
Abstract
Purpose
Primary Sjogren's syndrome (pSS) is a prevalent autoimmune disease. The immune dysregulation it causes often leads to the development of diffuse large B-cell lymphoma (DLBCL) in clinical practice. However, how it contributes to these two disorders at the molecular level is not yet known. This study explored the potential molecular mechanisms associated with the differences between DLBCL and pSS.
Patients and methods
Gene expression matrices from discovery cohort 1, discovery cohort 2, and the validation cohort were downloaded from the GEO and TCGA databases. Weighted gene coexpression network analysis (WGCNA) was performed to identify the coexpression modules of DLBCL and pSS in discovery cohort 1 and obtain shared genes. GO and KEGG enrichment analyses and PPI network analysis were performed on the shared genes. Immune-related genes (IRGs) were intersected with shared genes to obtain common genes. Afterward, common genes were identified via machine learning methods. The immune infiltration analysis, miRNA-TF-hub gene regulatory chart, gene interactions of the hub genes, and gene‒drug target analysis were performed. Finally, STAT1 was identified as a possible essential gene by the above analysis, and immune infiltration and GSEA pathway analyses were performed in the high- and low-expression groups in discovery cohort 2. The diagnostic efficacy of the hub genes was assessed in the validation cohort, and clinical samples were collected for validation.
Results
By WGCNA, one modular gene in each group was considered highly associated with the disease, and we obtained 28 shared genes. Enrichment analysis revealed shared genes involved in the viral response and regulation. We obtained four hub genes (ISG20, STAT1, TLR7, and RSAD2) via the algorithm. Hub genes and similar genes are primarily involved in regulating type I IFNs. The construction of a miRNA-TF-hub gene regulatory chart revealed that hsa-mir-155-5p, hsa-mir-146b-5p, hsa-mir-21-3p, and hsa-mir-126-3p play essential roles in both diseases. Hub genes were differentially expressed in B-cell memory according to immune infiltration analysis. Hub genes had a strong diagnostic effect on both diseases. STAT1 plays an essential role in immune cells in both diseases.
Conclusion
We identified hub susceptibility genes for DLBCL and pSS and identified hub genes and potential therapeutic targets that may act as biomarkers.
Similar content being viewed by others


Introduction
pSS is one of the most prevalent autoimmune conditions. The disease affects 61 per 100,000 people, is more prevalent in females than in males, and occurs more frequently after the age of 50 [1]. It primarily affects the exocrine glands, such as the salivary and lacrimal glands. Hence, the most prominent presentation signs are ocular and mouth dryness. pSS may influence nearly every organ system in the body and result in tiredness, depression, and impaired physical function, all of which harm quality of life [2]. Interstitial pneumonia, arthritis, vasculitis, nervous system involvement, and kidney involvement are systemic manifestations of pSS. The most severe complication of pSS is non-Hodgkin lymphoma (NHL), which occurs in 5% of patients [3]. DLBCL has been demonstrated to be a common NHL in pSS patients [4]. However, the underlying molecular biological mechanisms remain unclear.
Research [5, 6] has shown that excessive B cells in pSS patients cause immune complex deposits and increase DLBCL risk. Fourteen years from the diagnosis of pSS to DLBCL are needed. The five-year survival rate of DLBCL patients is 37.5%. pSS resulting from DLBCL is independent of prior treatment, and persistent stimulation by chronic antigens contributes to this severe complication [7]. In 75% of patients, DLBCL is the most common aggressive NHL [5]. Its histological structural features are divided into diffuse growth and structural destruction of the lymph nodes. DLBCL can be divided into activated B-cell (ABC) and germinal centre B-cell (GCB) subtypes. ABC DLBCL is characterized by activation of the NF-κB signalling pathway [8]. R-CHOP (rituximab plus cyclophosphamide, doxorubicin, vincristine, and prednisone) can be used to treat two-thirds of DLBCL patients in the clinic [9]. Approximately 30% to 40% of treated DLBCL cases recur, and 10% are primary refractory DLBCLs [10].
The aetiology of the onset of pSS is still unknown, and numerous variables are believed to contribute to disease development. Long noncoding RNAs (lncRNAs) are RNA transcripts with more than 200 nucleotides and no protein coding. LncRNAs are functional units that significantly affect the function and behaviour of cells [11]. MicroRNAs (miRNAs) are short RNAs consisting of approximately 20 nucleotides that regulate the transcription of target genes upon expression [12]. In addition, lncRNAs and miRNAs have been linked to the pathogenesis of pSS [13, 14]. Lymphocyte abnormalities, such as B-cell hyperactivation, the production of serum polyclonal gammaglobulinemia, increased free light chains, and increased autoantibodies, such as SSA and SSB, are responsible for the development of pSS, and patients have an increased risk of B-cell lymphoma [15]. CD4+ T [16], Th17 [17], and Treg cells [18] are associated with the development of pSS. Additionally, infections can also increase the risk of pSS. Hepatitis C virus (HCV), Epstein–Barr virus (EBV), cytomegalovirus (CMV), and human T-lymphotropic virus-1 (HTLV-1) may cause salivary gland infections and lead to pSS [19, 20].
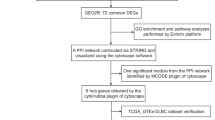
This study aimed to identify potential biomolecular mechanisms involved in the development of pSS into DLBCL through transcriptomics. We performed WGCNA, identification of hub genes, enrichment analysis, construction of PPI networks, miRNA-TF-hub gene regulatory maps, immune cell infiltration evaluation, and validation of the hub genes via datasets downloaded from public databases. Hub genes affecting both diseases were identified, and their molecular mechanisms and functions were investigated. We also speculate that STAT1 may play a vital role in both diseases. This study provides new insights into the analysis of pSS development in DLBCL patients at the B-cell level. The study flow chart is shown in Fig. 1.

Flow chart of the experimental design
Materials and methods
Data source and processing
The Gene Expression Omnibus (GEO) database contains high-throughput gene expression microarray data uploaded by researchers [21]. We designed the following search strategy to collect suitable datasets from the GEO database: first, the dataset must contain case and control groups, and the number of samples in each group must be more than ten to minimize errors in the WGCNA; second, the samples were derived from B cells, and transcriptomic analysis was performed; third, the dataset contained raw data for subsequent analysis. Finally, the datasets GSE12195 [22], GSE135809 [23], GSE56315 [24], and GSE199868 were adopted for the study. To analyse the impact of immune infiltration on both diseases, the DLBCL dataset of RNA sequencing (RNA-seq) data downloaded from The Cancer Genome Atlas (TCGA) database and the GSE154926 dataset were selected for subsequent analysis. Datasets with excessive differences in expression ranges were log2 normalized. Six datasets were annotated with gene symbols through platform information.
