
Abstract
Myocardial infarction (MI) is a prevalent form of ischemic heart disease, significantly contributing to heart disease-related deaths worldwide. This condition is primarily caused by myocardial ischemic-reperfusion injury (MIRI). Sirtuin 5 (SIRT5) is a desuccinylase known for its ability to reduce protein succinylation. Recent studies have highlighted the potential role of SIRT5 in various human diseases, including MIRI. This study aims to investigate the specific role of SIRT5 in modulating autophagy and cardiomyocyte death in a MIRI model, as well as to identify the downstream protein targets of SIRT5. Initially, we established a hypoxia/reoxygenation (H/R)-induced MIRI cell model to measure SIRT5 expression and assess its functions. Our results indicated that H/R induction led to a downregulation of SIRT5 expression, decreased autophagy, and increased cell death. Notably, overexpression of SIRT5 effectively promoted autophagy and inhibited cell death in the MIRI cell model. Mechanistically, SIRT5 was found to directly interact with the target of myb1 membrane trafficking protein (TOM1) at the K48 site, inducing its desuccinylation and stabilization. Further rescue assays revealed that TOM1 knockdown reversed the changes in autophagy and apoptosis caused by SIRT5 overexpression in the MIRI cell model. In vivo experiments demonstrated that SIRT5 alleviated myocardial injury in MI models. In conclusion, this study uncovers the role of SIRT5-mediated desuccinylation of TOM1 in regulating autophagy-related cell death in MIRI, providing new insights into potential therapeutic strategies for MI.
Similar content being viewed by others

Introduction
Myocardial infarction (MI) is a common ischemic heart disease and a leading cause of death worldwide [1]. Pathologically, MI is characterized by ischemic necrosis of myocardial tissues [2]. Early restoration of blood supply, known as reperfusion, is crucial for reducing infarct size and preserving cardiac function [3]. However, reperfusion often exacerbates myocardial tissue injury, a phenomenon referred to as myocardial ischemic-reperfusion injury (MIRI) [4, 5]. Autophagy is a conserved and tightly regulated intracellular catabolic process. It facilitates the degradation and recycling of damaged and dysfunctional macromolecules or organelles, thereby maintaining cellular homeostasis [6]. In the heart, autophagy plays a vital role in preserving cardiac function by executing various physiological processes. Abnormal autophagy, however, can negatively impact cardiac functions in various heart diseases, including cardiac hypertrophy [7], heart failure [8], and MIRI [9]. Therefore, understanding the specific molecular mechanisms that modulate autophagy-mediated MIRI is crucial for developing new insights and therapeutic strategies for MI treatment.
Succinylation is a post-translational modification that influences protein expression and stability [10]. Current research has highlighted the involvement of succinylation in various human diseases, including malignant tumors [11, 12] and stroke [13]. Studies have revealed the role of succinylation-mediated post-translational modifications in myocardial injury. For instance, MG53 ameliorates myocardial ischemia-reperfusion injury (MIRI) via succinylation [14], and the SIRT5-SDH-succinate pathway affects I/R-induced myocardial injury [15]. Sirtuin 5 (SIRT5) is a desuccinylase that catalyzes the removal of succinyl groups, thereby reducing protein succinylation. Recent studies have shown that SIRT5-mediated desuccinylation is associated with diverse biological activities, including fatty acid metabolism [16], mitochondrial metabolism [17], cardiomyocyte pyroptosis [18], and mitochondrial surveillance [19]. However, whether SIRT5-mediated protein desuccinylation can regulate autophagy-related cardiomyocyte death in MIRI remains unclear.
The purpose of this study is to investigate the specific role of SIRT5-mediated desuccinylation in modulating autophagy and cardiomyocyte death in a MIRI model and to explore the downstream protein targets of SIRT5.
