
Abstract
Background
Clematis species are attractive ornamental plants with a variety of flower colors and patterns. Heat stress is one of the main factors restricting the growth, development, and ornamental value of Clematis. Clematis lanuginosa and Clematis crassifolia are large-flowered and evergreen Clematis species, respectively, that show different tolerance to heat stress. We compared and analyzed the transcriptome of C. lanuginose and C. crassifolia under heat stress to determine the regulatory mechanism(s) of resistance.
Results
A total of 1720 and 6178 differentially expressed genes were identified from C. lanuginose and C. crassifolia, respectively. The photosynthesis and oxidation–reduction processes of C. crassifolia were more sensitive than C. lanuginose under heat stress. Glycine/serine/threonine metabolism, glyoxylic metabolism, and thiamine metabolism were important pathways in response to heat stress in C. lanuginose, and flavonoid biosynthesis, phenylalanine metabolism, and arginine/proline metabolism were the key pathways in C. crassifolia. Six sHSPs (c176964_g1, c200771_g1, c204924_g1, c199407_g2, c201522_g2, c192936_g1), POD1 (c200317_g1), POD3 (c210145_g2), DREB2 (c182557_g1), and HSFA2 (c206233_g2) may be key genes in the response to heat stress in C. lanuginose and C. crassifolia.
Conclusions
We compared important metabolic pathways and differentially expressed genes in response to heat stress between C. lanuginose and C. crassifolia. The results increase our understanding of the response mechanism and candidate genes of Clematis under heat stress. These data may contribute to the development of new Clematis varieties with greater heat tolerance.
Similar content being viewed by others


Background
Clematis L. (Ranunculaceae) are mainly perennial woody vines. There are many species, varieties, and flower patterns. The optimum growth conditions for Clematis generally involve cool to moderate temperatures. There are approximately 355 species of Clematis worldwide and more than 147 species in China [1, 2]. The horticultural Clematis varieties are mostly cultivated in Poland, Britain, and other European countries.
Clematis species also have medicinal value. Their chemical components include triterpenoid saponins, alkaloids, flavonoids, lignans, and steroids. Clematis spp. are plant sources of many pharmaceutical active ingredients [3,4,5]. Flavonoids isolated from Clematis aethusifolia showed moderate toxicity to five human solid tumor cell lines, including A-375 and SK-OV-3 [6]. The triterpene saponins isolated from Clematis aethusifolia inhibited the growth and development of Plutella xylostella in crops, and may have potential for insecticide development [7]. Clematis is also an important genus of garden greening vines that increases the diversity of vertical greening plants in gardens. They can be used as ground cover plants, potted plants, and garden greening plants, and are excellent ornamentals.
With global climate change and increased temperatures, extreme heat stress has become a problem that threatens plant growth and development. Heat stress can cause physiological, molecular, and biochemical changes in plants and interfere with the growth and development process of cells and the entire plant. Heat stress can limit the growth, metabolism, and yield of plants [8,9,10]. Heat stress affects seed germination and growth, leading to seed malformation and seed cell death. Heat stress also affects the development of flower organs [11, 12]. Some horticultural varieties of Clematis show symptoms such as leaf wilting and plant wilt after suffering from heat stress. Heat stress significantly restricts the ornamental uses and growth environment of Clematis. Therefore, understanding the heat resistance mechanism of Clematis and the breeding of heat-resistant varieties are important topics.
Plants can adapt to, or resist, heat stress by morphological changes, photosynthesis, protective enzyme activity, and osmotic substance regulation [13, 14]. Plant antioxidant activity is mainly completed by the enzymatic and non-enzymatic clearance systems. Enzymatic clearance systems include superoxide dismutase (SOD), catalase (CAT), and peroxidase (POD); non-enzymatic clearance systems include ascorbic acid (ASA), mannitol (MT), and vitamin E. When the plant suffers from heat stress, the balance of reactive oxygen radicals in the plant will be disrupted, reactive oxygen species (ROS) accumulation will occur, and the reactive oxygen scavenging system will help to reduce the damage caused by the stress [15,16,17]. The plant’s endogenous hormones will change to activate the mechanisms of plant stress resistance. Abscisic acid (ABA), salicylic acid (SA), and jasmonic acid (JA) can improve the tolerance of plants to heat stress [18,19,20]. Heat stress can lead to changes in plant secondary metabolites that act to reduce stress damage. The levels of tocopherol, flavonoids, phenylpropane and ascorbic acid precursors in the seeds of heat-resistant soybean genotypes were higher after heat stress [21]. The active flavonoid glycosides in Clematis lasiandra provide effective anti-tobacco mosaic virus (TMV) activity through the TMV-CP target [22]. Heat stress leads to upregulation of the oxylipin biosynthetic process and proline biosynthetic process in Agrostis stolonifera [23]. Activation of phenolic biosynthesis and inhibition of its oxidation promote the accumulation of phenols in plants, helping them to cope with heat shock stress [24].
Many transcription factors in plants are significantly upregulated or downregulated, and their expressions change the physiological and biochemical responses in plants to heat stress. The plant heat stress transcription factor family (HSFs) includes the most important transcription factors in plant responses to heat stress. They can induce the synthesis of heat shock proteins (HSPs) in plants to improve their heat tolerance [25,26,27]. AtHsfAs were the key transcription factors for Arabidopsis thaliana to increase its heat tolerance [28]. In the early phase of heat stress, AtHsfA1a and AtHsfA1b could upregulate HPSs and other related genes, and protect plants from cytotoxicity through the expression of related genes involved in osmotic regulation [29]. Silenced AtHsfA2 expression in Arabidopsis thaliana resulted the downregulation of the HSPs expression, and AtHsfA2 could protect plants against heat stress induced oxidative damage, organelle dysfunction and subsequent cell death [30].
The regulation mechanisms of transcription factors such as MADS, WRKY, MYB, bHLH, and other genes responding to heat stress signals have been studied [31,32,33]. OsMADS87 is a heat-sensitive gene regulating the seed size of rice, and it has the potential to improve rice heat tolerance [34]. AtWRKY39 could promote plant responses to heat stress through SA and JA mediated signaling pathways [35]. The Arabidopsis pif4 mutant was early flowering but did not show rapid extension of plant axes and leaf hyponasty under heat stress. This showed that PIF4 was an important component in heat stress response [36].
Transcriptome sequencing provides important information on gene expression patterns, functional genes, and regulatory mechanisms involved in plant abiotic stress [37, 38]. In OsMYB55 transgenic maize, a significant number of genes involved in responses to abiotic stresses, such as high temperature, dehydration, and oxidative stress, were upregulated [39]. Several potential genes associated with heat stress were isolated after transcriptional analysis of heat-sensitive and heat-resistant varieties of Chieh-qua. Among these, several genes of HSP, cytochrome P450, and bHLH transcription factor were specifically induced [40]. In the thermotolerant rice germplasm (SDWG005), a total of 3559 differentially expressed genes (DEGs) were identified by transcriptomic analysis. The agmatine-coumarin-acyltransferase gene OsACT in different germplasm may be involved in the heat resistance of SDWG005 [41].
Clematis lanuginose was introduced to Europe in the nineteenth century and is the parent of many early varieties of Clematis. Clematis crassifolia, which is an evergreen Clematis with many flowers in winter, is sensitive to high temperatures in summer. In this study we used transcriptome analysis to compare different heat stress periods on C. lanuginose and C. crassifolia. We compared and analyzed the DEGs under heat stress and observed response differences between C. lanuginose and C. crassifolia. while also screening potential heat stress response genes to cultivate Clematis varieties with strong heat resistance.
