
Abstract
Background
Transgenic (Tg) mice are widely used in biomedical research, and they are typically generated by injecting transgenic DNA cassettes into pronuclei of one-cell stage zygotes. Such animals often show unreliable expression of the transgenic DNA, one of the major reasons for which is random insertion of the transgenes. We previously developed a method called “pronuclear injection-based targeted transgenesis” (PITT), in which DNA constructs are directed to insert at pre-designated genomic loci. PITT was achieved by pre-installing so called landing pad sequences (such as heterotypic LoxP sites or attP sites) to create seed mice and then injecting Cre recombinase or PhiC31 integrase mRNAs along with a compatible donor plasmid into zygotes derived from the seed mice. PITT and its subsequent version, improved PITT (i-PITT), overcome disadvantages of conventional Tg mice such as lack of consistent and reliable expression of the cassettes among different Tg mouse lines, and the PITT approach is superior in terms of cost and labor. One of the limitations of PITT, particularly using Cre-mRNA, is that the approach cannot be used for insertion of conditional expression cassettes using Cre-LoxP site-specific recombination. This is because the LoxP sites in the donor plasmids intended for achieving conditional expression of the transgene will interfere with the PITT recombination reaction with LoxP sites in the landing pad.
Results
To enable the i-PITT method to insert a conditional expression cassette, we modified the approach by simultaneously using PhiC31o and FLPo mRNAs. We demonstrate the strategy by creating a model containing a conditional expression cassette at the Rosa26 locus with an efficiency of 13.7%. We also demonstrate that inclusion of FLPo mRNA excludes the insertion of vector backbones in the founder mice.
Conclusions
Simultaneous use of PhiC31 and FLP in i-PITT approach allows insertion of donor plasmids containing Cre-loxP-based conditional expression cassettes.
Similar content being viewed by others


Introduction
Mice in which foreign DNA is inserted into the genome are called “transgenic (Tg) mice.” Since the development by Gordon et al. in 1980, many Tg mice have been produced by microinjection of DNA into fertilized eggs, and used for functional analysis of various genes and creation of disease mouse models [1]. Over the years, other methods have been developed to create Tg mice, including infection of early embryos with retroviral vectors and creation of chimeric mice from implanted modified embryonic stem (ES) cells. Although the microinjection method is quite simple, the genomic loci where the transgenes are inserted and their copy numbers are unpredictable [2]. Gene expression can be greatly affected by a position effect, by the state of chromatin at the insertion site, and by the regulatory sequences present in the flanking genomic sequences [3]. In addition, repeat-induced gene silencing may occur when genes with multiple copies are inserted in tandem, and thus there may not be a positive correlation between copy number and gene expression level [4]. Furthermore, some Tg DNA sequences may be subject to epigenetic effects such as DNA methylation [5]. Because reproducibility and stability of gene expression often cannot be obtained even within the same strain, it becomes necessary to analyze multiple founder lines of Tg mice to confirm that the phenotype is consistent.
This problem can be avoided by using ES cell-mediated gene targeting to insert a single copy transgene at a defined genomic locus (targeted transgenesis). However, the ES cell-mediated method is labor intensive, time-consuming, and expensive [2]. Since about a decade, CRISPR genome editing technology has been used to perform targeted transgenesis via microinjection technique. Even though CRISPR-based approaches are routinely used for generating conditional knockout- and short knock-in- models that require insertion of cassettes of about 1 to 2kb [6,7,8,9], these approaches are still inefficient for inserting cassettes of several kilobases long [6, 7, 10]. The DNA repair process often results in additional lesions such as short insertions or deletion (indel) mutations in addition to the target Tg allele [11,12,13], and the necessity of longer homology arms for plasmid-based inserts requires additional cloning steps. Other than these methods, targeted transgenesis methods using site-specific recombination and integrase systems derived from bacteriophages and microinjection have been developed by our group and several others [14,15,16,17,18,19].
We previously developed a modified transgenesis method called pronuclear injection-based targeted transgenesis (PITT). The first version of PITT relied on recombinase-mediated cassette exchange (RMCE) using the Cre-LoxP site-specific recombination system [14]. To generate targeted Tg mice using the PITT method, it is necessary first to generate a mouse strain with recombinase recognition sequences such as LoxP at a defined region of the genome. Although this prerequisite step is time-consuming and expensive, once a seed mouse line is established, there is no need to handle ES cells; many different types of targeted Tg mice can be generated using only direct microinjection of zygotes from the seed mice [2]. Because the seed mice will need to contain short landing pad sequences of only a couple hundred bases, such models can also be easily generated via a CRISPR approach that will obviate the need for any ES cell-based approaches [12, 20].