Materials and methods
Establishment of animal model
Eight-weeks old Sprague-Dawley (SD) rats used for all animal experiments were provided by SJA Laboratory Animal Co., Ltd (Hunan, China) commercially. All procedures of animal study were performed after being approved by Institutional Animal Care and Use Committee of Nanjing Medical University (2023-12-13; IACUC-2312031). Rats were assigned into four groups with equal numbers (n = 5 per group): Sham, MIRI, MIRI + lvNC, and MIRI + lvSIRT5. Synthesis of lentivirus packaging SIRT5 expression vector (lvSIRT5) and negative control (lvNC) were completed by Hanheng Biotechnology Co., Ltd. (Shanghai, China). After lentivirus injection, rats in the later three groups were subjected to MIRI in accordance with methods described in a previous study [20]. In brief, rats were injected with 3% pentobarbital sodium (Sigma-Aldrich) for anesthesia. The left anterior descending coronary artery was then ligated with a braided silk suture to produce myocardial ischemia. Half an hour after ischemia, the myocardium was subjected to six-hours’ reperfusion. Rats underwent the same surgical procedures but without ligation of the left coronary artery were defined as Sham group. After that, all rats were euthanized for collection of their corresponding heart samples.
Triphenyltetrazolium chloride (TTC) staining
To assess myocardial infarction in MIRI rats, TTC staining was utilized. After sacrifice, hearts were quickly excised and washed in ice-cold saline. Transverse Sect. (2 mm thick) were prepared and immersed in 1% TTC solution (pH 7.4) at 37 °C for 20 min in the dark. TTC stains viable myocardium brick-red, while necrotic tissue remains unstained and appears white or pale. The sections were then fixed in 10% formalin, and digital images were captured. Infarct size was calculated as a percentage of the left ventricle using image analysis software, providing a quantitative measure of I/R-induced damage.
Haematoxylin and eosin (H&E) staining
Cardiac tissue was harvested from the four groups of rats mentioned above under pentobarbital sodium anesthesia (35 mg/kg, i.p.) and washed in PBS. The tissue was then fixed in 4% paraformaldehyde for 24 h and embedded in paraffin. Next, samples were sectioned into 4.0 μm slices and stained with H&E for 5 min. The stained sections were observed using a Zeiss confocal microscope (Oberkochen, Germany).
Cell culture and cell model establishment
Human cardiomyocytes (AC16 cells) were purchased from ATCC (Manassas, VA, USA) and cultured in DMEM supplemented with 10% fetal calf serum (FCS) and penicillin-streptomycin. The cells were maintained in a humidified incubator set at 37 °C with 5% CO2.
To establish the hypoxia/reoxygenation (H/R)-induced myocardial ischemia-reperfusion injury (MIRI) cell model, AC16 cells were incubated under hypoxic conditions (95% N2 + 5% CO2) for 3 h. Subsequently, the cells were transferred to a reoxygenation incubator (95% O2 + 5% CO2) for another 3 h. Cells cultured under normoxic conditions served as the control group.
Cell transfection and treatment
Short hairpin RNAs (shRNAs) targeting SIRT5 (shSIRT5) or TOM1 (shTOM1) as well as their corresponding negative controls (shNC) were all synthesized by Ribobio (Guangzhou, China). The whole length of SIRT5 was sub-cloned into the pcDNA3.1 vector to construct SIRT5 overexpression vector (named SIRT5), and the empty vector was defined as the negative control (named vector). All plasmids were transfected into AC16 cells using Lipofectamine 3000 transfection Kit (Invitrogen, Carlsbad, CA, USA).
To evaluate TOM1 protein stability under different conditions, AC16 cells with SIRT5 overexpression or knockdown were treated with 50 µg/ml of cyclohexane (CHX; MCE, NJ, USA) for 0, 6, 12, and 24 h.
RT-qPCR
Total RNA extracted from indicated AC16 cells using TRIzol™ reagent (Invitrogen) was then subjected to reverse transcription to generate cDNA with PrimeScript® 1st Strand Synthesis Kit (TaKaRa, Tokyo, Japan). QuantiTect SYBR® Green RT-PCR Kit (QIAGEN, Dusseldorf, Germany) was applied for performing the real‐time RT‐qPCR. Relative SIRT5 mRNA expression was determined by using 2−▵▵ct calculation method by normalizing to GAPDH.