Results
Effect of heat stress on plant growth
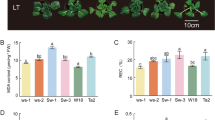
C. lanuginose had leaf tip wilting and withered new leaves 4 d after heat stress. C. crassifolia leaves turned yellow and wilted 2 d after heat stress (Fig. 1a). The net photosynthetic rate of C. lanuginose decreased by 51.21% at d 4 under heat stress, while the net photosynthetic rate of C. crassifolia decreased by 61.71% at d 1 and 90.87% at d 4 (Fig. 1b). The transpiration rate of C. lanuginose increased significantly after 2 d of heat stress, while the transpiration rate of C. crassifolia increased 265.14% at 1 d and decreased 94.25% at d 4 of heat stress (Fig. 1c).

Effect of heat stress on the morphology and photosynthetic parameters of C. lanuginose and C. crassifolia. a The phenotype of C. lanuginose and C. crassifolia leaves; b Net photosynthetic rate (Pn); c Transpiration rate (TR). Different letters indicate significant differences based on two-way ANOVA followed by Tukey’s multiple comparison (P ≤ 0.05)
Effect of heat stress on antioxidant enzyme activities and H2O2 level
The POD activity of C. crassifolia increased by 90.40% at d 1, and there was no significant difference in C. lanuginose during the heat stress (Fig. 2a). The SOD activity of C. lanuginose and C. crassifolia increased by 37.04% and 49.88% at d 1, respectively, and then gradually decreased (Fig. 2b). CAT activity of C. crassifolia was decreased by 29.87% at d 4 (Fig. 2c). The H2O2 levels of C. lanuginose and C. crassifolia increased by 30.63% and 109.62%, respectively, at d 4 (Fig. 2d).

Effect of heat stress on the POD, SOD, CAT activity and H2O2 levels of C. lanuginosa and C. crassifolia. a POD activity; b SOD activity; c CAT activity; d H2O2 level. Different letters indicate significant differences based on two-way ANOVA, followed by Tukey’s multiple comparison (P ≤ 0.05)
Transcriptome analyses of C. lanuginosa and C. crassifolia under heat stress conditions
The leaf tissues of C. lanuginosa and C. crassifolia were collected from HS d 0, HS d 1, HS d 2, and HS d 4, and from them we isolated total RNA and constructed eight cDNA libraries for transcriptome sequencing. The average number of clean reads was 70,390,475 and the average GC content was 46.76% in C. lanuginosa. In C. crassifolia, the average number of clean reads and GC content were 70,715,132 and 44.74%, respectively (Table 1). All the clean reads of cDNA libraries were assembled with the de-novo assembly method because Clematis does not have a reference genome sequence. A total of 540,495 transcripts and 395,844 unigenes were obtained after splicing (Table S1).
Annotation of the transcriptome
The gene function annotation results showed that a total of 89,933 genes were annotated to the NCBI non-redundant (NR) database, 35,990 genes were annotated to the Gene Ontology (GO) database, 5,098 genes were annotated to the Kyoto Encyclopedia of Genes and Genomes (KEGG) database, 81,459 genes were annotated to the evolutionary genealogy of genes: Non-supervised Orthologous Groups (eggNOG) database, and 75,064 genes were annotated to the Swiss-Prot database. Of these, 3,795 genes were annotated in all databases (Fig. 3a). According to the GO annotation analysis, there were 18,943, 17,841, 1,965, 1,227, and 304 genes with the molecular functions of catalytic activity, binding, transporter activity, structural molecule activity, and antioxidant activity, respectively (Fig. S1).

Annotation of the transcriptome. a Gene function annotations in five databases (NR, Swiss-Prot, GO, KEGG, eggNOG); b Homologous species distribution of the annotated in the NR database
According to sequence homology alignment results, 10,889 (12.11%) genes were homologous to Nelumbo nucifera; 8644 (9.61%) genes had significant hits for Vitis vinifera, followed by Hordeum vulgare subsp. vulgare (2481, 2.76%), Cajanus cajan (2055, 2.29%), Malus domestica (1699, 1.89%), Klebsormidium flaccidum (1636, 1.82%) and Beta vulgaris subsp. vulgaris (1585, 1.76%). A total of 60, 943 genes were homologous to other species (Fig. 3b).
Gene expression profiles of C. lanuginosa and C. crassifolia under heat stress
Many genes were differentially expressed in C. lanuginosa and C. crassifolia under heat stress. After 1, 2, and 4 d of heat stress treatment, 327, 536 and 209 genes were upregulated and 454, 255 and 500 genes were downregulated, respectively, in C. lanuginosa (Fig. 4a). In C. crassifolia, after 1 d of heat stress, 1727 genes were upregulated and 1469 genes were downregulated. After 2 and 4 d of heat stress, there were 810 and 1231 genes upregulated, 1586 and 1317 genes downregulated, respectively (Fig. 4b).

The number of up and down regulated differentially expressed genes (DEGs) and Venn diagram in C. lanuginosa and C. crassifolia. a The DEG number of C. lanuginosa; b The DEG number of C. crassifolia; c Venn diagrams for DEGs in the three comparison groups of C. lanuginosa; d Venn diagrams for DEGs in the three comparison groups of C. crassifolia; e Venn diagrams for DEGs between C. lanuginosa and C. crassifolia
Under the heat stress treatment, 94 genes were significantly differentially expressed at different times (HS d 1, HS d 2, and HS d 4) in C. lanuginosa, while 329 genes in C. crassifolia (Figs. 4c, d). Among all the differentially expressed genes, 421 (5.6%) genes were significantly differentially expressed in both C. lanuginosa and C. crassifolia, 1299 (17.4%) genes were specifically expressed in C. lanuginose and 5757 (77.0%) genes were uniquely expressed in C. crassifolia (Fig. 4e).
GO annotation of DEGs in C. lanuginosa and C. crassifolia under heat stress
Go enrichment analysis was performed on the DEGs of C. lanuginosa and C. crassifolia. In the comparison groups of HS d 1 vs. HS d 0, 314 (40.20%) and 809 (25.31%) DEGs were annotated into the GO terms in C. lanuginose and C. crassifolia, respectively. For HS d 2 vs. HS d 0 comparison groups, 173 (21.87%) and 628 (26.21%) DEGs were annotated in C. lanuginose and C. crassifolia, respectively. In the groups of HS d 4 vs. HS d 0, 320 (45.13%) and 625 (24.53%) DEGs in C. lanuginose and C. crassifolia were annotated, respectively (Fig. S2).
In C. lanuginosa, DEGs were mainly enriched in the terms involved in biological processes, such as single-organism process, oxidation–reduction process, and single-organism metabolic process. The oxidation reduction process, as well as the establishment of the localization and transport of DEGs, were observed in C. crassifolia (Fig. 5).

Enriched GO terms (Biological process) (P < 0.05) of DEGs. a C. lanuginosa; b C. crassifolia
For molecular functions, the genes were mostly enriched in oxidoreductase activity, tetrapyrrole binding, chlorophyll binding, and dioxygenase activity in C. lanuginosa. They were mainly enriched in oxidoreductase activity, catalytic activity, transferase activity, and kinase activity in C. crassifolia (Fig. 6). Within the cellular component category, the DEGs were commonly enriched in the terms of membrane, chloroplast, and plastid in C. lanuginosa. In C. crassifolia, DEGs were mainly concentrated in the membrane, intracellular membrane-bounded organelle, membrane-bound organelle, and mitochondrion (Fig. S3).