By using the PITT approach, we have generated a variety of Tg mice, including fluorescent gene-expressing mice [14, 21], tissue-specific gene-expressing mice [22, 23], and knockdown mice [14, 24], and have shown that transgene expression in these mice is highly reproducible and stable. Therefore, unlike Tg mice with randomly inserted transgenes, the mice generated by the PITT method have the advantage that multiple founder lines do not need to be generated, maintained, and analyzed.
Furthermore, we developed a seed mouse that allows use of multiple recombination systems, such as FLP-FRT and PhiC31 integrase, as well as Cre-LoxP. This modified PITT method was named improved PITT (i-PITT) and we demonstrated that simultaneous use of Cre-loxP-mediated recombination and PhiC31 integration significantly enhances the targeted insertion efficiency [15]. Although i-PITT system has a potential to insert a LoxP-flanked DNA cassette (commonly known as the ‘floxed cassette’) for the purpose of conditional expression, theoretically, this approach has yet to be demonstrated to generate conditional expression Tg mice. Specifically, transgene insertion by PITT using the Cre-LoxP system is not feasible for inserting a floxed cassette because the LoxP sites within the donor cassettes will be used up for Cre-mediated integration of the donor plasmid and therefore the recombined LoxP sites are unavailable for the conditional functionality of the transgene. To overcome this challenge, we devised an alternative strategy of PITT by using the PhiC31 and FLP-FRT systems.
Materials and methods
Mice
Inbred C57BL/6N and outbred MCH(ICR) mice were purchased from CLEA Japan Inc. (Tokyo, Japan). The seed mice (TOKMO-3) containing landing pads for targeted insertion of donor vectors were maintained as homozygotes with the inbred genetic background of C57BL/6N (Fig. 1A) [15]. NPHS2-CreERT2 mice (Tg(NPHS2-cre/ERT2)Mkas) [25] were mated with the Tg mouse conditionally expressing Maff (Condi-Maff; RBRC11275, generated in this study), to obtain Condi-Maff/NPHS2-CreERT2 mouse. The littermates containing only the Condi-Maff cassette were used as controls for assessing the conditional gene expression.

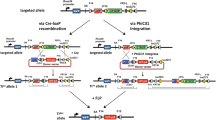
Schematic diagram of the insertion of a conditional expression cassette using the modified i-PITT method. Primers are shown in red (see Supplemental Table 1). The region shown as “STOP” consists of three SV40-pA sequences. F14, F15, FRT-L, FRT-R: mutant FRT sites. SA: splice acceptor. WPRE: woodchuck hepatitis virus posttranscriptional regulatory element. Lox2272: mutant LoxP. bGH-pA: bovine growth hormone polyadenylationsignal sequence. SV40-pA: Simian virus 40 polyadenylation signal sequence. GOI: gene of interest.CAG: hybrid construct consisting of the cytomegalovirus enhancer fused to the chicken beta-actin promoter. attB, attP, attR, attL: attachment sites for PhiC31 integrase. GTNOP: a cassette containing “eGFP-T2A-Neomycin resistant gene-hOCT4-PolyA” [15]
Mice were fed ad libitum under a 12:12 light and dark cycle, under the condition in specific pathogen-free (SPF). The animals were monitored daily, supplied with food. Non-transgenic mice were euthanized by cervical dislocation under anesthesia according to the Guidelines for the Care and Use of Animals for Scientific Purposes at Tokai University.
Plasmid construction
Using commercial gene synthesis services (GENEWIZ) and conventional restriction enzyme-based cloning steps, pBIE, pBIK, and pBIM, plasmids containing a conditional expression cassette with attB, mutant FRT, CAG promoter, STOP cassette (three polyA addition sites flanked by LoxP sequences), WPRE, and polyA sequences were constructed (Supplementary Fig. 1). The three polyA sequences in the STOP cassette region are derived from the sequence of Ai65 plasmid (Addgene #61,577) [26]. Genes of interest (GOI) were inserted into restriction enzyme sites of these vectors to generate donor vectors 1 to 11 (DV1 to 11) listed in Table 1. For example, Maff cDNA was inserted into the pBIE vector to make plasmid DV4, which was used to generate a Tg mouse with conditional expression of the Maff. We used a donor vector without WPRE sequences in Project 11 (Table 1). The pBER donor vector containing a promoter-less tdTomato-polyA cassette was used to determine optimal concentration of FLPo mRNA [15]. With this system, tdTomato transgene is expressed from endogenous Rosa26 promoter only when the pBER is inserted into the genome via PhiC31 integrase and/or FLP-FRT system.
