in responses related to oxidative stress. PLoS Genet. 2015;11(7):e1005373.
Tang W, Zhao Y, Zeng J, Li Z, Fu Z, Yang M, Zeng D, Chen X, Lai Z, Wang-Pruski G, et al. Integration of small RNA and transcriptome sequencing reveal the roles of miR395 and ATP sulfurylase in developing seeds of chinese Kale. Front Plant Sci. 2021;12:778848.
Siira SJ, Spåhr H, Shearwood A-MJ, Ruzzenente B, Larsson N-G, Rackham O, Filipovska A. LRPPRC-mediated folding of the mitochondrial transcriptome. Nat Commun. 2017;8(1): 1532.
Chujo T, Ohira T, Sakaguchi Y, Goshima N, Nomura N, Nagao A, Suzuki T. LRPPRC/SLIRP suppresses PNPase-mediated mRNA decay and promotes polyadenylation in human mitochondria. Nucleic Acids Res. 2012;40(16):8033–8047.
Liu X, Tan C, Cheng X, Zhao X, Li T, Jiang J. miR168 targets Argonaute1A mediated miRNAs regulation pathways in response to potassium deficiency stress in tomato. BMC Plant Biol. 2020;20(1):477.
Li W, Cui X, Meng Z, Huang X, Xie Q, Wu H, Jin H, Zhang D, Liang W. Transcriptional regulation of Arabidopsis MIR168a and argonaute1 homeostasis in abscisic acid and abiotic stress responses. Plant Physiol. 2012;158(3):1279–1292.
Zhou B, Kang Y, Leng J, Xu Q. Genome-wide analysis of the miRNA-mRNAs network involved in Cold Tolerance in Populus simonii × P. nigra. Genes (Basel). 2019;10(6).
Wen FL, Yue Y, He TF, Gao XM, Zhou ZS, Long XH. Identification of miR390-TAS3-ARF pathway in response to salt stress in Helianthus tuberosus L. Gene. 2020;738:144460.
Shiu SH, Bleecker AB. Plant receptor-like kinase gene family: diversity, function, and signaling. Sci STKE. 2001;2001(113):re22.
Furuya T, Matsuoka D, Nanmori T. Phosphorylation of Arabidopsisthaliana MEKK1 via Ca2+ signaling as a part of the cold stress response. J Plant Res. 2013;126(6):833–840.
Jung CG, Hwang SG, Park YC, Park HM, Kim DS, Park DH, Jang CS. Molecular characterization of the cold- and heat-induced Arabidopsis PXL1 gene and its potential role in transduction pathways under temperature fluctuations. J Plant Physiol. 2015;176:138–146.
Stone SL. The role of ubiquitin and the 26S proteasome in plant abiotic stress signaling. Front Plant Sci. 2014;5:135.
Zhao J, Zhao L, Zhang M, Zafar SA, Fang J, Li M, Zhang W, Li X. Arabidopsis E3 Ubiquitin Ligases PUB22 and PUB23 negatively regulate Drought Tolerance by targeting ABA receptor PYL9 for degradation. Int J Mol Sci. 2017;18(9).
Min HJ, Jung YJ, Kang BG, Kim WT. CaPUB1, a Hot Pepper U-box E3 ubiquitin ligase, confers enhanced cold stress tolerance and decreased Drought stress tolerance in Transgenic Rice (Oryza sativa L.). Mol Cells. 2016;39(3):250–257.
Cho SK, Chung HS, Ryu MY, Park MJ, Lee MM, Bahk YY, Kim J, Pai HS, Kim WT. Heterologous expression and molecular and cellular characterization of CaPUB1 encoding a hot pepper U-Box E3 ubiquitin ligase homolog. Plant Physiol. 2006;142(4):1664–1682.
Zhang JF, Xu YQ, Dong JM, Peng LN, Feng X, Wang X, Li F, Miao Y, Yao SK, Zhao QQ, et al. Genome-wide identification of wheat (Triticum aestivum) expansins and expansin expression analysis in cold-tolerant and cold-sensitive wheat cultivars. PLoS One. 2018;13(3):e0195138.
İncili ÇY, Arslan B, Çelik ENY, Ulu F, Horuz E, Baloglu MC, Çağlıyan E, Burcu G, Bayarslan AU, Altunoglu YC. Comparative bioinformatics analysis and abiotic stress responses of expansin proteins in Cucurbitaceae members: watermelon and melon. Protoplasma. 2022.
Zhang Y, Li D, Zhang D, Zhao X, Cao X, Dong L, Liu J, Chen K, Zhang H, Gao C, et al. Analysis of the functions of TaGW2 homoeologs in wheat grain weight and protein content traits. Plant J. 2018;94(5):857–866.
Semeniuk P, Moline HE, Abbott JA. A comparison of the Effects of ABA and an antitranspirant on Chilling Injury of Coleus, Cucumbers, and Dieffenbachia. Journal of the American Society for Horticultural Science. 1986;111(6):866–868.
Abdellatef E, Kamal NM, Tsujimoto H. Tuning beforehand: a foresight on RNA interference (RNAi) and in Vitro-Derived dsRNAs to enhance crop resilience to biotic and abiotic stresses. Int J Mol Sci. 2021;22(14).
Huang S, Li R, Zhang Z, Li L, Gu X, Fan W, Lucas WJ, Wang X, Xie B, Ni P, et al. The genome of the cucumber, Cucumis sativus L. Nat Genet. 2009;41(12):1275–1281.
Wu HJ, Ma YK, Chen T, Wang M, Wang XJ. PsRobot: a web-based plant small RNA meta-analysis toolbox. Nucleic Acids Res. 2012;40(W1):W22-W28.
Friedländer MR, Mackowiak SD, Li N, Chen W, Rajewsky N. miRDeep2 accurately identifies known and hundreds of novel microRNA genes in seven animal clades. Nucleic Acids Res. 2012;40(1):37–52.
Kanehisa M, Goto S. KEGG: kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 2000;28(1):27–30.
Kanehisa M, Furumichi M, Sato Y, Kawashima M, Ishiguro-Watanabe M. KEGG for taxonomy-based analysis of pathways and genomes. Nucleic Acids Res. 2023;51(D1):D587-d592.
Acknowledgements
We thank Novogene (Beijing, China) for helping sequencing cucumber miRNAs. We also thank three anonymous reviewers for their valuable comments and suggestions for improving an early version of the manuscript.
Funding
This research was financially supported by the National Natural Science Foundation of China (31772362 and 32072654) and the China Agriculture Research System of MOF and MARA (CARS-23).
Author information
Authors and Affiliations
Contributions
QM and MW conceived and designed the study. CXN and CHC contributed new reagents and analytical tools. MQ and CW performed experiments and data analysis. QM wrote the manuscript. YL and MW revised the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
The materials used in this experiment were provided by the Qingdao Academy of Agricultural Sciences, China. This article did not contain any studies with animals or humans performed by any of the authors. This study complied with institutional, national and international guidelines and legislation.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Ma, Q., Niu, C., Wang, C. et al. Effects of differentially expressed microRNAs induced by rootstocks and silicon on improving chilling tolerance of cucumber seedlings (Cucumis sativus L.). BMC Genomics 24, 250 (2023). https://doi.org/10.1186/s12864-023-09337-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12864-023-09337-x