Enriched GO terms (Molecular function) (P < 0.05) of DEGs. a C. lanuginosa; b C. crassifolia
KEGG annotation and unique DEGs in C. lanuginosa and C. crassifolia under heat stress
In C. lanuginosa, all the DEGs of the three comparison groups HS d 1 vs. HS d 0, HS d 2 vs. HS d 0, and HS d 4 vs. HS d 0 were annotated into 57, 35, and 86 KEGG pathways, respectively, and more DEGs were significantly distributed in 23 pathways including carbon metabolism, carbon fixation in photosynthetic organisms, glyoxylate and dicarboxylate metabolism. Under the condition of heat stress, there were 116, 124 and 128 KEGG pathways annotated by DEGS in C. crassifolia, respectively, significantly focusing on 23 pathways, such as oxidative phosphorylation, photosynthesis, plant hormone signal transduction, and phenylpropanoid biosynthesis (Fig. 7). We analyzed all the DEGs in C. lanuginosa and C. crassifolia, and 34 unique genes related to heat stress were observed in C. lanuginosa (Table S2). A total of 29 DEGs related to heat stress were independently expressed in C. crassifolia (Table S3).

KEGG enrichment analysis of DEGs (P < 0:05) in C. lanuginosa and C. crassifolia. a C. lanuginosa; b C. crassifolia
Identification of the heat shock protein, antioxidant enzyme, photosynthetic-related genes and transcription factors in C. lanuginosa and C. crassifolia under heat stess
To evaluate the potential regulation in C. lanuginosa and C. crassifolia under heat stress, we searched for transcription factors, heat shock protein, antioxidant enzyme and photosynthetic genes from the RNA-seq dataset. There were eight heat shock proteins, nine antioxidant enzyme genes and four photosynthesis-related genes showing the same expression trend, while HSP80 (c196872_g2), PODP7 (c204229_g1) and PsbY (c200811_g3) showed different expression trends in C. lanuginosa and C. crassifolia (Fig. 8a). In 27 candidate transcription factors, nine transcription factors showed a significant increase or reduced expression, including ASIL2 (c195867_g1), bHLH112 (c203571_g1), JAZ1 (c194555_g1), MYBR1 (c204139_g1), MYC2 (c208293_g1), TCP15 (c195225_g2), WRKY40 (c189120_g1), WRKY41 (c200654_g1), and WRKY51 (c187717_g1). Four transcription factors including DREB2 (c182557_g1), PIF3 (c209598_g2), WRKY7 (c206794_g2), and WRKY72 (c203219_g1) showed opposite expression trends in the two Clematis species (Fig. 8b).

Expression of transcription factors (TFs), heat shock protein, antioxidant enzyme, and photosynthesis-related genes of HS d 4 vs. HS d 0 in C. lanuginosa and C. crassifolia by RNA-seq dataset. a Gene expression of heat shock proteins, antioxidant enzyme and photosynthetic-related genes; b Gene expression of TFs
We selected some DEGs annotated as HSPS, antioxidant enzymes and transcription factors for q-PCR analysis. The results showed that the expression of six HSPS genes were upregulated in C. lanuginosa and C. crassifolia, but the expression trends were different. HSP17.8 (c176964_g1), HSP26.5 (c200771_g1), HSP70 (c204924_g1), HSP18.1 (c199407_g2) and HSP20 (c201522_g2) were significantly upregulated from d 1 to the end of heat stress, and HSP17 (c192936_g1) and HSFA2 (c206233_g2) were significantly upregulated on d 1. DREB2 (c182557_g1), POD1 (c200317_g1) and POD3 (c210145_g2) were significantly upregulated at 4 d of heat stress. JAZ1 (c194555_g1) was significantly downregulated from d 1 to d 3, and upregulated at d 4 (Fig. 9).

Fold change of gene expression in C. crassifolia. Bars indicate SE (n = 3). Different letters indicate significant differences based on one-way ANOVA followed by Tukey’s multiple comparison (P ≤ 0.05)
In C. crassifolia, HSP17 (c192936_g1), HSP17.8 (c176964_g1), HSP26.5 (c200771_g1), HSFA2 (c206233_g2), DREB2 (c182557_g1) all increased significantly after 2 d of heat stress, and HSP70 (c204924_g1), HSP18.1 (c199407_g2), HSP20 (c201522_g2) were significantly upregulated on the first day. POD1 (c200317_g1) and POD3 (c210145_g2) were significantly upregulated under heat stress (Fig. 10).

Fold change of gene expression in C. crassifolia. Bars indicate SE (n = 3). Different letters indicate significant differences based on one-way ANOVA followed by Tukey’s multiple comparison (P ≤ 0.05)
Discussion
C. lanuginosa and C. crassifolia showed distinct leaf damage phenotypes under heat stress (Fig. 1). Transcriptome data analysis indicated 1720 differentially expressed genes in C. lanuginosa, of which 1299 (75.52%) were specifically and significantly expressed in the leaf. There were 6178 DEGs, among which 5757 (93.19%) genes were significantly expressed only in C. crassifolia. These results indicate that the gene expression in C. crassifolia was more sensitive to heat stress (Fig. 4).
Photosynthesis is a plant physiological process sensitive to heat. Heat stress has negative effects on photosynthesis by destroying the electron transport chain, carbon metabolism and the Photosystem II (PSII) system oxygen releasing complex [42, 43]. Analysis of the net photosynthetic rate of C. lanuginosa and C. crassifolia showed that the net photosynthetic rate of C. crassifolia continued to decrease significantly under heat stress (Fig. 1b). During the initial stage, 2 d after heat stress in the GO enrichment analysis, photosynthesis light harvesting and the light reaction process were inhibited in C. lanuginosa and C. crassifolia. In the later stage of heat stress, the plants produced precursor metabolites and energy to regulate light capture and other functional genes and maintain photosynthesis (Figs. 5 and 6). Photosystem I (PSI) is a multiprotein complex composed of two large subunits of 82 kD and several small subunits of less than 20 kD in the plant chloroplast thylakoid membrane, which mediates the light-driven electron transfer from plastocyanin to Fd [44]. The genes encoding these subunits include PsaA—PsaL [45]. In the later period of heat stress, PsaH, PsaK, PsbY, PSI—F, and PSI—N genes were downregulated more in C. crassifolia, indicating that the inhibitory effect of heat stress on PSI in C. crassifolia was greater than that in C. lanuginose (Fig. 8).