Preparation of mRNA
PhiC31o mRNA was used as previously reported [15]. The pBBJ plasmid (Addgene #62,672) used for generating FLPo mRNA was linearized using XbaI digestion, and FLPo mRNA was transcribed in vitro using mMESSAGE mMACHINE T7 Ultra Kit (Ambion) followed by purification of the mRNA using MEGAclear Kit (Ambion). mRNA was filtered by passing through an Ultrafree-MC filter (HV; 0.45 μm pore size; #UFC30HV00; Millipore) before mixing it with the donor plasmids [15].
Microinjection
In the experiments to generate Tg mouse strains, donor vector DNA(s) (5–10 ng/µl in total), PhiC31o mRNA (7.5 ng/µl) and FLPo mRNA (11.3 ng/µl) were mixed together in EmbryoMax Injection Buffer (#MR-095–10 F; Millipore). The DNA/mRNA mixtures were stored at − 80 °C until use. For removal of the donor vector backbone in founder mice, FLPo mRNA solution was prepared at a concentration of 15 ng/µl in EmbryoMax Injection Buffer. To determine optimal concentration of FLPo mRNA, the concentrations of the donor vector pBER (10 ng/µl) and PhiC31o mRNA (7.5 ng/µl) were kept constant, while the FLPo mRNA concentration was tested from 0 to 33.8 ng/µl (Fig. 2).

Fluorescence-based evaluation of targeted integration of donor vector into the Rosa26 locus to evaluate optimal concentrations of FLPo mRNA. (A) Schematic diagram of the insertion of donor vector using the modified i-PITT method. The pBER donor vector DNA (containing a tdTomato-polyA cassette) and PhiC31o mRNA were used at 10 ng/ul and 7.5 ng/ul concentrations, respectively, along with different concentrations of FLPo mRNA (indicated above each image). Injections were performed at the zygote stage and red fluorescence was recorded at the blastocyst stage. (B) Fluorescence at the blastocyst stage. The numbers shown in the lower right corner of each photo are “number of zygotes that survived just after injection / number of embryos that developed to the blastocyst stage / number of normally developed eggs showing red fluorescence.” Scale bar: 100µm
Unfertilized oocytes isolated from superovulated female mice (C57BL/6N) were subjected to in vitro fertilization (IVF) with spermatozoa obtained from a homozygous TOKMO-3 male mouse. Microinjection of the DNA/mRNA mixture was performed into both the pronuclei and cytoplasm of in vitro fertilized eggs. The injected embryos were cultured until the blastocyst stage to assess insertion efficiencies (by observing red fluorescence that originates from the inserted tdTomato) or transferred into the uteri of pseudopregnant MCH(ICR) females to allow for their development. Offspring were genotyped to assess successful targeted transgenesis. Injection of FLPo mRNA (15 ng/µl) into the cytoplasm of in vitro fertilized eggs derived from founder mice or the offspring was performed to eliminate the vector portion from i-PITT mice to obtain the conditionalΔex (floxedΔex) allele (without extra sequence) (Fig. 1).
Detection of transgenes
Correct insertion of donor vectors into the Rosa26 locus, the site of pre-installed landing pad, was assessed by observing tissue samples under a fluorescence microscope and/or PCR-based genotyping of genomic DNA samples as described in Ohtsuka et al. 2015 [15]. For PCR detection of the transgene insertion in newborns, genomic DNA was isolated from the tail or ear using 40 to 50 µl of Allele-In-One Mouse Tail Direct Lysis Buffer (#ABP-PP-MT01500; Allele Biotechnology). PCR was performed in a total of 10 µl solution containing 2 x GC buffer I, 1 µl of the crude lysate, and the primer pair using TaKaRa Taq. For all experiments except 2 and 11, three primer sets viz. M273/M124 or M880/M026 or PP232/M274 were used for detection of targeted insertion allele, or targeted insertion allele with vector backbone, or detection of targeted insertion allele without vector backbone, respectively (Figs. 1 and 3A). For Projects 2 and 11, the GOI-specific and M1160 primers were used instead of PP232 primer (Supplementary Table 1). Nucleotide sequences of the junction were confirmed by sequencing.
Conditional expression of maff transgene
To achieve podocyte-specific transgene expression of Maff, Tg mice with a conditional expression cassette for Maff (Condi-Maff; RBRC11275) were mated with NPHS2-CreERT2 mice (Tg(NPHS2-cre/ERT2)Mkas) [25]. Intraperitoneal injections of 75 mg/kg tamoxifen for five consecutive days were performed into the resultant Condi-Maff/NPHS2-CreERT2 mouse and littermate controls (Condi-Maff cassette alone) at 33-weeks of age. Kidney specimens were prepared nine weeks after the injections.
Conditional Maff transgene expression was detected by immunohistochemistry. Kidney tissues obtained from Condi-Maff Tg mice were embedded in OCT compounds, and 6 μm frozen sections were prepared. Sections were fixed in 4% paraformaldehyde for 10 min and incubated in PBS containing 0.1% Triton X-100. The following antibodies were used: rabbit anti-Maff (1;100, Protein-tech, 12771-1-AP) and goat anti-Nephrin (1:100, R&D, AF3159).