Flag HA Co-IP
pcDNA3.1 vectors encoding Flag-SIRT5 and HA-TOM1 were constructed using standard molecular cloning techniques. Sequence verification was performed to confirm correct insertion and reading frame. At 70–80% confluence, AC16 cells were transfected with Flag-SIRT5 and HA-TOM1 plasmids, either individually or co-transfected, using lipofectamine-based reagent following the manufacturer’s instructions. Cells were incubated for 48 h post-transfection to allow protein expression. Next, transfected cells were harvested and lysed in ice-cold IP lysis buffer (containing protease and phosphatase inhibitors) using gentle agitation. Lysates were centrifuged to remove debris. Cleared lysates were incubated overnight at 4℃ with anti-Flag magnetic beads to pull down Flag-SIRT5 along with any interacting proteins. Control samples included beads incubated with lysate from cells expressing HA-TOM1 alone to assess nonspecific binding. After that, beads were washed extensively with lysis buffer to remove unbound proteins. Next, interacting proteins were eluted using Flag peptide or denaturing conditions, depending on downstream analysis requirements. Eluates were subjected to SDS-PAGE and transferred to PVDF membranes for Western blot analysis.
Determination of autophagic cells
The mCherry-GFP-LC3 adenovirus (Beyotime, Shanghai, China) was transfected into AC16 cells (1 × 105 cells/well) cultured in a 24-well plate at a multiplicity of infection (MOI) of 40 at 37 °C in a 5% CO2 incubator for 24 h. Following transfection, the cells were incubated with fresh medium for an additional 12 h at 37 °C. Thereafter, the cells underwent H/R treatment and transfection experiments. For imaging, three random fields per cell were selected under a confocal laser scanning microscope (Carl Zeiss, Jena, Germany) to quantify the number of autophagosomes (mCherry + GFP yellow) and autolysosomes (mCherry red) per cell.
Cell death detection
PI staining was used to evaluate cell death. Briefly, AC16 cells with indicated transfections or treatments were cultured in a 12-well plate at a density of 105 cells per well. Cells were then incubated in a solution mixed with DAPI and PI for 25 min. Finally, PI-positive cells were monitored and imaged under a fluorescence microscope.
Western blot
Total protein was extracted using RIPA lysis buffer (Beyotime, Beijing, China). Total protein (50 µg) was separated by 10% SDS-PAGE, followed by transferring onto a PVDF membrane. The membrane was then blocked with 5% milk in TBST, followed by incubation with primary antibodies against LC3BII/I (1: 2000), p62 (1: 1000), SIRT5 (1:1000), TOM1 (1:1000), GAPDH (1:1000) at 4 °C overnight. After that, the membrane was washed in TBST and then treated with the secondary antibody (1:5,000) for further incubation. All antibodies were purchased from Santa Cruz Biotechnology (Dallas, TX, USA). Protein bands were visualized by exposing to ECL (Beyotime).
The succinylation level was detected by western blot analysis using l-lactyllysine antibody (PTM Biolabs, Chicago, IL, USA) as mentioned previously [21].
Immunoprecipitation assay
AC16 cells were harvested by gentle trypsinization, washed in ice-cold PBS, and pelleted by low-speed centrifugation. The cell pellets were then lysed in ice-cold IP lysis buffer supplemented with protease inhibitors. Lysates were incubated on ice for 30 min and subsequently centrifuged at 16,000 x g for 15 min at 4 °C to remove cellular debris. The supernatants were incubated overnight at 4 °C with an anti-TOM1 antibody pre-bound to protein A/G agarose beads, allowing specific complex formation between the antibody and target protein. Following incubation, the bead-antibody-protein complex was thoroughly washed with cold lysis buffer to eliminate non-specifically bound proteins. The bound proteins were then eluted from the beads by boiling in reducing sample buffer for succinylation determination by Western blot assay.