Plants will accumulate ROS such as 1O2, O2−, H2O2, and OH− under heat stress, which will impair chloroplast and mitochondrial functions, subject the plant cells to oxidative damage, including lipid peroxidation, protein oxidation, and DNA damage. H2O2 is one of ROSs which is closely related to oxidative stress. It is derived from superoxide anion disproportionation, and the product of H2O2 has strong oxidation ability [46, 47]. During evolution, plants have developed an enzymatic antioxidant system to remove excess ROS, which is dominated by ascorbate peroxidase (APX), SOD, POD, and CAT. The antioxidant enzyme activities are positively correlated with the heat tolerance of plants [48,49,50]. SlMAPK3 was a negative regulator of thermotolerance in Solanum lycopersicum, slmapk3 mutants have higher activities and transcript levels of POD, SOD, CAT, and APX than wild type plants [51]. There was no significant change in the content of H2O2 in C. lanuginosa, which might be related to the maintenance of antioxidant enzyme activity. In C. crassifolia, the increase of SOD and POD activities on d 1 and d 2 maintained ROS balance at the early stage of heat stress (Fig. 2). Gene enrichment in the oxidation reduction process improved the ability of C. lanuginosa and C. crassifolia to eliminate reactive oxygen species so that the plants could maintain the short-term balance of ROS. The biological processes related to establishment of localization, transport, and oxidation reduction process were significantly enriched in C. crassifolia at d 1 under heat stress (Figs. 5 and 6). However, the activities of CAT, POD, and SOD of C. crassifolia relatively decreased after 4 d, and the H2O2 content increased significantly at d 4, indicating that the protective enzyme system had a strong time dependence in response to heat stress in C. crassifolia (Fig. 2d).
Flavonoids have a variety of biological functions including antioxidant, antiviral, auxin transport, and antimicrobial [52, 53]. The increase in flavonoid substances in some plant species can enhance their ability to resist biological and abiotic stress [54]. Heat stress reduced the fertilization success of Ipomoea purpurea, and flavonoids could ameliorate the adverse effects of heat stress on fertilization and early seed maturation [55]. In C. lasiandra, three flavonoids including kaempferol 3-O-α-L-rhamnopyranoside, 34isovitexin 6''-O-E-ρ-coumarate, and 35quercetin 3-O-β-D-glucopyranuronide showed higher anti-TMV active compared with ningnanmycin, especially 34isovitexin 6''-O-E-ρ-coumarate could directly fracture TMV particles into small fragments combining with the fusion phenomena. The DEGs of C. lanuginosa and C. crassifolia were annotated in the KEGG classification of flavone and flavonol biosynthesis and flavonoid biosynthesis, suggesting that flavonoids played important role in the response of heat stress (Fig. 7).
Amino acids are involved in protein synthesis. Plants can promote the synthesis of proteins involved in photosynthesis, enzymatic antioxidant system and stress signals by accumulating amino acids under heat stress. These can also protect the lipids in thylakoid membranes from damage [56, 57]. The accumulation of glyoxylic acid can affect the expression of modified proteins or stress-related genes in plants under high light and heat stress [58]. Thiamine plays an important role in metabolic pathways such as glycolysis, nicotinamide adenine dinucleotide phosphate (NADPH) and adenosine-triphosphate (ATP) synthesis, and is activated as an enzyme cofactor in plants responding to abiotic stress [23, 59, 60]. In the KEGG analysis of C. lanuginosa, genes related to glycine, serine, and threonine metabolism, glyoxylate and dicarboxylate metabolism, protein processing in the endoplasmic reticulum and thiamine metabolism classificantions were expressed differentially compared to C. crassifolia (Fig. 7a). This indicates that the accumulation of amino acids and the activation of thiamine metabolic pathways may be important in the response of C. lanuginosa to heat stress. A previous study confirmed that heat stress can promote the accumulation of arginine and proline content in C. crassifolia [61]. The differentially expressed genes in C. crassifolia were specifically upregulated in phenylalanine metabolism, arginine/proline metabolism and the flavonoid biosynthesis classificantions. These may be important pathways for C. crassifolia to respond to heat stress (Fig. 7b). Heat shock proteins are highly conserved proteins in plants that have anti-stress effects. Based on their molecular size, they are mainly classified into HSP110, HSP90, HSP70, HSP60 and sHSPs [62, 63]. Under heat stress, heat shock proteins can bind to other proteins as molecular chaperones to maintain protein homeostasis, repair denatured proteins, and assist in protein transport. After heat stress treatment, the significantly upregulated heat shock proteins in C. lanuginosa and C. crassifolia were mainly concentrated in sHSPs, including the six genes HSP17, HSP17.8, HSP18.1, HSP20, HSP26.5, and HSP70 (Figs. 8a, 9 and 10). This finding was similar to the upregulation of small heat shock proteins in the transcriptomes of C. apiifolia under heat stress [64]. The chaperone activity of small heat shock proteins involves passively mediating the synthesis and release of substrates without using ATP [65]. Also, the small heat shock proteins have a cross protection function and can adjust the membrane fluidity, interact with thylakoid membrane and reduce plasma membrane fluidity to maintain cell homeostasis [66, 67]. In the annotation analysis of DEGs in C. lanuginosa and C. crassifolia, many DEGs were involved in the composition of the cell membrane, chloroplast thylakoid membrane, thylakoid membrane and plastid thylakoid membrane. These results indicated that the upregulated expression of these six sHSPs genes may protect the stability of the Clematis membranous system.
Transcription factors are important in the signal transduction process of plants in response to stress [68]. The increase or decrease in the expression of transcription factors can regulate downstream gene expression while also transmitting and amplifying the stress signals. HSF, WRKY, MADS, bZIP, MYB, bHLH, AP2/EREBP, NAC, and other transcription factors genes are closely related to plant abiotic stress responses [69, 70]. In Arabidopsis, hsfa2 mutants were more sensitive to heat stress, and silencing AtHsfA2 resulted in downregulation of HSPs gene expression [71, 72]. Similarly, we observed that HSFA2 (c206233_g2) was significantly upregulated in the early and late stages of heat stress in C. crassifolia (Figs. 6b and 10), suggesting that HSFA2 may play an important role in the heat response of C. crassifolia. CvHSF30-2 was a transcription factor induced by heat stress in Clematis vitalba, which improved the heat tolerance of C. vitalba by increasing the expression of HSPs [73]. Upregulation of HSF30 (c194517_g1) was also observed in C. lanuginosa and C. crassifolia (Fig. 8; Fig. S5). JAZ is a negative regulator of the jasmonic acid (JA) signal response pathway. Exogenous application of meJA can enhance the heat tolerance of wheat, while HSFA1b can regulate the expression of the JA synthesis gene AtOPR3 in Arabidopsis thaliana by combining heat shock elements (HSE) [74]. JAZ1 (c194555_g1) was significantly downregulated during heat stress in C. lanuginosa and C. crassifolia (Figs. 8b, 9 and 10), suggesting that the JAZ1 transcription factor may regulate jasmonic acid signaling pathways to adapt to heat stress. DREB2 plays an important role in heat stress, and its over-expression can improve plant heat tolerance [75]. WRKY72 in rice can be upregulated by heat stress, and may be involved in a variety of plant biological processes [76]. Soybean studies indicate that PIF3 may be a potential target gene for regulating weed tolerance in soybean [77]. The c182557_g1 gene was annotated as encoding DREB2. The c182557_g1 was upregulated in C. lanuginosa and C. crassifolia, but its expression patterns were different (Fig. 9; Fig. 10). PIF3 (c209598_g2) and WRKY72 (c203219_g1) were upregulated in C. lanuginosa and C. crassifolia, respectively (Fig. 8b). However, the regulatory roles of these genes in response to heat stress in Clematis remains to be verified. This study provides a reference for further analyzing the molecular regulatory mechanism of Clematis in response to heat stress and the breeding of Clematis cultivars with increased heat tolerance.