Results
Development of a system to generate tg mice for conditional gene expression
Transgene insertion using PhiC31-mediated integration alone does not allow subsequent removal of the extra vector backbone, and the backbone has prokaryote-derived sequences that can hinder reliable gene expression [14]. To solve these issues, we used a combination of the PhiC31 and FLP-FRT system, which could increase the insertion efficiency, and at the same time it allowed vector backbone deletion (Fig. 2A) [15]. The optimal concentration of PhiC31o integrase mRNA was previously standardized [15], and thus we examined the optimal concentration of only FLPo mRNA. The FLPo mRNA was set to final concentrations ranging from 0 to 34 ng/µl and mixed with the pBER donor vector carrying the red fluorescent gene tdTomato and 7.5ng/ul of PhiC31o integrase mRNA. The mixture was microinjected into the pronucleus and cytoplasm of fertilized eggs derived from TOKMO-3 mice [15]. After culturing embryos to the blastocyst stage, the success of donor insertion was examined by observing red fluorescence. In the first set of experiments, the insertion efficiency using PhiC31-mediated integration alone was low. For instance, of the 24 zygotes that survived just after injection 17 developed to blastocyst stage and only one of these showed red fluorescence, indicative of correct insertion) (Fig. 2B, Supplementary Table 2). In contrast, FLPo mRNA injection together with PhiC31o mRNA generated embryos showing red fluorescence when 8.4 ng/µl and 16.9 ng/µl FLPo mRNA were used (generated 5 and 3 red fluorescent blastocysts, respectively) (Fig. 2B, Supplementary Table 2). This suggest that the use of FLPo mRNA in combination with the PhiC31 system improves insertion efficiency compared to the experiments that used only PhiC31 integrase (1/24). Based on this result, we decided to use 11.3 ng/µl of FLPo mRNA, which is approximately in the midpoint of the two concentrations (8.4 ng/µl and 16.9 ng/µl), for all subsequent experiments, to generate live offspring.
The above experiment was repeated four more times to test if co-injection of PhiC31o and FLPo mRNAs produce consistent results. Even though we did not see statistically significant differences between PhiC31o alone or combination of PhiC31o and FLPo the insertion efficiency seems to be slightly higher when both mRNAs were injected (8/73 [11.0%, 0.0-23.5% in each experiment] in 33.8 ng/µl, 6/82 [7.3%, 0.0-18.8% in each experiment] in 16.9 ng/µl, 9/85 [10.6%, 0.0-23.8% in each experiment] in 8.4 ng/µl, 14/95 [14.7%, 0.0-23.8% in each experiment] in 4.2 ng/µl) than when only PhiC31 integrase was used (7/87 [8.0%, 4.5–12.5% in each experiment]) (Supplementary Table 2).

Examples of PCR genotyping analyses. Genotypes of each offspring (IDs indicated by numbers) were verified using three different primer sets (A). (B) A representative example of genotyping of offspring from Project 9 (Table 1). The PCR-positive offspring are indicated in red. (C) Example genotyping of pups obtained by injecting FLPo mRNA into fertilized eggs derived from offspring #56 with extra vector sequence obtained in Project 6 (Table 2). The PCR-positive offspring are indicated in red. N: Negative control. Full sized images and other raw data files relevant to this figure are included in Supplementary Information Figs. 2A-C and 3, and supplementary information files AT1054.TIF, AT1058.TIF and AT1061.TIF and AT0446.TIF
Next, we designed and constructed several donor vectors for conditional gene expression (Supplementary Fig. 1). These cassettes enable conditional gene expression via the Cre-LoxP system. A target gene, downstream of a stop sequence (3 x polyA) placed between two LoxP sites, would be expressed after deletion of the stop sequence by the Cre-LoxP site-specific recombination. We generated a plasmid vector named pBIE (with attB and a pair of FRT sequences) that can insert the target DNA cassette (CAG promoter– LoxP– 3 x polyA– LoxP– WPRE– polyA) using the PhiC31 and FLP-FRT systems described above. Insertion cassettes included reporter genes such as eGFP and mCherry (pBIK and pBIM, respectively) as well as various GOIs.