Weighted gene coexpression network analysis
Weighted gene coexpression network analysis (WGCNA) can divide highly related genes into several gene modules and analyse the module eigengene (ME) in correlation with external traits [25]. Absolute median differences were calculated for the GSE12195 and GSE135809 datasets, and the top 5000 genes were selected for WGCNA. The R-based (version 4.2.2) WGCNA package plots the sample clusters and removes outlier samples. There were no outliers in the DLBCL group, and two outlier samples were removed from the pSS group. Next, a scale-free grid was drawn, and a suitable soft threshold (β) was calculated via the pickSoftThreshold function, with β = 3 in the DLBCL group and β = 16 in the pSS group. Modules with > 75% similarity were combined to plot the clustering results. Finally, the clinical traits of the two groups were collated, and correlations between ME and clinical traits were calculated.
Identification and analysis of genes common to DLBCL and pSS
Based on the results of WGCNA, shared genes were identified by plotting Venn diagrams through selected MEs with high correlation coefficients. The clusterProfiler package in R was used for Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) enrichment analysis of the shared genes [26, 27]. p < 0.05 was considered statistically significant, and the results were visualized.
The String database is a database used to study protein interactions and construct protein‒protein interaction (PPI) networks. PPI networks of the shared genes were created via the String database. The genes were visualized via Cytoscape software (version 3.9.1), and the importance of the common genes was analysed via the CytoNCA plugin betweenness (BC) algorithm.
Identification and analysis of hub genes
IRGs were downloaded from the ImmPort database to explore the immunological aspects of both diseases. Shared genes were intersected with IRGs to obtain common genes. The common genes were analysed via the last absolute shrinkage and selection operator (LASSO) logistic regression and support vector machine recursive feature elimination (SVM-RFE) machine learning methods. The results of the two methods were combined to obtain the hub genes. The Lasso analysis parameters were alpha = 1 and nlambda = 100, and lambda.min was chosen as the best lambda.
GeneMANIA is a website that can analyse gene functions and identify similar genes [28]. Related gene analysis of the hub genes was performed via GeneMANIA. The DGIdb website [29] can be used to explore drugs that act with target genes.
Construction of a regulatory chart for miRNA-TF-hub genes
The Human microRNA Disease Database (HMDD, Version: 3.2) [30] provides access to disease-associated miRNAs. The shared miRNAs associated with the two diseases were accessed by searching the database above. The miRNAs of the hub genes were predicted via the TarBase database (V.3.0). The miRNAs obtained from both pathways were intersected to obtain key miRNAs for diseases affected by the hub genes. NETWORKanalyst is an online website that can plot gene regulatory networks [31]. The JASPAR database on this website was searched for transcription factors (TFs) with a high correlation with hub genes. These miRNAs and TFs were selected, and a miRNA-TF-hub gene regulatory network was generated.
Immune cell infiltration evaluation
To further analyse the importance of immune cells in both diseases, an immune infiltration evaluation was performed via the CIBERSORT package in R. CIBERSORT is a deconvolution algorithm that evaluates related genes [32]. The discovery cohort 2 was used to calculate the ratio of samples to 22 immune cells. The CIBERSORT results were retained at P < 0.05. Spearman's statistical approach was used to calculate immune cell–immune cell and hub gene–immune cell correlations. The ggplot2 package was used to visualize the results.
STAT1 evaluation
Differentially expressed gene (DEG) analysis of the validation cohort was performed via the limma package in R. We performed log2 transformation on the microarray datasets and normalized them via the normalizeBetweenArrays function (Supplementary Fig. 1). A P value < 0.05 and ∣log2fold change (log2FC) ∣ > 0.5 were considered to be associated with DEGs. The same genes were identified in discovery cohort 1 and the validation cohort. Taken together, the above data show that STAT1 plays a crucial role in disease. Single-gene gene set enrichment analysis (GSEA) pathway enrichment analysis was performed on STAT1. The dataset's top quartile of STAT1 expression was set as the high-expression group, and the remaining quartile was set as the low-expression group.
Immunoinfiltration analysis was performed in discovery cohort 2. The samples in each dataset were divided into high and low groups based on the median STAT1 expression cut-off. Analyses of the differences in the expression of STAT1 between immune cells in the high and low groups and the correlation of STAT1 with immune cells were performed. With the GSVA package in R, the datasets were subjected to single-sample GSEA (ssGSEA) according to the grouping information. The high and low groups were analysed for correlations with the abundances of 28 immune cells, and p values were calculated via the Wilcoxon rank-sum test and Spearman's rank correlation test.
Validation of the hub genes
Validation of hub genes in the validation cohort. The diagnostic efficacy of the hub genes in disease was assessed by plotting receiver operating characteristic (ROC) diagnostic curves. Next, we used the Wilcoxon rank-sum test to compare the gene expression of the hub genes between DLBCL and pSS patients to compare whether the expression of the hub genes was significantly different.
The Second Hospital of Shanxi Medical University Ethics Committee approved the experimental protocol, and all participants signed informed consent forms and agreed to participate in this study. A total of 9 pSS patients (mean age of 44.67 ± 2.52 years), DLBCL patients (mean age of 47.00 ± 3.00 years), and healthy controls (mean age of 42.67 ± 2.52 years) were included. Patients with symptoms of dry mouth and dry throat lasting for more than three months who were positive for anti-SSA/anti-SSB antibodies were clinically diagnosed with pSS and classified into the pSS group. Patients confirmed as DLBCL through cellular immunohistochemistry analysis of tissue pathology sections were classified into the DLBCL group. The healthy control group met the following criteria: normal results for complete blood count, urinalysis, liver function, kidney function, blood glucose, and blood lipid tests; and no history of cancer, chronic diseases, acute diseases, recent medication, major surgery, or infectious diseases.