Conclusions
In this study, the transcriptomes of C. lanuginosa and C. crassifolia were assembled. A total of 1,720 and 6,178 DEGs were identified from C. lanuginosa and C. crassifolia, respectively. DEGs enrichment of metabolic pathways and gene expression analysis showed that glycine/serine/threonine metabolism, glyoxylic metabolism and thiamine metabolism were important pathways in the response to heat stress in C. lanuginosa. Flavonoid metabolism, phenylalanine metabolism, and arginine/proline metabolism were the key pathways in C. crassifolia. Several candidate genes that may be involved in the response of C. lanuginosa and C. crassifolia to heat stress were identified, and these indicated that C. lanuginosa and C. crassifolia have different response strategies to heat stress.
Methods
Plant materials and growth conditions
The Clematis lanuginosa Lindl. and Clematis crassifolia Benth. used in this study were provided by the Zhejiang Institute of Subtropical Crops, Wenzhou, Zhejiang Province, China. It was identified by Professor Jian Zheng. These species were preserved in National Clematis Germplasm Resource Center, Wenzhou, Zhejiang Province, China (C. lanuginose voucher code: W-2016–43; C. crassifolia voucher code: W-2016–58). C. lanuginosa and C. crassifolia plants were grown in the Zhejiang Institute of Subtropical Crops, China. Healthy, two-year-old plants were grown in a grown chamber under 25/20 °C (16:8 h (L:D) photoperiod); 65% humidity) conditions for two weeks. After two weeks of pretreatment, C. lanuginosa and C. crassifolia plants were transferred to a growth chamber for cultivation at 45/40 °C temperature and 16:8 h (L:D) photoperiod. The heat stress treatment duration was 4 d. During the treatment period, 500 ml of water was given to each plant every 2 d, to ensure sufficient soil moisture. The leaves sampled before the heat stress were labelled as “HS 0d,” and those after heat stress were labelled as HS 1d, HS 2d, and HS 4d. Experimental treatments were repeated three times. All methods, including plant experimental research, were in compliance with the relevant guidelines, regulations and legislation.
Leaf gas exchange parameters
Healthy and fully developed leaves were randomly chosen for photosynthetic parameter measurements, using LI-6400 XT portable photosynthesis system (Li-Cor Inc., Lincoln, NE, USA), and equipped with a 6400–18 RGB LED light source. The measurements were carried out from 9:00 to 11:00 am, the photosynthetic photon flux density was 1200 μmol m−2 s−1, the CO2 concentration was 400 ppm, and the relative humidity was 65%.
Determination of Superoxide Dismutase (SOD), Catalase (CAT), Peroxidase (POD) activity and H2O2 content
For peroxidase enzyme activity analysis, fresh leaves (0.1 g) were ground in liquid nitrogen and suspended in 8.0 ml solution containing 50 mM phosphate buffer (pH 7.4). The homogenate was centrifuged 15 min (10,000 rpm) at 4 °C, and then the supernatant was collected to obtain crude enzymes.
SOD activity was analyzed by measuring the inhibition rate of the enzyme to O2− produced. SOD activity was determined at 550 nm in absorbance after 40 min of reaction at 37 °C. One-unit of SOD activity (U) was defined as the amount of enzyme that resulted in 50% inhibition of reduction of nitrite in 1 ml of reaction solution.
CAT activity was determined by the hydrolysis reaction of H2O2 with CAT, and the yellow MA-H2O2 complex was generated by adding ammonium molybdate to quickly stop the reaction. CAT activity was calculated at 405 nm. One-unit was defined as the amount of enzyme that resulted in the decomposition of 1 µmol H2O2 per second in 1.0 g fresh tissue.
POD activity was measured at 470 nm by catalyzing H2O2 based on the change of absorbance. One-unit was defined as the amount of enzyme that resulted in the change of 0.01 at 470 nm per minute by 1.0 g fresh tissues in the reaction system. Leaf tissue amounting to 0.2 g was finely ground with 25 mL acetone and homogenized at 0 °C; the content was calculated using H2O2 as the standard [78].
The H2O2 content was measured according to the method described by Patterson [79]. A 0.2 g amount of leaf tissue was finely ground finely homogenized with 25 ml of acetone at 0 °C. The H2O2 content was calculated using H2O2 as the standard.
RNA extraction, cDNA library construction, and Illumina sequencing
Total RNA from different leaf samples of C. lanuginosa and C. crassifolia was extracted using TRIZOL reagent (Takara, Beijing, China). The concentration and purity of the total RNA was tested with an Agilent 2100 Bioanalyzer. The mRNA from total RNA was purified by the polyA structure unique to mRNA and mRNA with the polyA structure was enriched by Oligo(dT) magnetic beads. The first strand of cDNA was synthesized using 6-base random primers and reverse transcriptase using mRNA as a template. The second strand of cDNA was synthesized using the first strand of cDNA as template. The chain-specific library was established, and the quality of the library was detected by Agilent 2100 Bioanalyzer. Next Generation Sequencing (NGS) was used to perform paired-end (PE) sequencing (Illumina HiSeq X-Ten, San Diego, CA, USA; Sequencing company: Personalbio, HangZhou, China).
There was no reference genome in the transcriptome sequencing of Clematis, so Trinity software (r20140717) was used to splice clean reads to obtain the transcript for subsequent analysis. Trinity is a De Novo assembly software for transcriptome splicing, splicing high-quality sequences based on the DBG (De Bruijn Graph) splicing principle [80]. The longest Transcript under each gene was extracted as the representative sequence of the gene and the transcript and unigene sequences were statistically analyzed.
Gene function annotation
NR, Swiss-Prot, eggNOG and KEGG databases (www.kegg.jp/kegg/kegg1.html) were used to annotate all the unigenes (E value < 1.0 e−5) [81,82,83]. GO annotation was performed through Blast2GO based on NR annotation results. Based on the above comparison results, protein functional annotation information of unigenes was obtained.
Differentially expressed genes
Gene expression were analyzed using the FPKM (Fragments Per Kilobase of exon model per Million mapped reads) method. The criteria for screening DEG were p-value ≤ 0.05, false discovery rate (FDR) < 0.001, Fold Change ≥ 2, or Fold Change ≤ 0.5. Subsequently, GO and KEGG databases were used to analyze the main functions and metabolic pathways of the DEGs.
Quantitative real-time PCR of genes in different time under heat stress
Total RNA was extracted from leaves and the cDNA was synthesized using the Revert Aid RT Kit (Thermo Scientific, Waltham, MA, USA), Primers designed with Primer Premier 5.0 are shown in Table S5. The qRT-PCR experiment was carried out using an ABI PRISM 7500 Real-time PCR System (Applied Biosystems, Foster City, CA, USA) and AceQ qPCR SYBR Green Master Mix (Vazyme, Nanjing, Jiangsu Province, China). The PCR-PCR reaction system was as follows: 95 °C for 5 min, followed by 40 cycles of 95 °C for 15 s, and 60 °C for 30 s. Each sample was repeated three times, and the internal normalizations was GAPDH gene. Each primer pair was validated the specificity by melt curve analysis, and the gene expression levels were calculated by the 2−△△Ct method.
Statistical analysis
Data were analyzed by one-way or two-way ANOVA using the SPSS 10 program (SPSS Inc., Chicago, IL, USA). Different letters on the histograms between different treatments indicate a significant difference at P ≤ 0.05.
Availability of data and materials
The raw transcriptome data have been deposited at the NCBI Sequence Read Archive with accession number PRJNA702123 (https://www.ncbi.nlm.nih.gov/sra/PRJNA702123) and PRJNA751406 (https://www.ncbi.nlm.nih.gov/sra/PRJNA751406), respectively.