Insertion of conditional expression cassettes by the modified i-PITT method
Various vectors containing expression cassettes ranging from 3.3 to 9.7 kb (overall vector size from 5.6 to 12.0 kb) were mixed to a final concentration of 5–10 ng/µl, along with 7.5 ng/µl of PhiC31o integrase mRNA and 11.3 ng/µl of FLPo mRNA, and microinjected the solution into pronucleus and cytoplasm of fertilized eggs obtained from TOKMO-3 mice. Genotyping of pups obtained in a total of 11 projects revealed that founder mice with expression cassettes inserted at the Rosa26 locus were obtained for all projects (Fig. 3A and B, Supplementary Fig. 2). The overall insertion efficiency was 13.7% (53/388) per live-born pups, and 2.2% (53/2440) per injected eggs (Table 1). Among the 53 pups that contained the targeted transgene, 24 (45.3%) had an insertion allele without vector backbone (termed as conditionalΔex allele) by FLP-FRT recombination. Four of the 24 pups (with conditionalΔex allele) also had conditionalex allele containing vector backbone sequence, indicating that these mice were mosaic for both alleles.
We previously demonstrated that it is possible to obtain up to three different Tg mouse lines in one injection session by injecting several donor vectors simultaneously in the i-PITT method [15]. Two of our 11 different i-PITT experiments contained more than one donor vector (Table 1). Two out of the four pups obtained in project 1 (injected with DV1 and DV2 plasmids) had a cassette insertion of DV1, of which one of the mice also had a DV2 cassette at the same time. This indicates that one of the cassettes may have been inserted at a random genomic location or that the two cassettes were combined by intermolecular recombination via mutant FRT sequences and then inserted into the Rosa26 locus by the PITT method. The other two pups had only the DV2 cassette inserted. Because the offspring with the DV1 cassette did not reproduce, another injection experiment with only DV1 was performed to obtain the desired Tg mouse line (Project 8). For the Project 3, only one offspring with a cassette of DV4 was obtained, and there were no offspring with DV5. Therefore, another injection experiment with only DV5 was performed to obtain the target Tg (Project 5). From these results, we conclude that although it may be possible to obtain multiple types of Tg founders in one injection session by mixing multiple vectors, performing individual injections to obtain multiple Tg lines may be more practical for PhiC31o and FLPo mRNA recombination.
Example of incorrect junctional sequence of the donor constructs
Among the 53 animals that contained the desired targeted insertion of donor plasmid, 51 had accurate recombination (96.2%), and only two mice had minor inaccuracies (3.8%) in their 5’ junctions (Fig. 4A). In the offspring #720 from Project 4, the 5’ end of the attR sequence was missing from the 3’ region of the splice acceptor sequence, and a portion of the attB sequence was inserted between them (Fig. 4B). In the offspring #65 from Project 7, the “F14-attR-F14” sequence in the conditionalex allele (floxedex allele) was duplicated (Fig. 4A). Nevertheless, we could generate the correct conditionalΔex allele (floxedΔex allele) by repairing the inaccurate allele, such as #65 after FLP recombination. These results indicate that i-PITT would be a better approach for creating targeted transgenic mice because it is more accurate. Further, minor inaccuracies in recombination could be repaired using FLP recombination to generate the correct conditionalΔex allele.

Example of incorrect junction sequences obtained after targeted integration. (A) The expected 5’ junctional architecture in the conditionalex allele (floxedex allele) is shown as “correct”. Diagramed below are the incorrect 5’ junctional sequences identified in #720 (from Project 4) and #65 (from Project 7). (B) ClustalW alignment of expected (shown as “correct”) and incorrect (shown as “#720”) sequences. The region from splice acceptor (SA) to F14 are shown
Efficiency of removing extra vector sequences
It is not uncommon that vector sequences also get inserted with the i-PITT method. Among the 53 animals generated in this work that contained the insert, 29 (55%) contained vector sequences (Table 1; Fig. 3B). The architecture of the donor plasmids allows removal of vector sequences via FLP recombination, which can be achieved in one of three ways: (1) by including FLPo mRNA in the injection solution, (2) by introducing FLPo mRNA via injection into the cytoplasm of fertilized zygotes obtained from the founder Tg mice (to remove the vector backbone in F1 offspring), or (3) by breeding the founder mice with FLP Tg mice [27]. The first approach saves time and resources. We showed that vector sequences can be excluded in 84% (65/77) of offspring using the second approach (Table 2; Fig. 3C, Supplementary Fig. 3).
Conditional expression of transgene
The main goal of this work was to enable insertion of constructs containing floxed cassettes using the PITT method. Given our experience with improved PITT (i-PITT) that the combination of integrases (PhiC31) and recombinases (Cre) significantly enhances efficiency [15], we reasoned that excluding Cre and adding FLP instead in i-PITT approach should achieve insertion of floxed cassettes. We successfully achieved this by developing Tg mice containing a floxed allele, targeted to the Rosa26 locus. Conditional expression of GOIs was confirmed by mating the Condi-Maff Tg mouse line with NPHS2-CreERT2 which enables podocyte-specific CreERT2 expression. In the kidney of double Tg mice (Condi-Maff/NPHS2-CreERT2), Maff protein was expressed in podocytes after administration of tamoxifen. This confirms that the Tg mouse generated in this study elicit Cre-mediated conditional expression function as intended (Fig. 5).