Statistical analysis
Experimental data obtained from three independent samples were graphed as mean ± standard deviation. According to group number, statistical analysis of all data was completed using SPSS v17.0 (IBM, Chicago, IL, USA) with two different methods (student’s t test for two groups, and one-way ANOVA for multiple groups). P < 0.05 was a threshold of being statistically significant.
Results
SIRT5 is downregulated in H/R-indued MIRI cell model
Initially, AC16 cells underwent hypoxia/reoxygenation (H/R) to mimic the myocardial ischemia-reperfusion injury (MIRI) cell model. After H/R induction, autophagy and cell death conditions in AC16 cells were monitored. Observations of mCherry-GFP-LC3 fluorescence indicated a significant decrease in GFP-LC3-positive autophagosomes in H/R-injured AC16 cells (Fig. 1A). Western blot analysis revealed that H/R-induced AC16 cells exhibited elevated levels of p62 and a decreased LC3BII/I ratio (Fig. 1B). PI-positive cell detection suggested that cell death was promoted in AC16 cells post-H/R induction (Fig. 1C). Subsequently, changes in SIRT5 expression were evaluated by measuring both mRNA and protein levels. RT-qPCR and Western blot results demonstrated a notable decrease in SIRT5 mRNA and protein expression in H/R-induced AC16 cells (Fig. 1D-E). These findings collectively indicate that SIRT5 is downregulated in H/R-induced injured cardiomyocytes.

SIRT5 is downregulated in H/R-indued MIRI cell model. (A) mCherry-GFP-LC3 fluorescence was detected to show the autophagy condition in cardiomyocytes before or after H/R induction. (B) The autophagy-related protein markers were assessed by western blot in cardiomyocytes before or after H/R induction. (C) PI positive cells were detected to observe the cell death before or after H/R induction. D-E. SIRT5 expression changes were evaluated through measuring both mRNA and protein levels with RT-qPCR and western blot. **P < 0.01: H/R vs. CON
Overexpression of SIRT5 promotes autophagy while inhibits cell death of MIRI cell model
To analyze whether SIRT5 is involved in autophagy and cell death in the H/R-induced MIRI cell model, a vector expressing SIRT5 was transfected into model cells. The overexpression efficiency was confirmed by RT-qPCR (Fig. 2A). Analysis of mCherry-GFP-LC3 fluorescence showed that SIRT5 overexpression promoted autophagy, which was otherwise blocked by H/R induction in cardiomyocytes (Fig. 2B). Additionally, SIRT5 overexpression prevented H/R-induced cell death (Fig. 2C). Next, autophagy-related protein markers were detected. As shown in Fig. 2D, the elevated level of p62 and the decreased LC3BII/I ratio caused by H/R induction were reversed by SIRT5 overexpression. These data suggest that SIRT5 plays a crucial role in promoting autophagy to repair myocardial cell damage induced by H/R.

Overexpression of SIRT5 promotes autophagy while inhibits cell death of MIRI cell model. (A) The vector expressing SIRT5 was transfected into model cells and the overexpression efficiency was indicated by RT-qPCR data. (B) The autophagy was detected in H/R induced cardiomyocytes after SIRT5 overexpression through mCherry-GFP-LC3 fluorescence analysis. (C) Cell death in H/R induced cardiomyocytes after SIRT5 overexpression. (D) The autophagy-related protein markers were assessed by western blot in H/R induced cardiomyocytes after SIRT5 overexpression. **P < 0.01: H/R vs. CON; ##P < 0.01: H/R + SIRT5 vs. H/R + vector
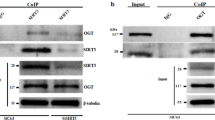
SIRT5 interacts with TOM1 and decreases the succinylation of TOM1 protein at site K48
Since SIRT5 can exert its role in various biological processes through succinylation modification, we further analyzed the correlation between SIRT5 and its downstream target, TOM1. We measured the levels of TOM1 and succinylated TOM1 in cardiomyocytes with either SIRT5 overexpression or knockdown. The results showed that SIRT5 overexpression increased TOM1 levels while reducing succinylated TOM1 levels. Conversely, SIRT5 silencing led to opposite results (Fig. 3A). The physical interaction between SIRT5 and TOM1 was confirmed by both exogenous (Fig. 3B) and endogenous (Fig. 3C) co-immunoprecipitation (CO-IP). We then predicted the potential site of TOM1 succinylation and identified the K48 site (Fig. 3D). To verify whether K48 was the actual succinylation site, we mutated the K48 site (K48R) and performed further Western blot analysis. The results indicated that mutating the K48 site abolished the effects of SIRT5 overexpression or knockdown on TOM1 and succinylated TOM1 levels (Fig. 3E). Finally, we investigated whether SIRT5-mediated desuccinylation altered TOM1 protein stability in cycloheximide (CHX)-treated cardiomyocytes. The results suggested that SIRT5 overexpression hampered CHX-induced TOM1 protein destabilization, while SIRT5 knockdown had the opposite effect (Fig. 3F). Collectively, these findings confirm that SIRT5 enhances TOM1 protein expression by interacting with it to induce its desuccinylation.