Two millilitres of fresh peripheral blood was collected from the patients, and the hub genes were detected via quantitative real-time PCR (qPCR) via the SYBR Green I dye method. Total RNA was extracted from the cell pellets (1 × 106) via the TRIzol reagent (Ambion) according to the manufacturer's instructions. cDNA synthesis was performed via the PrimeScript II RTase Kit (TAKARA). The qPCRs were conducted on a CFX-Connect 96 Real-Time PCR Detection System (Bio-Rad). Each 20-µL reaction mixture contained 10 µL of SYBR FAST qPCR Master Mix (KAPA Biosystems), 0.5 µM forward and reverse primers (sequences provided in the supplementary file), 1 µL of cDNA template, and 8 µL of DNase/RNase-free water. The thermal cycling conditions were 95 °C for 3 min, followed by 40 cycles of 95 °C for 5 s, 56 °C for 10 s, and 72 °C for 25 s. Melt curve analysis was performed from 65 °C to 95 °C, increasing by 0.5 °C every 5 s. Data analysis was conducted via Bio-Rad CFX Manager software. The relative expression levels were calculated via the ΔΔCt method, with β-actin serving as the internal control gene. All the samples were tested in triplicate. The differences in the expression of the hub genes between the pSS, DLBCL, and control groups were analysed to validate the hub genes derived from this study.
Statistical analysis
R software (version 4.2.2) and GraphPad Prism (version 9.5) were used for data processing and statistical analysis. The differences in the gene expression of the hub genes between the DLBCL and pSS samples were compared via the Wilcoxon signed-rank test. ROC analysis of the hub genes was performed via the pROC package of R. In addition, we used one-way ANOVA to determine the differences between the pSS, DLBCL, and control groups via GraphPad Prism. P < 0.05 was considered statistically significant.
Results
Grouping of the datasets
GSE12195 and GSE135809 were used as discovery cohort 1; TCGA and GSE154926 were used as discovery cohort 2; and GSE56315 and GSE199868 were used as the validation cohort. Details of the dataset are displayed in Table 1.
Shared genes between DLBCL and pSS patients
WGCNA was used to analyse the datasets for both diseases. The DLBCL group was divided into four colour modules, and the pSS group was divided into six colour modules (Fig. 2 A, D). A heatmap of correlations between traits and modules was then generated, containing correlations and significance values (p values) of module genes with traits (Fig. 2 B, E). A total of 2945 MEs were selected for the turquoise modules in DLBCL patients, and 130 MEs were selected for the black modules in pSS patients (Fig. 2 C, F). Both groups of MEs were positively correlated with disease and negatively correlated with controls, and both were significantly different. The two ME groups were intersected to obtain 28 shared genes affecting both diseases (Fig. 2 G).

Weighted gene coexpression network analysis. A Cluster dendrogram of coexpressed genes in DLBCL; one colour block represents a class of genes. B Module–trait relationships in DLBCL that contain the corresponding correlation and p value. Red indicates a high correlation, and blue indicates a low correlation. C Scatterplot describing the relationship between gene significance and module membership in the turquoise module of DLBCL. D Cluster dendrogram of coexpressed genes in pSS; one colour block represents a class of genes. E Module–trait relationships in pSS that contain the corresponding correlation and p value. Red indicates high correlation, and blue indicates low correlation. F Scatterplot describing the relationship between gene significance and module membership in the black module of pSS. G Genes shared between the turquoise ME of DLBCL and the black ME of pSS
GO and KEGG enrichment analysis; construction of the PPI network
GO and KEGG analyses were performed on the shared genes. GO analysis revealed that these genes are involved mainly in viral defence, regulation, and interferon (IFN) production (Fig. 3 A, B). KEGG pathway analysis revealed that these genes are associated with several viruses (Fig. 3 C, D). According to related studies [19, 20], these viruses are involved in the disease development of pSS and DLBCL.

GO and KEGG enrichment analyses of the shared genes. A, B GO enrichment analysis of the shared genes. The dotted area represents the number of paths. Red indicates a low P value, and blue indicates a high P value. C, D KEGG enrichment analysis of the shared genes. The dotted area represents the number of paths. Red indicates a low P value, and blue indicates a high P value
The STRING database results were imported into Cytoscape after satisfying the enrichment p value: < 1.0e-16, interaction score > 0.4, and exclusion of individual genes. The result of the PPI network, which included 16 nodes with 96 edges, was obtained (Fig. 4 A).

Analysis of shared genes. A PPI network diagram of the shared genes. A darker colour indicates a stronger genetic correlation. B Common genes between the shared genes and the IRGs. The appropriate copyright permission of these KEGG images was obtained and used in this study
Identification of hub genes
Seven common genes were obtained by intersecting shared genes with IRGs (Fig. 4 B). All seven genes were involved in related immune responses. The hub genes were screened via LASSO logistic regression and SVM-RFE analyses (Supplementary Fig. 2), and four hub genes were ultimately obtained: ISG20, STAT1, TLR7, and RSAD2 (Fig. 5 A).

Analysis of the hub genes. A Hub genes from the intersection of LASSO and SVM-RFE; B Hub gene coexpression network. The lines represent the associated paths between two genes. The size of the gene represents importance. C Regulatory network of miRNA-TF-hub genes. Hub genes are indicated in red, miRNAs in blue, and TFs in green
Gene interactions of hub genes and gene‒drug target analysis
We used the GeneMANIA website to search for similar genes among the hub genes and to analyse their functions (Fig. 5 B). The results revealed that 77.64% of the hub genes physically interacted with each other, and the main functions of the genes were type I IFN response and virus regulation. A total of 6 drugs associated with STAT1 and 12 drugs associated with TLR7 were obtained from DGIdb (Table 2).
Construction of the regulatory map of the miRNA-TF-hub genes
A total of 54 miRNAs associated with DLBCL and 16 miRNAs associated with pSS were obtained from the HMDD. Eight miRNAs jointly affected both diseases. We then used the TarBase database (V.3.0) to predict miRNAs via the use of hub genes and obtained a total of 76 miRNAs associated with the hub genes. The above miRNAs were intersected to obtain four miRNAs that affect both diseases through hub genes, including hsa-mir-155-5p, hsa-mir-146b-5p, hsa-mir-21-3p, and hsa-mir-126-3p. A review of the relevant literature revealed that hsa-mir-155-5p did not significantly change in pSS but was elevated in DLBCL. The other three miRNAs are elevated in both diseases. Nine highly associated TFs with hub genes were identified via the NetworkAnalyst website, and miRNA-TF-hub gene regulatory charts were generated (Fig. 5 C).
Immune cell infiltration evaluation
The changes in immune cells in DLBCL and pSS patients were analysed via the CIBERSORT algorithm to explore the level of immune cell infiltration in both diseases. In DLBCL, B-cell memory accounted for a large proportion, and plasma cells accounted for the largest proportion in pSS (Fig. 6 A, D). An examination of immune cell correlations revealed a significant association between immune cells in both disorders (Fig. 6 B, E). STAT1 expression in B-cell memory was negatively correlated with DLBCL but positively correlated with pSS. ISG20 expression in resting memory CD4 T cells was negatively correlated with both diseases. Conversely, RSA2 expression in B-cell memory was correlated with both diseases. In contrast, the expression of TLR7 in memory B cells and regulatory T cells (Tregs) was oppositely associated with both diseases and identical in M1 macrophages (Fig. 6 C, F).