Podocyte-specific expression of Maff protein in Condi-Maff mouse. Representative immunofluorescence micrographs of kidney sections from Condi-Maff mice (without [left] or with [right] NPHS2-CreERT2 transgene) after administration of tamoxifen, stained with antibodies against Maff (green) and Nephrin (red). Scale bars = 50 μm
Discussion
This study represents further development of targeted transgenesis technology in mice to promote reproducible gene expression using site-specific recombination and integrase systems [14, 15]. We previously demonstrated that simultaneous use of multiple recombination systems (for example, Cre and PhiC31) can improve insertion efficiency [15]. Transgenic animals containing Cre-LoxP-based conditional gene expression cassettes are widely used in biomedical research. However, because Cre cannot be used for insertion of the LoxP-containing cassettes, we used the combination of PhiC31 and FLP to insert the construct containing LoxP sequences. The combination of these recombinant systems had an average insertion efficiency of 13.7% (a range of 6.9–18.0% among 11 different projects), compared to 10 to 30% (up to 62%) when Cre and PhiC31 were used together [15]. Among the 53 Tg mice generated, 51 (96.2%) were correctly recombined and only 2 (3.8%) were incorrectly recombined. Inaccurate recombination events occur in almost all genetic engineering methods including the CRISPR-based approaches [29,28,30]. Considering that insertions through inaccurate recombination occur more commonly using CRISPR approaches, i-PITT approach offers as better approach. As a general practice, it is necessary to confirm the accuracy of the insert by sequencing the junctions and confirm that the GOI expresses as expected.
Introduction of FLP in the i-PITT microinjection step enabled the removal of the extra sequence of the plasmid donor that gets inserted into the mouse genome. In fact, the extra sequence was successfully removed in half of the targeted founder mice. In addition, we could easily get rid of the extra sequence (if it was remained in some founder mice) by reinjection of FLPo mRNA. Co-injection of FLPo in i-PITT step will have the advantage of not only saving the time and effort of injecting FLPo to remove the extra sequence, but it also allows the creation of a strain (F1) by mating with FLPe Tg mice [27].
CRISPR-based approaches, which are widely adapted, use different types of donor DNAs such as ssDNA [31], plasmid DNA [32], or AAV vectors [33]. Although efficiency of CRISPR-based approaches, particularly using ssDNA donors, are generally high, inaccurate insertions such as missing fragments or duplication of some segments are more frequent when the size of the insert is longer than a few kilobases [34]. When plasmid DNA is used in CRISPR approaches, it is known to insert relatively accurately, but the efficiency is often lower than or comparable with that of the i-PITT method [35]. In addition, some vector backbones also get inserted using the CRISPR approach [36]. The homology arms used are often long in CRISPR-based approaches, which makes construction of plasmid donors time-consuming and in some cases makes it challenging to genotype [35]. Also, some loci are generally hard to amplify (for example Rosa26 which is GC rich). Recently, a knock-in method using AAV vectors called CRISPR-READI has been reported to be very efficient [33]. However, the packaging limit of AAV vectors is only up to about 5 kb including homology arms. On the other hand, up to 15 kb sequences can be inserted using i-PITT. In addition, the insertion-junctions are invariably accurate, and the insertion fragments are almost always fully intact. The fact that all but one of the inserts in this experiment were larger than 4 kb suggests that these Tg cannot be produced by knock-in using AAV.
Data availability
All data generated and analyzed in this work are available in this published article and Supplementary data. Plasmids constructed in this study are available from Addgene (www.addgene.org) or the corresponding author on reasonable request. The Condi-Maff mice (C57BL/6-Gt(ROSA)26Sor < tm2(CAG-Egr1)Motoj>) produced and analyzed in this study are available from RIKEN BRC (RBRC11275).
References
Gordon JW, Scangos GA, Plotkin DJ, Barbosa JA, Ruddle FH. Genetic transformation of mouse embryos by microinjection of purified DNA. Proc Natl Acad Sci U S A. 1980;77:7380–4.
Ohtsuka M, Miura H, Sato M, Kimura M, Inoko H, Gurumurthy CB. PITT: pronuclear injection-based targeted transgenesis, a Reliable transgene expression method in mice. Exp Anim. 2012;61:489–502.
Allshire RC, Javerzat J-P, Redhead NJ, Cranston G. Position effect variegation at fission yeast centromeres. Cell. 1994;76:157–69.
Garrick D, Fiering S, Martin DIK, Whitelaw E. Repeat-induced gene silencing in mammals. Nat Genet. 1998;18:56–9.