SIRT5 interacts with TOM1 and decreases the succinylation of TOM1 protein at site K48. A. The level of TOM1 and succinylated TOM1 in was separately measured cardiomyocytes with SIRT5 overexpression or knockdown by western blot. B-C. The interaction between SIRT5 and TOM1 was demonstrated by exogenous and endogenous co-IP. D. The potential site of TOM1 succinylation was K48 site. E. The effects of SIRT5 overexpression or knockdown on the level of TOM1 or succinylated TOM1 were detected by western blot after K48 site was mutated (K48R). F. The effects of SIRT5 overexpression or knockdown on the protein stability of TOM1 were detected in CHX-treated cardiomyocytes by western blot. **P < 0.01: SIRT5 vs. vector; ##P < 0.01: shSIRT5 + vs. shNC
Silencing of TOM1 reverses the tendencies of autophagy and cell death altered by SIRT5 overexpression in MIRI cell model
Rescue assays were performed in the H/R-induced cell model to demonstrate the role of the SIRT5/TOM1 axis in mediating autophagy and cell death. TOM1 expression was interfered with before all experiments, as validated by RT-qPCR (Fig. 4A). By analyzing mCherry-GFP-LC3 fluorescence, we determined that the autophagy recovery induced by SIRT5 overexpression in the H/R-induced MIRI cell model was inhibited again after TOM1 knockdown (Fig. 4B). Similarly, the reduction in cell death due to SIRT5 overexpression in the H/R-induced MIRI cell model was reversed following TOM1 knockdown (Fig. 4C). Lastly, changes in autophagy-related protein markers were observed under the same conditions. The results indicated that the decreased level of p62 and the increased LC3BII/I ratio, both induced by SIRT5 overexpression, were reversed after TOM1 knockdown (Fig. 4D). Therefore, we conclude that SIRT5 increases TOM1 expression to induce autophagy and suppress myocardial damage in MIRI.

Silencing of TOM1 reverses the tendencies of autophagy and apoptosis altered by SIRT5 overexpression in MIRI cell model. (A) TOM1 expression was interfered, as validated by RT-qPCR. **P < 0.01: shTOM1 + vs. shNC. (B) The effect of TOM1 silencing on SIRT5 overexpression-induced autophagy was determined by analyzing mCherry-GFP-LC3 fluorescence. (C) The cell death prevented by SIRT5 overexpression in H/R-induced MIRI cell model was further augmented by after TOM1 knockdown. (D) The changes in autophagy-related protein markers were observed through western blot analysis. **P < 0.01: H/R vs. CON. ##P < 0.01: H/R + shSIRT5 + shTOM1 vs. H/R + shSIRT5 + shNC
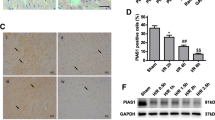
SIRT5 alleviates myocardial injury in in vivo MI models
The role of SIRT5 in ameliorating myocardial injury was further validated using an in vivo MIRI model. TTC staining results showed that SIRT5 overexpression significantly decreased the infarct area of myocardial tissues in MIRI rats (Fig. 5A). HE staining results indicated no apparent pathological changes in the Sham group. In contrast, myocardial tissues in the MIRI group exhibited significant fractures of myocardial fibers, which were loosely and irregularly arranged. These pathological changes were alleviated following SIRT5 overexpression (Fig. 5B). Additionally, protein levels of SIRT5, TOM1, and the LC3II/I ratio, which were decreased in the MIRI model, were restored after SIRT5 overexpression. Conversely, levels of succinylated TOM1 and p62, which were increased in the MIRI model, were reduced following SIRT5 overexpression (Fig. 5C). Based on these findings, we conclude that SIRT5 ameliorates myocardial injury in MIRI animal models.