Immune infiltration evaluation of DLBCL and pSS patients. A The ratio of immune cells in DLBCL. Each column represents a patient. B Association among immune cells of DLBCL. Blue represents a positive correlation, and red represents a negative correlation. C Association between hub genes and immune cells in DLBCL. Blue represents a negative correlation, and red represents a positive correlation. D The ratio of immune cells in pSS patients. One column represents a patient. E Association among immune cells in pSS patients. Blue represents a positive correlation, and red represents a negative correlation. F Association between hub genes and immune cells in pSS patients. Blue represents a negative correlation, and red represents a positive correlation
STAT1 assessment, GSEA pathway enrichment analysis, immune infiltration assessment, and ssGSEA
The analysis validated the common DEGs between the two diseases. A total of 128 DEGs were obtained, and it was discovered that both the DEGs and the hub genes shared STAT1 and USP18 (Fig. 7). An analysis of the PPI network chart and the miRNA-TF-hub gene regulatory map revealed that STAT1 plays an essential role in all of these genes. Therefore, STAT1 may play a vital role in the development of DLBCL from pSS. Next, single-gene GSEA pathway enrichment analysis of STAT1 was performed for both disease groups. The potential role of STAT1 in this disease was investigated. In DLBCL, the STAT1 high-expression group was involved in the lysosome, glycosaminoglycan degradation, and chemokine signalling pathways (Supplementary Figs. 3 A-C), while the low-expression group was involved in base excision repair, RNA degradation, and glycosylphosphatidylinositol (GPI) anchor biosynthesis (Supplementary Figs. 3 D-F). In pSS, the STAT1 high-expression group was involved in homologous recombination, pyruvate metabolism, and the citrate cycle (Supplementary Figs. 3G-I), and the low-expression group was involved in drug metabolism, other enzymes, ECM receptor interactions, and the MAPK signalling pathway (Supplementary Figs. 3 J-L).

DEG analysis. A Volcano plot of DLBCL. Red represents upregulated genes. Blue represents downregulated genes; B Heatmap of DLBCL patients; C Volcano plot of pSS patients. Red represents upregulated genes. Blue represents downregulated genes. D Heatmap of pSS patients. E Intersection of DEGs and shared genes
To better analyse the importance of STAT1 in the immune microenvironment of DLBCL and pSS patients, the samples were divided into STAT1 high- and low-expression groups, and CIBERSORT was used to calculate the differences in expression between the subpopulations of immune cells. The results revealed that the four cell types expressed in DLBCL patients were significantly different from the seven immune cell types expressed in pSS patients (Fig. 8 A, G). To demonstrate the correlation of these cells with STAT1, we generated scatter plots. We found that activated memory CD4 T cells, M1 macrophages, activated memory CD4 T cells, and STAT1 were significantly positively correlated with DLBCL, whereas memory B cells were significantly negatively correlated with DLBCL (Fig. 8 C-F). In pSS, activated memory CD4 T cells, naïve CD4 T cells, follicular helper T cells, gamma delta T cells, memory B cells, and M1 macrophages were positively correlated with STAT1, but resting Mast cells were negatively correlated with STAT1 (Fig. 8 I-O). Common immune cells include memory B cells, activated memory CD4 T cells, and M1 macrophages. The infiltration levels of 28 immune cells in the high- and low-STAT1 expression groups were analysed via the ssGSEA algorithm. The data revealed that STAT1 expression was significantly different in activated CD4 T cells, activated dendritic cells, central memory CD4 T cells, central memory CD8 T cells, effector memory CD8 T cells, eosinophils, immature dendritic cells, MDSCs, natural killer cells, natural killer T cells, plasmacytoid dendritic cells, regulatory T cells, T follicular cells, T cells, eosinophils, immature dendritic cells, MDSCs, natural killer cells, natural killer T cells, plasmacytoid dendritic cells, regulatory T cells, T follicular helper cells, type 1 T helper cells, and type 2 T helper cells (Fig. 8B, H).

Immune cell infiltration evaluation via STAT1 and ssGSEA. A Comparison of gene expression between the high-STAT1 expression group and low-STAT1 expression group in DLBCL; B ssGSEA in DLBCL; (C-F) correlation scatter plot between STAT1 expression and four immune cell types (B memory cells, M1 macrophages, CD4 memory T cells, and CD8 T cells); (G) comparison of gene expression between the high- and low-STAT1 expression groups in pSS; and (I-O) correlation scatter plot between STAT1 expression and seven immune cell types (B memory T cells, M1 macrophages, resting Mast cells, activated memory T cells, naïve CD4 T cells, follicular helper T cells, and gamma delta T cells)
Verification of the hub genes
In the validation cohort, the diagnostic efficacy of the hub genes in disease was verified by plotting ROC diagnostic curves. In both groups, the hub genes had good diagnostic efficacy. We found that the area under the ROC curve (AUC) of STAT1 was greater than 0.7 in both groups (Fig. 9A, B). A comparative gene expression chart revealed that the number of hub genes was greater in both pSS and DLBCL patients (Fig. 9 C). Validation by qPCR revealed statistically significant differences in hub genes between the two diseases, which was consistent with previous analyses (Fig. 9 D).

Validation of the hub genes. A Diagnostic efficacy of hub genes in DLBCL; (B) diagnostic efficacy of hub genes in pSS; (C) differentiation of hub gene expression in pSS and DLBCL; (D) qPCR validation of hub genes. *p < 0.05; **p < 0.01; ***p < 0.001; ****p < 0.0001
Discussion
This study analysed the pSS and DLBCL datasets published in the GEO and TCGA databases (GSE12195, GSE135809, GSE154926, GSE56315, and GSE199868). Based on the WGCNA, the modular genes for each disease most associated with the clinical trait were selected. The genes shared by both diseases were machine learned, and four hub genes were selected: ISG20, STAT1, TLR7, and RSAD2. These four genes may play essential roles in the development of pSS and DLBCL. Finally, STAT1 is presumed to be highly important.
T and B cells in pSS patients infiltrate the salivary and lacrimal glands in large numbers and then secrete many cytokines (e.g., IFN-γ, TNF-α, and IL-6). A small number of patients will develop DLBCL [33]. The role of the immune system in DLBCL is reflected mainly in the tumour microenvironment and immune escape mechanism [34]. According to previous reports, risk factors for the development of lymphoma in pSS patients include permanent salivary gland lymphocytic infiltration, lymphadenopathies, decreased C4 complement component levels, the presence of autoimmune antibodies, and lymphocytopenia [35]. Dysregulation of the immune system plays an essential role in the occurrence and development of pSS and DLBCL.