Change history
05 April 2022
A Correction to this paper has been published: https://doi.org/10.1186/s12870-022-03566-0
Abbreviations
- ABA:
-
Abscisic acid
- APX:
-
Ascorbate peroxidase
- ATP:
-
Adenosine-triphosphate
- CAT:
-
Catalase
- DBG:
-
De Bruijn Graph
- DEG:
-
Differentially expressed gene
- eggNOG:
-
Evolutionary genealogy of genes: non-supervised orthologous groups
- FPKM:
-
Fragments per kilobase of exon model per million mapped reads
- GO:
-
Gene ontology
- H2O2 :
-
Hydrogen peroxide
- HSF:
-
Heat stress transcription factor
- HSP:
-
Heat shock protein
- JA:
-
Jasmonic acid
- KEGG:
-
Kyoto encyclopedia of genes and genomes
- NADPH:
-
Nicotinamide adenine dinucleotide phosphate
- NGS:
-
Next generation sequencing
- NR:
-
NCBI non-redundant
- Pn:
-
Net photosynthetic rate
- POD:
-
Peroxidase
- PSI:
-
Photosystem I
- PSII:
-
Photosystem II
- ROS:
-
Reactive oxygen species
- SA:
-
Salicylic acid
- SOD:
-
Superoxide dismutase
- TMV:
-
Anti-tobacco mosaic virus
- TR:
-
Transpiration rate
References
Liu Z, Shao W, Shen Y, Ji M, Chen W, Ye Y, Shen Y. Characterization of new microsatellite markers based on the transcriptome sequencing of Clematis finetiana. Hereditas. 2018;155(1):1–9.
Ma X, Qian R, Zhang X, Hu Q, Liu H, Zheng J. Contrasting growth, physiological and gene expression responses of Clematis crassifolia and Clematis cadmia to different irradiance conditions. Sci Rep. 2019;9(1):1–12.
Chawla R, Kumar S, Sharma A. The genus Clematis (Ranunculaceae): Chemical and pharmacological perspectives[J]. J Ethnopharmacol. 2012;143(1):116–50.
Mimaki Y, Yokosuka A, Hamanaka M, Sakuma C, Yamori T, Sashida Y. Triterpene Saponins from the Roots of Clematis chinensis. J Nat Prod. 2004;67(9):1511–6.
Pan LL, Wang XL, Zhang QY, Luo XL, Xu P, Liu SY, Liu XH. Boehmenan, a lignan from the Chinese medicinal plant Clematis armandii, induces apoptosis in lung cancer cells through modulation of EGF-dependent pathways. Phytomedicine. 2016;23(5):468–76.
Chang Y, Zhang P, Zhang X, Chen J, Rausch WD, Gula A, Bao B. Cytotoxic activities of flavonoids from a traditional Mongolian medicinal herb Clematis aethusifolia Turcz. Nat Prod Res. 2017;31(10):1223–7.
Tian X, Li Y, Hao N, Su X, Du J, Hu J, Tian X. The antifeedant, insecticidal and insect growth inhibitory activities of triterpenoid saponins from Clematis aethusifolia Turcz against Plutella xylostella (L.). Pest Manag Sci. 2021;77(1):455–63.
Crafts-Brandner SJ, Salvucci ME. Sensitivity of photosynthesis in a C4 plant, maize, to heat stress. Plant Physiol. 2002;129(4):1773–80.
Hasanuzzaman M, Nahar K, Alam M, Roychowdhury R, Fujita M. Physiological, biochemical, and molecular mechanisms of heat stress tolerance in plants. Int J Mol Sci. 2013;14(5):9643–84.
Schöffl F, Prandl R, Reindl A. Molecular responses to heat stress. Molecular responses to cold, drought, heat and salt stress in higher plants. 1999;83:93.
Chauhan H, Khurana N, Nijhavan A, Khurana JP, Khurana P. The wheat chloroplastic small heat shock protein (sHSP26) is involved in seed maturation and germination and imparts tolerance to heat stress. Plant Cell Environ. 2012;35(11):1912–31.
Jagadish SK. Heat stress during flowering in cereals–effects and adaptation strategies. New Phytol. 2020;226(6):1567–72.
Hu S, Ding Y, Zhu C. Sensitivity and responses of chloroplasts to heat stress in plants. Front Plant Sci. 2020;11:375.
Hemantaranjan A, Bhanu AN, Singh MN, Yadav DK, Patel PK, Singh R, Katiyar D. Heat stress responses and thermotolerance. Adv Plants Agric Res. 2014;1(3):1–10.
Gill SS, Tuteja N. Reactive oxygen species and antioxidant machinery in abiotic stress tolerance in crop plants. Plant Physiol Biochem. 2010;48(12):909–30.
Ahmad P, Jaleel CA, Salem MA, Nabi G, Sharma S. Roles of enzymatic and nonenzymatic antioxidants in plants during abiotic stress. Crit Rev Biotechnol. 2010;30(3):161–75.
Zandalinas SI, Balfagón D, Arbona V, Gómez-Cadenas A. Modulation of antioxidant defense system is associated with combined drought and heat stress tolerance in citrus. Front Plant Sci. 2017;8:953.
Ahammed GJ, Li X, Zhou J, Zhou YH, Yu JQ. Role of hormones in plant adaptation to heat stress. Plant hormones under challenging environmental factors. 2016. Springer, Dordrecht, 1–21.
Balfagón D, Sengupta S, Gómez-Cadenas A, Fritschi FB, Azad RK, Mittler R, Zandalinas SI. Jasmonic acid is required for plant acclimation to a combination of high light and heat stress. Plant Physiol. 2019;181(4):1668–82.
Rai KK, Pandey N, Rai SP. Salicylic acid and nitric oxide signaling in plant heat stress. Physiol Plant. 2020;168(2):241–55.
Chebrolu KK, Fritschi FB, Ye S, Krishnan HB, Smith JR, Gillman JD. Impact of heat stress during seed development on soybean seed metabolome. Metabolomics. 2016;12(2):28.
Li Y, Ye S, Hu Z, Hao N, Bo X, Liang H, Tian X. Identification of anti‐TMV active flavonoid glycosides and their mode of action on virus particles from Clematis lasiandra Maxim. Pest Manag Sci. 2021;7(11):5268–77.
Xu Y, Huang B. Comparative transcriptomic analysis reveals common molecular factors responsive to heat and drought stress in Agrostis stolonifera. Sci Rep. 2018;8(1):1–12.
Rivero RM, Ruiz JM, Garcıa PC, Lopez-Lefebre LR, Sánchez E, Romero L. Resistance to cold and heat stress: accumulation of phenolic compounds in tomato and watermelon plants. Plant Sci. 2001;160(2):315–21.
Scharf KD, Berberich T, Ebersberge I, Nover L. The plant heat stress transcription factor (Hsf) family: structure, function and evolution. Biochim Biophys Acta (BBA)-Gene Regul Mech. 2012;1819(2):104–19.
Nover L, Scharf KD, Gagliardi D, Vergne P, Czarnecka-Verner E, Gurley WB. The Hsf world: classification and properties of plant heat stress transcription factors. Cell Stress Chaperones. 1996;1(4):215.
Kumsta C, Chang JT, Schmalz J, Hansen M. Hormetic heat stress and HSF-1 induce autophagy to improve survival and proteostasis in C. elegans. Nat Commun. 2017;8(1):1–12.