SIRT5 alleviates myocardial injury in in vivo MI models. (A) TTC staining for infarct area of myocardial tissues in four different rat models. (B) HE staining results for pathology of myocardial tissues in four different rat models. (C) The levels of SIRT5, TOM1, TOM1-succ, and autophagic markers (LC3II/1 and p62) was measured by western blot in myocardial tissues obtained from four groups of rat models
Discussion
MIRI is a primary cause of cardiac death in MI patients [22]. Previous studies have demonstrated that autophagy can protect against myocardial I/R by regulating cardiomyocyte death [23]. For example, berbamine pre-treatment induces autophagy, providing protection against I/R-induced heart injury [24]. Similarly, ALDH2 has been identified as a therapeutic target for MIRI due to its role in promoting autophagy during ischemia and reperfusion [25].Our study found that autophagy was inhibited, while cell death increased in H/R-induced cardiomyocytes, indicating that autophagy-induced suppression of cardiomyocyte death was impaired in the MIRI cell model.
SIRT5 is a desuccinylase that functions by catalyzing the removal of succinylation, thereby inhibiting protein succinylation. Evidence has revealed the potential roles of SIRT5 in various human heart diseases [26, 27]. Importantly, SIRT5’s impact on I/R-induced myocardial injury has been previously documented [15]. However, whether SIRT5 affects autophagy in myocardial ischemia-reperfusion injury (MIRI) through the desuccinylation of its target proteins remains unclear. In this study, we confirmed that SIRT5 was significantly downregulated in the MIRI cell model. To analyze SIRT5’s involvement in autophagy and cell death in the H/R-induced MIRI cell model, we overexpressed SIRT5 and conducted various detections. Our experimental results indicated that SIRT5 overexpression promoted autophagy, which was previously blocked by H/R induction in cardiomyocytes, and prevented H/R-induced cell death. Further measurements of autophagy-related protein markers revealed that the increased level of p62 and the decreased ratio of LC3BII/I caused by H/R induction were reversed by SIRT5 overexpression. These findings confirm the role of SIRT5 in inducing autophagy, thereby promoting the repair of myocardial cell damage induced by H/R.
Mechanistically, SIRT5 regulates protein desuccinylation to exert its functions across different disease models. For instance, SIRT5 mediates the desuccinylation of ACOX1, decreasing its activity in hepatocellular carcinoma development [28]. In the context of intervertebral disc degeneration, SIRT5 protects mitochondrial homeostasis via the desuccinylation modification of AIFM1 [29]. Additionally, SIRT5 enhances cellular antioxidant defense by promoting the desuccinylation of IDH2 [30] and eliminates reactive oxygen species (ROS) through desuccinylation-mediated activation of SOD1 [31]. However, the interaction between SIRT5 and TOM1 had not been revealed until now. This study demonstrates that SIRT5 overexpression increases the level of TOM1 while reducing the level of succinylated TOM1, indicating a regulatory interaction between SIRT5 and TOM1.