The signal transducer and activator of transcription (STAT) family contains seven TFs: STAT1, STAT2, STAT3, STAT4, STAT5a, STAT5b, and STAT6. It is involved in embryonic development, the immune response, cell growth, and cell death [36]. STAT1 can induce and stimulate type I, II, and III IFN responses and is an important component of the pathogenic immune response [37]. In B cells and monocytes from patients with pSS, the sensitivity of STAT1 activation signals is increased such that patients have increased IFN in their blood [38]. Several studies have revealed an essential role for STAT1 in pSS [39,40,41]. Wu et al. [42] reported that paeoniflorin-6'-O-benzene sulfonate (CP-25) inhibited the JAK1-STAT1/2-CXCL13 signalling pathway and blocked B-cell invasion of the salivary glands, which could alleviate the symptoms of pSS. A recent genome-wide association study revealed a strong genetic correlation between STAT1 and Sjögren syndrome [43]. Jacob's study demonstrated that STAT1 enhances IFN signalling, leading to chronic activation of T and B lymphocytes in pSS patients, and suggested that increased STAT1 expression is a biomarker [44]. The IFN pathway is a key pathogenic pathway in pSS, and Chen et al. reported that activation of the JAK/STAT1 pathway can increase transcription of the BAFF promoter region, thus enhancing the role of IFN in pSS [40, 45]. B-aggressive lymphoma-1 protein (BAL1) and the ADP-ribosyltransferase BAL1/ARTD9 are thought to be associated with DLBCL. Rosalba [46] demonstrated that BAL1 stimulates STAT1 phosphorylation and inhibits the IFNγ-STAT1-IRF1-p53 axis, leading to proliferation and drug resistance in DLBCL. Currently, miR130b inhibits the IFNAR1/p-STAT1 axis to downregulate OX40L expression [47]. STAT1 plays a key role in the immune escape mechanism in DLBCL. By regulating the STAT1 signalling pathway, the recognition and clearance of tumours by the immune system can be enhanced. Combination therapy with immune checkpoint inhibitors (such as PD-1/PD-L1 inhibitors) and STAT1 inhibitors is expected to improve the effectiveness of immunotherapy [48, 49]. In addition, studies have shown that inhibiting the JAK-STAT signalling pathway can alleviate and improve the dryness symptoms of pSS patients and reduce the immune evasion and cell therapy resistance of DLBCL tumour cells [49, 50]. These results suggest a critical role for STAT1 in both pSS and DLBCL.
The ligand‒receptor interaction of IFN activates the JAK‒STAT signalling pathway, resulting in the upregulation of a gene cluster called the interferon-stimulated gene (ISG) [51]. Interferon-stimulated gene 20 kDa protein (ISG20) is a nuclease active type I and type II interferon-inducible protein that inhibits a broad spectrum of viruses [52]. ISG20 has RNase enzymatic properties that can directly degrade viral RNA [53]. The family of Toll-like receptors (TLRs) are key pattern recognition receptors (PRRs) that play a central role in the subsequent development of various immune responses [54]. TLR7 has been closely linked to autoimmune diseases, and several studies [55,56,57] have demonstrated that TLR7 plays an essential role in lupus. In pSS, the number of TLR7-expressing B cells is increased, and patients exhibit type I and type II IFN features [41] such as thrombocytopenia [58]. TLR7 is elevated in patients' salivary and parotid gland tissues, leading to hypofunction [59]. Radical s-adenosyl methionine domain-containing 2 (RSAD2) is a protein of IFN that is involved in antiviral activity and associated with endoplasmic reticulum function [60]. RSAD2 is upregulated in B cells from pSS patients and is positively correlated with interleukin-10 (IL-10). Inhibition of the NF-κb signalling pathway by knocking down RSAD2 reduces B-cell overactivity in pSS patients [61]. In addition, TLR7 upregulation leads to the upregulation of downstream RSAD2 in pSS patients [62].
All of the hub genes were found to be associated with IFN, which is an antiviral cytokine that responds to viral attacks by modulating the body's immune response [63]. IFNs are divided into three categories: type I, type II, and type III IFNs. It has been demonstrated [64] that IFN causes immune dysregulation, leading to pSS. Type I IFN overexpression is thought to play an essential role in pSS [65]. Plasmacytoid dendritic cells (pDCs) are thought to be specialist producers of type I IFN [66], and monocytes respond to type I IFN. pDCs can increase the expression of TLR7 and TLR9 and are more susceptible to activation by stimuli. The biological effect of IFN is that STAT1 and 2 are activated and form dimers after IFN binds to the corresponding receptors. Next, it binds to interferon-regulatory factors (IRFs) and transcribes and activates ISGs [67]. In addition, B-cell activating factor (BAFF) also responds to type I and type II IFNs. In pSS, BAFF amplifies autoimmune antibodies and is a major driver of lymphoma escape [15, 68]. These findings suggest a critical role for IFN in pSS. IFN-β also inhibits DLBCL via tumour necrosis factor-related apoptosis-inducing ligand (TRAIL) [69], and high expression of IFN-γ is associated with a protective effect against disease [70].
Because of the apparent antiviral effect of IFN, we also investigated the causes of pSS and DLBCL. Some of the literature has demonstrated the involvement of EBV in the pathogenesis of pSS [71] and that EBV is also a cause of DLBCL [72]. We believe that EBV may play a role in the progression of pSS to DLBCL, but further studies are needed.
Although pSS progresses to DLBCL, few studies have demonstrated a common molecular mechanism. This study analysed the hub genes involved in pSS development into DLBCL from a bioinformatics perspective, aiming to explore the underlying molecular mechanisms of this disease. In addition, we explored the relevance of immune cell profiles and hub genes in immune cells in DLBCL and pSS patients via the CIBERSORT and ssGSEA algorithms. It is speculated that STAT1 may be an essential gene in both diseases.
Limitations
There are many limitations associated with the current study. First, this study relies on bioinformatics analysis of public datasets, which could generate some inconsistencies in the results. The public datasets used were from European populations, and it remains unclear whether the conclusions apply universally. Moreover, larger clinical samples need to be collected to validate the accuracy of the results. In addition, this study focused only on blood hub gene differences in patients and did not further explore other differences. Finally, more molecular experiments are needed to explore the mechanisms involved.