Tiwari LD, Khungar L, Grover A. AtHsc70-1 negatively regulates the basal heat tolerance in Arabidopsis thaliana through affecting the activity of HsfAs and Hsp101. Plant J. 2020;103(6):2069–83.
Busch W, Wunderlich M, Schöffl F. Identification of novel heat shock factor-dependent genes and biochemical pathways in Arabidopsis thaliana. Plant J. 2005;41(1):1–14.
Zhang L, Li Y, Xing D, Gao C. Characterization of mitochondrial dynamics and subcellular localization of ROS reveal that HsfA2 alleviates oxidative damage caused by heat stress in Arabidopsis. J Exp Bot. 2009;60(7):2073–91.
Ohama N, Sato H, Shinozaki K, Yamaguchi-Shinozaki K. Transcriptional regulatory network of plant heat stress response. Trends Plant Sci. 2017;22(1):53–65.
Jacob P, Brisou G, Dalmais M, Thévenin J, van der Wal F, Latrasse D, Bendahmane A. The Seed Development Factors TT2 and MYB5 Regulate Heat Stress Response in Arabidopsis. Genes. 2021;12(5):746.
Wang Z, Shen Y, Yang X, Pan Q, Ma G, Bao M, Ning G. Overexpression of particular MADS-box transcription factors in heat-stressed plants induces chloroplast biogenesis in petals. Plant, Cell Environ. 2019;42(5):1545–60.
Chen C, Begcy K, Liu K, Folsom JJ, Wang Z, Zhang C, Walia H. Heat stress yields a unique MADS box transcription factor in determining seed size and thermal sensitivity. Plant Physiol. 2016;171(1):606–22.
Li S, Zhou X, Chen L, Huang W, Yu D. Functional characterization of Arabidopsis thaliana WRKY39 in heat stress. Mol Cells. 2010;29(5):475–83.
Koini MA, Alvey L, Allen T, Tilley CA, Harberd NP, Whitelam GC, Franklin KA. High temperature-mediated adaptations in plant architecture require the bHLH transcription factor PIF4. Curr Biol. 2009;19(5):408–13.
Fragkostefanakis S, Simm S, Paul P, Bublak D, SCHARF KD, Schleiff E. Chaperone network composition in Solanum lycopersicum explored by transcriptome profiling and microarray meta‐analysis. Plant Cell Environ. 2015;38(4):693–709.
Xu Y, Magwanga RO, Cai X, Zhou Z, Wang X, Wang Y, Liu F. Deep transcriptome analysis reveals reactive oxygen species (ROS) network evolution, response to abiotic stress, and regulation of fiber development in cotton. Int J Mol Sci. 2019;20(8):1863.
Casaretto JA, El-Kereamy A, Zeng B, Stiegelmeyer SM, Chen X, Bi YM, Rothstein SJ. Expression of OsMYB55 in maize activates stress-responsive genes and enhances heat and drought tolerance. BMC Genom. 2016;17(1):1–15.
Wang M, iang B, Liu W, Lin YE, Liang Z, He X, Peng Q. Transcriptome analyses provide novel Insights into heat stress responses in Chieh-Qua (Benincasa hispida Cogn. var. Chieh-Qua How). Int J Mol Sci. 2019;20(4):883.
Liu G, Zha Z, Cai H, Qin D, Jia H, Liu C, Jiao C. Dynamic transcriptome analysis of anther response to heat stress during anthesis in thermotolerant rice (Oryza sativa L.). Int J Mol Sci. 2020;21(3):1155.
Allakhverdiev SI, Kreslavski VD, Klimov VV, Los DA, Carpentier R, Mohanty P. Heat stress: an overview of molecular responses in photosynthesis. Photosynth Res. 2008;98(1):541–50.
Wingler A, Lea PJ, Quick WP, Leegood RC. Photorespiration: metabolic pathways and their role in stress protection. Philos Trans R Soc Lond B Biol Sci. 2000;355(1402):1517–29.
Havaux M. Short-term responses of photosystem I to heat stress. Photosynth Res. 1996;47(1):85–97.
Obokata J, Mikami K, Hayashida, Nakamura M, Sugiura M. Molecular heterogeneity of photosystem I (psaD, psaE, psaF, psaH, and psaL are all present in isoforms in Nicotiana spp.). Plant Physiol. 1993;102(4):1259–67.
Kissen R, Øverby A, Winge P, Bones AM. Allyl-isothiocyanate treatment induces a complex transcriptional reprogramming including heat stress, oxidative stress and plant defence responses in Arabidopsis thaliana. BMC Genom. 2016;17(1):1–26.
Yin Y, Qin K, Song X, Zhang Q, Zhou Y, Xia X, Yu J. BZR1 transcription factor regulates heat stress tolerance through FERONIA receptor-like kinase-mediated reactive oxygen species signaling in tomato. Plant Cell Physiol. 2018;59(11):2239–54.
Prerostova S, Dobrev PI, Kramna B, Gaudinova A, Knirsch V, Spichal L, Vankova R. Heat acclimation and inhibition of cytokinin degradation positively affect heat stress tolerance of Arabidopsis. Front Plant Sci. 2020;11:87.
Medina E, Kim SH, Yun M, Choi WG. Recapitulation of the function and role of ROS generated in response to heat stress in plants. Plants. 2021;10(2):371.
Panchuk II, Volkov RA, Schöffl F. Heat stress-and heat shock transcription factor-dependent expression and activity of ascorbate peroxidase in Arabidopsis. Plant Physiol. 2002;129(2):833–53.
Yu W, Wang L, Zhao R, et al. Knockout of SlMAPK3 enhances tolerance to heat stress involving ROS homeostasis in tomato plants. BMC Plant Biol. 2019;19:354.
Hernández MM, Pesquera-Alegría C, Manso-Martínez C, Menéndez CM. Antioxidant capacity and flavanol composition of seed extracts from a Grenache× Tempranillo population: Effect of sex and color. Ind Crops and Prod. 2021;161.
Yarmolinsky L, Huleihel M, Zaccai M, Ben-Shabat S. Potent antiviral flavone glycosides from Ficus benjamina leaves. Fitoterapia. 2012;83(2):362–7.
Khan AL, Kang SM, Dhakal KH, Hussain J, Adnan M, Kim JG, Lee IJ. Flavonoids and amino acid regulation in Capsicum annuum L. by endophytic fungi under different heat stress regimes. Sci Hortic 2013, 155, 1–7.
Coberly LC, Rausher MD. Analysis of a chalcone synthase mutant in Ipomoea purpurea reveals a novel function for flavonoids: amelioration of heat stress. Mol Ecol. 2003;12(5):1113–24.
Wang GP, Li F, Zhang J, Zhao MR, Hui Z, Wang W. Overaccumulation of glycine betaine enhances tolerance of the photosynthetic apparatus to drought and heat stress in wheat. Photosynthetica. 2010;48(1):30–41.
Simon-Sarkadi L, Kocsy G, Várhegyi Á, Galiba G, de Ronde JA. Genetic manipulation of proline accumulation influences the concentrations of other amino acids in soybean subjected to simultaneous drought and heat stress. J Agric Food Chem. 2005;53(19):7512–7.
Suzuki N, Mittler R. Reactive oxygen species and temperature stresses: a delicate balance between signaling and destruction. Physiol Plant. 2006;126(1):45–51.
Rapala-Kozik M, Wolak N, Kujda M, Banas AK. The upregulation of thiamine (vitamin B 1) biosynthesis in Arabidopsis thaliana seedlings under salt and osmotic stress conditions is mediated by abscisic acid at the early stages of this stress response. BMC Plant Biol. 2012;12(1):1–14.