Ashe A, Morgan DK, Whitelaw NC, Bruxner TJ, Vickaryous NK, Cox LL, et al. A genome-wide screen for modifiers of transgene variegation identifies genes with critical roles in development. Genome Biol. 2008;9:R182.
Miura H, Quadros RM, Gurumurthy CB, Ohtsuka M. Easi-CRISPR for creating knock-in and conditional knockout mouse models using long ssDNA donors. Nat Protoc. 2018;13:195–215.
Ohtsuka M, Sato M, Miura H, Takabayashi S, Matsuyama M, Koyano T, et al. i-GONAD: a robust method for in situ germline genome engineering using CRISPR nucleases. Genome Biol. 2018;19:25.
Gurumurthy CB, Sato M, Nakamura A, Inui M, Kawano N, Islam MA, et al. Creation of CRISPR-based germline-genome-engineered mice without ex vivo handling of zygotes by i-GONAD. Nat Protoc. 2019;14:2452–82.
Quadros RM, Miura H, Harms DW, Akatsuka H, Sato T, Aida T, et al. Easi-CRISPR: a robust method for one-step generation of mice carrying conditional and insertion alleles using long ssDNA donors and CRISPR ribonucleoproteins. Genome Biol. 2017;18:92.
Gurumurthy CB, Kent Lloyd KC. Generating mouse models for biomedical research: Technological advances. Dis Model Mech. 2019;12:dmm029462.
Shin HY, Wang C, Lee HK, Yoo KH, Zeng X, Kuhns T, et al. CRISPR/Cas9 targeting events cause complex deletions and insertions at 17 sites in the mouse genome. Nat Commun. 2017;8:1–10.
Quadros RM, Harms DW, Ohtsuka M, Gurumurthy CB. Insertion of sequences at the original provirus integration site of mouse ROSA26 locus using the CRISPR/Cas9 system. FEBS Open Bio. 2015;5:191–7.
Boroviak K, Fu B, Yang F, Doe B, Bradley A. Revealing hidden complexities of genomic rearrangements generated with Cas9. Sci Rep. 2017;7:1–8.
Ohtsuka M, Ogiwara S, Miura H, Mizutani A, Warita T, Sato M, et al. Pronuclear injection-based mouse targeted transgenesis for reproducible and highly efficient transgene expression. Nucleic Acids Res. 2010;38:e198.
Ohtsuka M, Miura H, Mochida K, Hirose M, Hasegawa A, Ogura A, et al. One-step generation of multiple transgenic mouse lines using an improved pronuclear injection-based targeted transgenesis (i-PITT). BMC Genomics. 2015;16:274.
Ohtsuka M, Miura H, Nakaoka H, Kimura M, Sato M, Inoko H. Targeted transgenesis through pronuclear injection of improved vectors into in vitro fertilized eggs. Transgenic Res. 2012;21:225–6.
Ohtsuka M, Miura H, Hayashi H, Nakaoka H, Kimura M, Sato M, et al. Improvement of pronuclear injection-based targeted transgenesis (PITT) by iCre mRNA-mediated site-specific recombination. Transgenic Res. 2013;22:873–5.
Tasic B, Hippenmeyer S, Wang C, Gamboa M, Zong H, Chen-Tsai Y, et al. Site-specific integrase-mediated transgenesis in mice via pronuclear injection. Proc Natl Acad Sci U S A. 2011;108:7902–7.
Low BE, Hosur V, Lesbirel S, Wiles MV. Efficient targeted transgenesis of large donor DNA into multiple mouse genetic backgrounds using bacteriophage Bxb1 integrase. Sci Rep. 2022;12:5424.
Harms DW, Quadros RM, Seruggia D, Ohtsuka M, Takahashi G, Montoliu L, et al. Mouse genome editing using the CRISPR/Cas system. Curr Protoc Hum Genet. 2014;83:15.7.1–27. https://doi.org/10.1002/0471142905.hg1507s83.
Ohtsuka M, Miura H, Gurumurthy CB, Kimura M, Inoko H, Yoshimura S, et al. Fluorescent transgenic mice suitable for multi-color aggregation chimera studies. Cell Tissue Res. 2012;350:251–60.
Tamura S, Yasuoka Y, Miura H, Takahashi G, Sato M, Ohtsuka M. Thy1 promoter activity in the rosa26 locus in mice: lessons from dre-rox conditional expression system. Exp Anim. 2020;69:287–94.
Tsuchida J, Matsusaka T, Ohtsuka M, Miura H, Okuno Y, Asanuma K, et al. Establishment of nephrin reporter mice and use for chemical screening. PLoS ONE. 2016;11:e0157497.