Conclusions
We identified hub genes (ISG20, STAT1, TLR7, and RSAD2) involved in the progression of pSS to DLBCL. The analysis revealed that the hub genes are primarily involved in the viral response and regulation and are associated with IFN. STAT1 may play a vital role in the development of both diseases, and these findings may provide new insight for subsequent studies. Additionally, future work involves studying the association of STAT1 with DLBCL and pSS through cell and animal experiments and further exploring the mechanisms involving STAT1.
Availability of data and materials
No datasets were generated or analysed during the current study.
Abbreviations
- pSS:
-
Primary Sjogren's syndrome
- DLBCL:
-
Diffuse large B-cell lymphoma
- GEO:
-
Gene Expression Omnibus
- RNA-seq:
-
RNA sequencing
- TCGA:
-
The Cancer Genome Atlas Program
- WGCNA:
-
Weighted Gene Co-Expression Network Analysis
- GO:
-
Gene Ontology
- KEGG:
-
Kyoto Encyclopedia of Genes and Genomes
- PPI:
-
Protein–protein interaction
- IRGs:
-
Immune-related genes
- LASSO:
-
Least absolute shrinkage and selection operator
- SVM-RFE:
-
Support vector machine recursive feature elimination
- GSEA:
-
Gene Set Enrichment Analysis
- NHL:
-
Non-Hodgkin lymphoma
- ABC:
-
Activated B-cell
- GCB:
-
Germinal centre B-cell
- lncRNAs:
-
Long noncoding RNAs
- miRNAs:
-
MicroRNAs
- HCV:
-
Hepatitis C virus
- EBV:
-
Epstein-Barr virus
- CMV:
-
Cytomegalovirus
- HTLV-1:
-
Human T-lymphotropic virus-1
- HMDD:
-
Human microRNA Disease Database
- TF:
-
Transcription Factors
- ssGSEA:
-
Single sample GSEA
- qPCR:
-
Quantitative Real-time PCR
- AUC:
-
Area under the ROC curve
- STAT:
-
Signal Transducer and Activator of Transcription
- IFN:
-
Interferon
- CP-2:
-
5Paeoniflorin-6'-O-benzene sulfonate
- BAL:
-
1B-aggressive lymphoma-1 protein
- ISG:
-
Interferon-stimulated gene
- TLR:
-
Toll-like receptors
- PRRs:
-
Pattern recognition receptors
- RSAD2:
-
Radical s-adenosyl methionine domain-containing 2
- pDCs:
-
Plasmacytoid dendritic cells
- IRFs:
-
Interferon-regulatory factors
- BAFF:
-
B cell activating factor
- TRAIL:
-
Tumor necrosis factor-related apoptosis-inducing ligand
References
Qin B, Wang J, Yang Z, et al. Epidemiology of primary Sjögren’s syndrome: a systematic review and meta-analysis. Ann Rheum Dis. 2015;74(11):1983–9.
Negrini S, Emmi G, Greco M, et al. Sjögren’s syndrome: a systemic autoimmune disease. Clin Exp Med. 2022;22(1):9–25.
Stefanski AL, Tomiak C, Pleyer U, Dietrich T, Burmester GR, Dörner T. The diagnosis and treatment of Sjögren’s Syndrome. Dtsch Arztebl Int. 2017;114(20):354–61.
Chiu YH, Chung CH, Lin KT, et al. Predictable biomarkers of developing lymphoma in patients with Sjögren syndrome: a nationwide population-based cohort study. Oncotarget. 2017;8(30):50098–108.
Teras LR, DeSantis CE, Cerhan JR, Morton LM, Jemal A, Flowers CR. 2016 US lymphoid malignancy statistics by World Health Organization subtypes. CA Cancer J Clin. 2016;66(6):443–59.
Mavragani CP. Mechanisms and new strategies for primary Sjögren’s syndrome. Annu Rev Med. 2017;68:331–43.
Nocturne G, Virone A, Ng WF, et al. Rheumatoid factor and disease activity are independent predictors of lymphoma in primary Sjögren’s Syndrome. Arthritis Rheumatol (Hoboken, NJ). 2016;68(4):977–85.
Tam CS, Trotman J, Opat S, et al. Phase 1 study of the selective BTK inhibitor zanubrutinib in B-cell malignancies and safety and efficacy evaluation in CLL. Blood. 2019;134(11):851–9.
Wang L, Li LR, Young KH. New agents and regimens for diffuse large B cell lymphoma. J Hematol Oncol. 2020;13(1):175.
Patriarca A, Gaidano G. Investigational drugs for the treatment of diffuse large B-cell lymphoma. Expert Opin Investig Drugs. 2021;30(1):25–38.
Bridges MC, Daulagala AC, Kourtidis A. LNCcation: lncRNA localization and function. J Cell Biol. 2021;220(2):e202009045.
Bartel DP. MicroRNAs: genomics, biogenesis, mechanism, and function. Cell. 2004;116(2):281–97.
Shi H, Cao N, Pu Y, Xie L, Zheng L, Yu C. Long non-coding RNA expression profile in minor salivary gland of primary Sjögren’s syndrome. Arthritis Res Ther. 2016;18(1):109.
Gao Q, Ye Z, Liu T, et al. Hsa-miR-3202 attenuates Jurkat cell infiltration via MMP2 in primary Sjögren’s syndrome. J Oral Pathol Med. 2022;51(9):818–28.
Nocturne G, Mariette X. B cells in the pathogenesis of primary Sjögren syndrome. Nat Rev Rheumatol. 2018;14(3):133–45.
Hong X, Meng S, Tang D, et al. Single-Cell RNA Sequencing Reveals the Expansion of Cytotoxic CD4+ T Lymphocytes and a Landscape of Immune Cells in Primary Sjögren’s Syndrome. Front Immunol. 2020;11:594658.
Verstappen GM, Corneth OBJ, Bootsma H, Kroese FGM. Th17 cells in primary Sjögren’s syndrome: Pathogenicity and plasticity. J Autoimmun. 2018;87:16–25.
Alunno A, Carubbi F, Bistoni O, et al. T Regulatory and T Helper 17 Cells in Primary Sjögren’s Syndrome: Facts and Perspectives. Mediators Inflamm. 2015;2015:243723.
Utomo SW, Putri JF. Infections as risk factor of Sjögren’s syndrome. Open Access Rheumatol. 2020;12:257–66.
Mofors J, Arkema EV, Björk A, et al. Infections increase the risk of developing Sjögren’s syndrome. J Intern Med. 2019;285(6):670–80.
Barrett T, Wilhite SE, Ledoux P, et al. NCBI GEO: archive for functional genomics data sets–update. Nucleic ACIDS res. 2013;41(Database issue):D991-995.