Moreover, SIRT5 was found to directly interact with TOM1. The potential site of TOM1 succinylation was predicted and identified as K48. Upon mutating the K48 site, the effects of SIRT5 overexpression or knockdown on the levels of TOM1 and succinylated TOM1 were abolished. Protein stability assays revealed that SIRT5 overexpression hampered the cycloheximide (CHX)-induced destabilization of TOM1, whereas SIRT5 knockdown enhanced TOM1 destabilization. This confirmed that SIRT5 interacts with TOM1 and decreases the succinylation of TOM1 at the K48 site to stabilize the protein. Rescue assays demonstrated that the autophagy recovered by SIRT5 overexpression in the H/R-induced MIRI cell model was inhibited again when TOM1 expression was knocked down. Additionally, TOM1 knockdown augmented the cell death that was prevented by SIRT5 overexpression in the H/R-induced MIRI cell model. These results collectively indicate that SIRT5-mediated desuccinylation of TOM1 at the K48 site is crucial for stabilizing TOM1, promoting autophagy, and protecting cardiomyocytes from death in the context of MIRI.
In conclusion, our study validated that SIRT5 induces autophagy to prevent cell death in MIRI. Additionally, SIRT5 ameliorated myocardial injury in MIRI rat models. Mechanistically, SIRT5 enhances TOM1 protein expression through desuccinylation-mediated stabilization. These research findings suggest that SIRT5-mediated protein desuccinylation is a potential therapeutic target for MIRI.
Data availability
The datasets used and/or analyzed during the current study are available from the corresponding author on reasonable request.
References
Ketchum ES, et al. The Seattle Post myocardial infarction model (SPIM): prediction of mortality after acute myocardial infarction with left ventricular dysfunction. Eur Heart J Acute Cardiovasc Care. 2014;3(1):46–55.
Barnett R. Acute myocardial infarction. Lancet. 2019;393(10191):2580.
De Villiers C, Riley PR. Mouse models of myocardial infarction: comparing permanent ligation and ischaemia-reperfusion. Dis Model Mech. 2020;13(11).
Blankenberg S, Neumann JT, Westermann D. Diagnosing myocardial infarction: a highly sensitive issue. Lancet. 2018;392(10151):893–4.
Isaaz K, Gerbay A. Deferred stenting in acute ST elevation myocardial infarction. Lancet. 2016;388(10052):1371.
Nakai A, et al. The role of autophagy in cardiomyocytes in the basal state and in response to hemodynamic stress. Nat Med. 2007;13(5):619–24.
Cao DJ, et al. Histone deacetylase (HDAC) inhibitors attenuate cardiac hypertrophy by suppressing autophagy. Proc Natl Acad Sci U S A. 2011;108(10):4123–8.
Ghosh R, Pattison JS. Macroautophagy and chaperone-mediated autophagy in Heart failure: the known and the unknown. Oxid Med Cell Longev. 2018;2018:p8602041.
Fu H, Li X, Tan J. NIPAAm-MMA nanoparticle-encapsulated visnagin ameliorates myocardial ischemia/reperfusion injury through the promotion of autophagy and the inhibition of apoptosis. Oncol Lett. 2018;15(4):4827–36.
Zhang Z, et al. Identification of lysine succinylation as a new post-translational modification. Nat Chem Biol. 2011;7(1):58–63.
Tong Y, et al. SUCLA2-coupled regulation of GLS succinylation and activity counteracts oxidative stress in tumor cells. Mol Cell. 2021;81(11):2303–e23168.
Bai W, et al. Protein succinylation associated with the progress of hepatocellular carcinoma. J Cell Mol Med. 2022;26(22):5702–12.
Lian J, et al. Succinylation modification: a potential therapeutic target in stroke. Neural Regen Res. 2024;19(4):781–7.
Wang Y, et al. MG53 alleviates hypoxia/reoxygenation-induced cardiomyocyte injury by succinylation and ubiquitination modification. Clin Exp Hypertens. 2023;45(1):2271196.
Liu L, et al. Exogenous nicotinamide adenine dinucleotide administration alleviates ischemia/reperfusion-induced oxidative injury in isolated rat hearts via Sirt5-SDH-succinate pathway. Eur J Pharmacol. 2019;858:172520.