Miura H, Inoko H, Tanaka M, Nakaoka H, Kimura M, Gurumurthy CB, et al. Assessment of artificial miRNA architectures for higher knockdown efficiencies without the undesired effects in mice. PLoS ONE. 2015;10:e0135919.
Yokoi H, Kasahar M, Mukoyama M, Mori K, Kuwahara K, Fujikura J, et al. Podocyte-specific expression of tamoxifen-inducible cre recombinase in mice. Nephrol Dial Transpl. 2010;25:2120–4.
Madisen L, Garner AR, Shimaoka D, Chuong AS, Klapoetke NC, Li L, et al. Transgenic mice for intersectional targeting of neural sensors and effectors with high specificity and performance. Neuron. 2015;85:942–58.
Kanki H, Suzuki H, Itohara S. High-efficiency CAG-FLPe deleter mice in C57BL/6J background. Exp Anim. 2006;55:137–41.
Gurumurthy’ CB, Saunders’ TL, Ohtsuka’ M. Designing and generating a mouse model: frequently asked questions. J Biomed Res. 2021;35:76–90.
Clark JF, Dinsmore CJ, Soriano P. A most formidable arsenal: genetic technologies for building a better mouse. Genes Dev. 2020;34:1256–86.
Gurumurthy CB, O’Brien AR, Quadros RM, Adams J, Alcaide P, Ayabe S, et al. Reproducibility of CRISPR-Cas9 methods for generation of conditional mouse alleles: a multi-center evaluation. Genome Biol. 2019;20:171.
Miura H, Gurumurthy CB, Sato T, Sato M, Ohtsuka M. CRISPR/Cas9-based generation of knockdown mice by intronic insertion of artificial microRNA using longer single-stranded DNA. Sci Rep. 2015;5:12799.
Aida T, Chiyo K, Usami T, Ishikubo H, Imahashi R, Wada Y, et al. Cloning-free CRISPR/Cas system facilitates functional cassette knock-in in mice. Genome Biol. 2015;16:87.
Chen S, Sun S, Moonen D, Lee C, Lee AYF, Schaffer DV, et al. CRISPR-READI: efficient generation of Knockin mice by CRISPR RNP Electroporation and AAV Donor infection. Cell Rep. 2019;27:3780–e37894.
Tanaka M, Yokoyama K, Hayashi H, Isaki S, Kitatani K, Wang T, et al. CRISPR-KRISPR: a method to identify on-target and random insertion of donor DNAs and their characterization in knock-in mice. Genome Biol. 2022;23(1):228. https://doi.org/10.1186/s13059-022-02779-8.
Zhang X, Li T, Ou J, Huang J, Liang P. Homology-based repair induced by CRISPR-Cas nucleases in mammalian embryo genome editing. Protein Cell. 2022;13:316–35.
Higashitani Y, Horie K. Long-read sequence analysis of MMEJ-mediated CRISPR genome editing reveals complex on-target vector insertions that may escape standard PCR-based quality control. Sci Rep. 2023;13:1–14.
Acknowledgements
We thank the staff of Support Center for Medical Research and Education in Tokai University for sequencing.
Funding
This work was supported by JSPS KAKENHI Grant Number JP16H04685 to MO, by 2017–2018 Tokai University School of Medicine Project Research to MM, by JSPS KAKENHI Grant Number JP19K07393, The Nakajima Foundation and The Takeda Science Foundation to YO, by JSPS KAKENHI Grant Number JP20H03716, by JST CREST Grant Number JPMJCR19H5, and by AMED Grant Number JP22ama221 and JP21fk0108556h0001 to AKo.
Author information
Authors and Affiliations
Contributions
HM and MO designed the study and analyzed data. HM, AN, AKu, MM, SO, YO, KH, KT, and MO performed experiments. SK was involved in design of the construct. AKo, MM, SLPS, CCM, and CBG assisted in study design and interpretation of results. HM, CBG and MO drafted and CCM and SLPS contributed to revising the manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
All animal experiments were performed in accordance with institutional guidelines and were approved by the Institutional Animal Care and Use Committee (permit nos. 165009, 171003, 182023, 182032, 193009, 193010, 204019 and 215009) at Tokai University. This study was carried out in compliance with the ARRIVE guidelines.
Consent for publication
Not applicable.
Competing interests
HK has received grant support from Kyowa Kirin and honoraria from Chugai Pharmaceutical, Kissei Pharmaceutical, Kyowa Kirin, Ono Pharmaceutical, and Sanwa Kagaku Kenkyusho.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Miura, H., Nakamura, A., Kurosaki, A. et al. Targeted insertion of conditional expression cassettes into the mouse genome using the modified i-PITT. BMC Genomics 25, 568 (2024). https://doi.org/10.1186/s12864-024-10250-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12864-024-10250-0