Compagno M, Lim WK, Grunn A, et al. Mutations of multiple genes cause deregulation of NF-kappaB in diffuse large B-cell lymphoma. Nature. 2009;459(7247):717–21.
Inamo J, Suzuki K, Takeshita M, et al. Identification of novel genes associated with dysregulation of B cells in patients with primary Sjögren’s syndrome. Arthritis Res Ther. 2020;22(1):153.
Dybkær K, Bøgsted M, Falgreen S, et al. Diffuse large B-cell lymphoma classification system that associates normal B-cell subset phenotypes with prognosis. J Clin Oncol. 2015;33(12):1379–88.
Langfelder P, Horvath S. WGCNA: an R package for weighted correlation network analysis. BMC Bioinformatics. 2008;9:559.
Kanehisa M, Furumichi M, Sato Y, Kawashima M, Ishiguro-Watanabe M. KEGG for taxonomy-based analysis of pathways and genomes. Nucleic Acids Res. 2023;51(D1):D587-d592.
Kanehisa M, Goto S. KEGG: kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 2000;28(1):27–30.
Franz M, Rodriguez H, Lopes C, et al. GeneMANIA update 2018. Nucleic Acids Res. 2018;46(W1):W60-w64.
Freshour SL, Kiwala S, Cotto KC, et al. Integration of the Drug-Gene Interaction Database (DGIdb 4.0) with open crowdsource efforts. Nucleic Acids Res. 2021;49(D1):D1144-d1151.
Huang Z, Shi J, Gao Y, et al. HMDD v3.0: a database for experimentally supported human microRNA-disease associations. Nucleic Acids Res. 2019;47(D1):D1013–7.
Zhou G, Soufan O, Ewald J, Hancock REW, Basu N, Xia J. NetworkAnalyst 3.0: a visual analytics platform for comprehensive gene expression profiling and meta-analysis. Nucleic Acids Res. 2019;47(W1):W234-w241.
Chen B, Khodadoust MS, Liu CL, Newman AM, Alizadeh AA. Profiling tumor infiltrating immune cells with CIBERSORT. Methods Mol Biol (Clifton, NJ). 2018;1711:243–59.
Verstappen GM, Pringle S, Bootsma H, Kroese FGM. Epithelial-immune cell interplay in primary Sjögren syndrome salivary gland pathogenesis. Nat Rev Rheumatol. 2021;17(6):333–48.
Autio M, Leivonen SK, Brück O, Karjalainen-Lindsberg ML, Pellinen T, Leppä S. Clinical impact of immune cells and their spatial interactions in diffuse large B-Cell Lymphoma Microenvironment. Clin Cancer Res. 2022;28(4):781–92.
Gorodetskiy VR, Probatova NA, Vasilyev VI. Characteristics of diffuse large B-cell lymphoma in patients with primary Sjögren’s syndrome. Int J Rheum Dis. 2020;23(4):540–8.
Butturini E, de CarcereriPrati A, Mariotto S. Redox Regulation of STAT1 and STAT3 Signaling. Int J Mol Sci. 2020;21(19):7034.
Toubiana J, Okada S, Hiller J, et al. Heterozygous STAT1 gain-of-function mutations underlie an unexpectedly broad clinical phenotype. Blood. 2016;127(25):3154–64.
Pertovaara M, Silvennoinen O, Isomäki P. Cytokine-induced STAT1 activation is increased in patients with primary Sjögren’s syndrome. Clin Immunol (Orlando, Fla). 2016;165:60–7.
Ha YJ, Choi YS, Kang EH, et al. Increased expression of interferon-λ in minor salivary glands of patients with primary Sjögren’s syndrome and its synergic effect with interferon-α on salivary gland epithelial cells. Clin Exp Rheumatol. 2018;36 Suppl 112(3):31–40.
Zheng Q, Liu L, Wang B, He Y, Zhang M, Shi G. Phosphorylated signal transducer and activator of transcription proteins 1 in salivary glandular tissue: an important histological marker for diagnosis of primary Sjögren’s syndrome. RMD Open. 2023;9(1):e002694.
Imgenberg-Kreuz J, Sandling JK, Björk A, et al. Transcription profiling of peripheral B cells in antibody-positive primary Sjögren’s syndrome reveals upregulated expression of CX3CR1 and a type I and type II interferon signature. Scand J Immunol. 2018;87(5):e12662.
Wu H, Chen X, Gu F, et al. CP-25 alleviates antigen-induced experimental Sjögren’s syndrome in mice by inhibiting JAK1-STAT1/2-CXCL13 signaling and interfering with B-cell migration. Lab Investig J Tech Methods Pathol. 2021;101(8):1084–97.
Casares-Marfil D, Martínez-Bueno M, Borghi MO, et al. A genome-wide association study suggests new susceptibility loci for primary antiphospholipid syndrome. Arthritis Rheumatology (Hoboken, NJ). 2024. https://doi.org/10.1002/art.42947.
Ritter J, Szelinski F, Aue A, et al. Elevated unphosphorylated STAT1 and IRF9 in T and B cells of primary sjögren’s syndrome: Novel biomarkers for disease activity and subsets. J Autoimmun. 2024;147: 103243.
Chen X, Li M, Li H, Liu M, Su J, Ji Y. Implications of IFNγ SNP rs2069705 in primary Sjögren’s syndrome: transcriptional activation and B cell infiltration. Am J Physiol Cell Physiol. 2024;326(5):C1494-c1504.
Camicia R, Bachmann SB, Winkler HC, et al. BAL1/ARTD9 represses the anti-proliferative and pro-apoptotic IFNγ-STAT1-IRF1-p53 axis in diffuse large B-cell lymphoma. J Cell Sci. 2013;126(Pt 9):1969–80.
Sun R, Zhang PP, Weng XQ, et al. Therapeutic targeting miR130b counteracts diffuse large B-cell lymphoma progression via OX40/OX40L-mediated interaction with Th17 cells. Signal Transduct Target Ther. 2022;7(1):80.
Camicia R, Winkler HC, Hassa PO. Novel drug targets for personalized precision medicine in relapsed/refractory diffuse large B-cell lymphoma: a comprehensive review. Mol Cancer. 2015;14:207.
Xu X, Zhang Y, Lu Y, et al. CD58 alterations govern antitumor immune responses by inducing PD-L1 and IDO in diffuse large B-cell lymphoma. Cancer Res. 2024;84 (13):2123–40.
Charras A, Arvaniti P, Le Dantec C, et al. JAK inhibitors and oxidative stress control. Front Immunol. 2019;10:2814.