Park J, et al. SIRT5-mediated lysine desuccinylation impacts diverse metabolic pathways. Mol Cell. 2013;50(6):919–30.
Wang G, et al. Regulation of UCP1 and mitochondrial metabolism in Brown Adipose tissue by reversible succinylation. Mol Cell. 2019;74(4):844–e8577.
Wei C, et al. SIRT5-related lysine demalonylation of GSTP1 contributes to cardiomyocyte pyroptosis suppression in diabetic cardiomyopathy. Int J Biol Sci. 2024;20(2):585–605.
Chang X, et al. SIRT5-Related desuccinylation modification contributes to Quercetin-Induced Protection against Heart failure and high-glucose-prompted cardiomyocytes injured through regulation of mitochondrial quality surveillance. Oxid Med Cell Longev. 2021;2021:p5876841.
Yu L, et al. Protective effect of berberine against myocardial ischemia reperfusion injury: role of Notch1/Hes1-PTEN/Akt signaling. Apoptosis. 2015;20(6):796–810.
Sadhukhan S, et al. Metabolomics-assisted proteomics identifies succinylation and SIRT5 as important regulators of cardiac function. Proc Natl Acad Sci U S A. 2016;113(16):4320–5.
Mozaffarian D, et al. Heart Disease and Stroke Statistics-2016 update: a Report from the American Heart Association. Circulation. 2016;133(4):e38–360.
Aviv Y, et al. Regulation of autophagy in the heart: you only live twice. Antioxid Redox Signal. 2011;14(11):2245–50.
Zheng Y, et al. Berbamine postconditioning protects the heart from ischemia/reperfusion injury through modulation of autophagy. Cell Death Dis. 2017;8(2):e2577.
Ma H, et al. Aldehyde dehydrogenase 2 (ALDH2) rescues myocardial ischaemia/reperfusion injury: role of autophagy paradox and toxic aldehyde. Eur Heart J. 2011;32(8):1025–38.
Zheng D, et al. SIRT5 alleviates hepatic ischemia and reperfusion injury by diminishing oxidative stress and inflammation via elevating SOD1 and IDH2 expression. Exp Cell Res. 2022;419(2):113319.
Hershberger KA, et al. Sirtuin 5 is required for mouse survival in response to cardiac pressure overload. J Biol Chem. 2017;292(48):19767–81.
Chen XF et al. SIRT5 inhibits peroxisomal ACOX1 to prevent oxidative damage and is downregulated in liver cancer. EMBO Rep. 2018;19(5).
Mao J, et al. SIRT5-related desuccinylation modification of AIFM1 protects against compression-induced intervertebral disc degeneration by regulating mitochondrial homeostasis. Exp Mol Med. 2023;55(1):253–68.
Zhou L, et al. SIRT5 promotes IDH2 desuccinylation and G6PD deglutarylation to enhance cellular antioxidant defense. EMBO Rep. 2016;17(6):811–22.
Lin ZF, et al. SIRT5 desuccinylates and activates SOD1 to eliminate ROS. Biochem Biophys Res Commun. 2013;441(1):191–5.
Acknowledgements
Not applicable.
Funding
This study was supported by the National Natural Science Foundation of China (82070476) and Joint Funds for the innovation of science and Technology, Fujian province (2019Y9057).
Author information
Authors and Affiliations
Contributions
ZL conceived the study; ZL and ZZ conducted the experiments; XD analyzed the data; ZL was a major contributor in writing the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
The study was approved by the Institutional Animal Care and Use Committee of Nanjing Medical University (2023-12-13; IACUC-2312031). All experiments were performed in accordance with ARRIVE guidelines.
Consent for publication
All authors approved the final manuscript and the submission to this journal.
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Li, Z., Zheng, Z. & Dai, X. SIRT5 induces autophagy and alleviates myocardial infarction via desuccinylation of TOM1. BMC Cardiovasc Disord 24, 464 (2024). https://doi.org/10.1186/s12872-024-04120-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12872-024-04120-6