Valifard M, Mohsenzadeh S, Niazi A, Moghadam A. Phenylalanine ammonia lyase isolation and functional analysis of phenylpropanoid pathway under salinity stress in “Salvia” species. Aust J Crop Sci. 2015;9(7):656–65.
Hu Q, Qian R, Zhang Y, Zhang X, Ma X, Zheng J. Physiological and Gene Expression Changes of Clematis crassifolia and Clematis cadmia in Response to Heat Stress. Front Plant Sci. 2021;12:421.
Vierling E. The roles of heat shock proteins in plants. Annu Rev Plant Biol. 1991;42(1):579–620.
Waters ER, Vierling E. Plant small heat shock proteins–evolutionary and functional diversity. New Phytol. 2020;227(1):24–37.
Gao L, Ma Y, Wang P, Wang SA, Yang R, Wang Q, Li Y. Transcriptome profiling of Clematis apiifolia: insights into heat-stress responses. DNA Cell Biol. 2017;36(11):938–46.
Lee GJ, Vierling E. A small heat shock protein cooperates with heat shock protein 70 systems to reactivate a heat-denatured protein. Plant Physiol. 2000;122(1):189–98.
Arena MP, Capozzi V, Longo A, Russo P, Weidmann S, Rieu A, Fiocco D. The phenotypic analysis of Lactobacillus plantarum shsp mutants reveals a potential role for hsp1 in cryotolerance. Front Microbiol. 2019;10:838.
Rütgers M, Muranaka LS, Schulz-Raffelt M, Thoms S, Schurig J, Willmund F, Schroda M. Not changes in membrane fluidity but proteotoxic stress triggers heat shock protein expression in Chlamydomonas reinhardtii. Plant, Cell Environ. 2017;40(12):2987–3001.
Khan SA, Li MZ, Wang SM, Yin HJ. Revisiting the role of plant transcription factors in the battle against abiotic stress. Int J Mol Sci. 2018;19(6):1634.
Liu XH, Lyu YS, Yang W, Yang ZT, Lu SJ, Liu JX. A membrane-associated NAC transcription factor OsNTL3 is involved in thermotolerance in rice. Plant Biotechnol J. 2020;18(5):1317–29.
Zhang X, Wollenweber B, Jiang D, Liu F, Zhao J. Water deficits and heat shock effects on photosynthesis of a transgenic Arabidopsis thaliana constitutively expressing ABP9, a bZIP transcription factor. J Exp Bot. 2008;59(4):839–48.
Charng YY, Liu HC, Liu NY, Hsu FC, Ko SS. Arabidopsis Hsa32, a novel heat shock protein, is essential for acquired thermotolerance during long recovery after acclimation. Plant Physiol. 2006;140(4):1297–305.
Schramm F, Ganguli A, Kiehlmann E, Englich G, Walch D, Von Koskull-Döring P. The heat stress transcription factor HsfA2 serves as a regulatory amplifier of a subset of genes in the heat stress response in Arabidopsis. Plant Mol Biol. 2006;60(5):759–72.
Wang R, Mao C, Jiang C, Zhang L, Peng S, Zhang Y, Ming F. One Heat Shock Transcription Factor Confers High Thermal Tolerance in Clematis Plants. Int J Mol Sci. 2021;22(6):2900.
Tian X, Wang F, Zhao Y, Lan T, Yu K, Zhang L, Xin M. Heat shock transcription factor A1b regulates heat tolerance in wheat and Arabidopsis through OPR3 and jasmonate signalling pathway. Plant Biotechnol J. 2020;18(5):1109.
Wu Z, Liang J, Zhang S, Zhang B, Zhao Q, Li G, Yi M. A canonical DREB2-type transcription factor in lily is post-translationally regulated and mediates heat stress response. Front Plant Sci. 2018;9:243.
Song Y, Chen L, Zhang L, Yu D. Overexpression of OsWRKY72 gene interferes in the abscisic acid signal and auxin transport pathway of Arabidopsis. J Biosci. 2010;35(3):459–71.
Horvath DP, Hansen SA, Moriles-Miller JP, Pierik R, Yan C, Clay DE, Clay SA. RNAseq reveals weed-induced PIF3-like as a candidate target to manipulate weed stress response in soybean. New Phytol. 2015;207(1):196–210.
Grabherr MG, Haas BJ, Yassour M, Levin JZ, Thompson DA, Amit I, Regev A. Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat Biotechnol. 2011;29(7):644–52.
Zheng J, Ma X, Zhang X, Hu Q, , Qian R. Salicylic acid promotes plant growth and salt-related gene expression in Dianthus superbus L. (Caryophyllaceae) grown under different salt stress conditions. Physiol Mol Biol Plants. 2018;24(2):231–8.
Patterson BD, Payne LA, Chen YZ, Graham D. An inhibitor of catalase induced by cold in chilling-sensitive plants. Plant Physiol. 1984;76(4):1014–8.
Kanehisa M, Goto S. KEGG: Kyoto Encyclopedia of Genes and Genomes. Nucleic Acids Res. 2000;28:27–30.
Kanehisa M. Toward understanding the origin and evolution of cellular organisms. Protein Sci. 2019;28:1947–51.
Kanehisa M, Furumichi M, Sato Y, Ishiguro-Watanabe M, Tanabe M. KEGG: integrating viruses and cellular organisms. Nucleic Acids Res. 2021;49:D545–51.
Acknowledgements
Not applicable.
Funding
All sources of funding for the research reported are declared here. The research was supported by Key Research and Development Program of Zhejiang Province (2019C02036); Zhejiang Science and Technology Major Program on Agricultural New Variety Breeding (2021C02071-6); Zhejiang Province Public Welfare Project (LGN20C150001); Zhejiang Provincial Scientific and Technological Cooperation Project (2021SNLF021); Wenzhou Major Scientific and Technological Innovation Projects (ZS2020002); Zhejiang Province Rare and Endangered Wild Animals and Plants Rescue and Protection Action Project—In Situ Protection and Artificial Breeding of Clematis tientaiensis.
Author information
Authors and Affiliations
Contributions
RQ and QH performed the experiments, analyzed data and wrote the manuscript; XM collected samples; XZ and HL analyzed data and assisted with writing; YY assisted in the analysis of plant physiological indicators; JZ guided the operation of the experiment; HG revised the manuscript. All authors have read and agreed on the final version of the manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
The original online version of this article was revised: authors would like to correct Affiliation 1 details.
Supplementary Information
Additional file 1: Fig. S1.
Gene Ontology classification and KEGG analysis of the transcriptome. Fig. S2. Enriched GO terms of DEGs. Fig. S3. Enriched GO terms (cellular component) (P < 0.05) of DEGs. Fig. S4. Clustering analysis of gene expression.
Additional file 2: Table S1.
Statistics of splicing results. Table S2. Annotation of unique DEGs related to heat stress in C. lanuginose. Table S3. Annotation of unique DEGs related to heat stress in C. crassifolia. Table S4. The GenBank number of the unigenes. Table S5. Primer sequences for gene expression analysis.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Qian, R., Hu, Q., Ma, X. et al. Comparative transcriptome analysis of heat stress responses of Clematis lanuginosa and Clematis crassifolia. BMC Plant Biol 22, 138 (2022). https://doi.org/10.1186/s12870-022-03497-w
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12870-022-03497-w