Deymier S, Louvat C, Fiorini F, Cimarelli A. ISG20: an enigmatic antiviral RNase targeting multiple viruses. FEBS Open Bio. 2022;12(6):1096–111.
Stadler D, Kächele M, Jones AN, et al. Interferon-induced degradation of the persistent hepatitis B virus cccDNA form depends on ISG20. EMBO Rep. 2021;22(6):e49568.
Nguyen LH, Espert L, Mechti N, Wilson DM 3rd. The human interferon- and estrogen-regulated ISG20/HEM45 gene product degrades single-stranded RNA and DNA in vitro. Biochemistry. 2001;40(24):7174–9.
Akira S, Uematsu S, Takeuchi O. Pathogen recognition and innate immunity. Cell. 2006;124(4):783–801.
Satterthwaite AB. TLR7 Signaling in Lupus B Cells: New Insights into Synergizing Factors and Downstream Signals. Curr Rheumatol Rep. 2021;23(11):80.
Majer O, Liu B, Kreuk LSM, Krogan N, Barton GM. UNC93B1 recruits syntenin-1 to dampen TLR7 signalling and prevent autoimmunity. Nature. 2019;575(7782):366–70.
Brown GJ, Cañete PF, Wang H, et al. TLR7 gain-of-function genetic variation causes human lupus. Nature. 2022;605(7909):349–56.
Zhang S, Qu J, Wang L, et al. Activation of Toll-Like Receptor 7 Signaling Pathway in Primary Sjögren’s Syndrome-Associated Thrombocytopenia. Front Immunol. 2021;12:637659.
Punnanitinont A, Kasperek EM, Kiripolsky J, Zhu C, Miecznikowski JC, Kramer JM. TLR7 agonism accelerates disease in a mouse model of primary Sjögren’s syndrome and drives expansion of T-bet(+) B cells. Front Immunol. 2022;13:1034336.
Yogarajah T, Ong KC, Perera D, Wong KT. RSAD2 and AIM2 Modulate Coxsackievirus A16 and Enterovirus A71 Replication in Neuronal Cells in Different Ways That May Be Associated with Their 5’ Nontranslated Regions. J Virol. 2018;92(6):e01914-17.
Zhu H, Zheng J, Zhou Y, Wu T, Zhu T. Knockdown of RSAD2 attenuates B cell hyperactivity in patients with primary Sjögren’s syndrome (pSS) via suppressing NF-κb signaling pathway. Mol Cell Biochem. 2021;476(5):2029–37.
Maria NI, Steenwijk EC, IJpma AS, et al. Contrasting expression pattern of RNA-sensing receptors TLR7, RIG-I and MDA5 in interferon-positive and interferon-negative patients with primary Sjögren’s syndrome. Ann Rheum Dis. 2017;76(4):721–30.
Wang W, Xu L, Su J, Peppelenbosch MP, Pan Q. Transcriptional Regulation of Antiviral Interferon-Stimulated Genes. Trends Microbiol. 2017;25(7):573–84.
Bodewes ILA, Björk A, Versnel MA, Wahren-Herlenius M. Innate immunity and interferons in the pathogenesis of Sjögren’s syndrome. Rheumatology (Oxford). 2021;60(6):2561–73.
Brkic Z, Versnel MA. Type I IFN signature in primary Sjögren’s syndrome patients. Expert Rev Clin Immunol. 2014;10(4):457–67.
Fitzgerald-Bocarsly P, Dai J, Singh S. Plasmacytoid dendritic cells and type I IFN: 50 years of convergent history. Cytokine Growth Factor Rev. 2008;19(1):3–19.
Del Papa N, Minniti A, Lorini M, et al. The Role of Interferons in the Pathogenesis of Sjögren’s Syndrome and Future Therapeutic Perspectives. Biomolecules. 2021;11(2):251.
Vincent FB, Saulep-Easton D, Figgett WA, Fairfax KA, Mackay F. The BAFF/APRIL system: emerging functions beyond B cell biology and autoimmunity. Cytokine Growth Factor Rev. 2013;24(3):203–15.
Oehadian A, Koide N, Mu MM, et al. Interferon (IFN)-beta induces apoptotic cell death in DHL-4 diffuse large B cell lymphoma cells through tumor necrosis factor-related apoptosis-inducing ligand (TRAIL). Cancer Lett. 2005;225(1):85–92.
Marangon AV, Colli CM, Cardozo DM, et al. Impact of SNPs/Haplotypes of IL10 and IFNG on the Development of Diffuse Large B-Cell Lymphoma. J Immunol Res. 2019;2019:2137538.
Maślińska M. The role of Epstein-Barr virus infection in primary Sjögren’s syndrome. Curr Opin Rheumatol. 2019;31(5):475–83.
Vockerodt M, Yap LF, Shannon-Lowe C, et al. The Epstein-Barr virus and the pathogenesis of lymphoma. J Pathol. 2015;235(2):312–22.
Acknowledgements
The authors thank the GEO database researchers for providing the data and algorithm developers.
Disclosure
The authors declare that the research was conducted without any commercial or financial relationships that could be construed as a potential conflict of interest.
Informed consent
Informed consent from publicly available databases is not applicable. Informed consent for the validation experiment in this study was obtained from and agreed to by all subjects.
Funding
This research was funded by the General Program of the National Natural Science Foundation of China (NSFC) (Item number: 201601D102067); Soft Science Research in Shanxi Province (Item number: 2017041038–3); Shanxi Basic Research Program (Item number: YDZJSX20231A063).
Author information
Authors and Affiliations
Contributions
The authors confirm their contribution to the paper as follows: project concept and idea formulation: YX, JH; data collection: YX, ZF; Analysis, interpretation, validation and visualization of results: YX, JH; draft manuscript preparation: YX; manuscript review & editing: SL. All authors reviewed the results and approved the final version of the manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
The data and methods of the study were performed according to the ethical standards of the Second Hospital of Shanxi Medical University, and all participants agreed to participate in this study. This study was approved by the Ethics Committee of the Second Hospital of Shanxi Medical University which was carried out in accordance with the Declaration of Helsinki (protocol code: (2023)YX(161), date of approval: 27th April 2023).
Consent for publication
All study participants have given their consent for publication.
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Xu, Y., Han, J., Fan, Z. et al. Transcriptomics explores potential mechanisms for the development of Primary Sjogren’s syndrome to diffuse large B-cell lymphoma in B cells. BMC Immunol 25, 53 (2024). https://doi.org/10.1186/s12865-024-00646-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12865-024-00646-8