
Abstract
Background
The highly expressed surface antigen 1 (SAG1)-related sequence (SRS) proteins of T. gondii tachyzoites, as a widespread zoonotic parasite, are critical for host cell invasion and represent promising vaccine targets. In this study, we employed a computer-aided multi-method approach for in silico design and evaluation of TgVax452, an epitope-based candidate vaccine against T. gondii tachyzoite-specific SRS proteins.
Methods
Using immunoinformatics web-based tools, structural modeling, and static/dynamic molecular simulations, we identified and screened B- and T-cell immunodominant epitopes and predicted TgVax452’s antigenicity, stability, safety, adjuvanticity, and physico-chemical properties.
Results
The designed protein possessed 452 residues, a MW of 44.07 kDa, an alkaline pI (6.7), good stability (33.20), solubility (0.498), and antigenicity (0.9639) with no allergenicity. Comprehensive molecular dynamic (MD) simulation analyses confirmed the stable interaction (average potential energy: 3.3799 × 106 KJ/mol) between the TLR4 agonist residues (RS09 peptide) of the TgVax452 in interaction with human TLR4, potentially activating innate immune responses. Also, a dramatic increase was observed in specific antibodies (IgM and IgG), cytokines (IFN-γ), and lymphocyte responses, based on C-ImmSim outputs. Finally, we optimized TgVax452’s codon adaptation and mRNA secondary structure for efficient expression in E. coli BL21 expression machinery.
Conclusion
Our findings suggest that TgVax452 is a promising candidate vaccine against T. gondii tachyzoite-specific SRS proteins and requires further experimental studies for its potential use in preclinical trials.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Background
Toxoplasma gondii (T. gondii) is a cosmopolitan coccidian parasite responsible for toxoplasmosis, a disease with significant public health and veterinary implications [1]. This genotypically diverse protozoan primarily uses Felidae (Felis catus) as definitive hosts, while other warm-blooded animals such as birds and mammals can also harbor the infection [2]. T. gondii can spread through various routs including (i) congenital [3], transfusion-transmitted infections by tachyzoites [4], organ transplants [5], and consumption of undercooked meat containing bradyzoites [6]. Approximately, one-third of the global human population has been exposed to this parasite [7], which can cause severe clinical outcomes such as chorioretinitis, hydrocephaly, and abortion in fetuses [8], and encephalitis in immunocompromised individuals [9] (Fig. 1). In veterinary contexts, T. gondii infections in livestock, particularly sheep and goats, result in abortion and stillbirth, affecting economic productivity [10].

T. gondii life cycle. (1) Unsporulated oocysts are shed in the cat’s faeces, as definitive hosts of the infection. (2) The sporulated and infective oocysts are formed in the environment. The intermediate host stage includes humans and other warm-blooded animals such as rodents, birds, and livestock. (3) Animals become infected after ingesting contaminated food or water or by direct contact with the cat’s infected faeces. The oocysts release sporozoites that invade cells in various tissues such as muscle, brain, and eyes. Then sporozoites differentiate into tachyzoites, and so as the immune system of the host responds to the infection, some tachyzoites differentiate into bradyzoites and form tissue cysts. (4) Cats can be infected via a prey-predator transmission system after consuming intermediate hosts like birds and/or rodents or directly by ingestion of sporulated oocysts. (5) Next, the tissue cysts are ingested and the sexual phase of T. gondii’s life cycle begins again. (6) Humans can become infected after eating undercooked meat of livestock harbouring bradyzoite cysts. (7) Moreover, humans can become infected after ingestion of sporulated oocysts in the environment. (7.1–7.3) T. gondii can also be transmitted vertically from mother to fetus during pregnancy or through organ transplantation or blood transfusion. Created with BioRender.com
Despite numerous studies exploring herbal extracts and nanoformulations [11, 12], there are currently no safe and broad-spectrum therapeutics that target both acute and chronic T. gondii infections [13]. Current treatments, such as antifolate drugs, are effective only against the acute tachyzoite stage and often come with significant side effects [13]. Therefore, alternative preventive measures, including vaccination, are crucial to limit parasite propagation independently of chemotherapeutic agents [14].
Research over the last three decades has explored various vaccination platforms [15], but these attempts have largely failed to eliminate tissue cysts or block vertical transmission in humans [16]. The only commercial vaccine available is Toxovax®, a live vaccine used in breeding female sheep to stimulate active immunity against the S48 strain of T. gondii [17].
Immunity against T. gondii primarily relies on T helper 1 (Th1) cell-mediated responses, characterized by the production of interleukin-12 (IL-12) and interferon-γ (IFN-γ), which are crucial for tachyzoite clearance [18]. T. gondii presents a rich source of antigenic compounds detectable by the immune cells, including surface-expressed antigens (SAGs) [19], and a wide array of excretory-secretory proteins such as micronemes (MICs), rhoptries (ROPs) and dense granule molecules (GRAs) [20,21,22,23]. The parasite’s surface is the most accessible part for the immune components to be recognized and interact with. It is covered with a lot of glycosylphosphatidylinositol (GPI)-linked proteins that are similar in structure and function to the immunogenic surface protein SAG1 [24]. SAG1-related sequence (SRS) proteins, including SAG1 (SRS29B), SRS1 (SRS29A), SAG2A, SRS2, SRS3 (SRS51), SAG3 (SRS57), SRS20A, SRS25, SRS52A, and SRS67, are prominent in the tachyzoite stage and are believed to play roles in host cell adhesion and immune evasion [25].
Designing an efficient vaccine against T. gondii involves targeting these immunogenic SRS proteins to activate both innate and adaptive immune responses [26]. Multi-epitope-based vaccines (MEBV) designed using state-of-the-art in silico approaches offer promising solutions [27, 28]. These modalities facilitate vaccine development by enhancing specificity, bypassing undesirable immune responses, and providing durable immunity in a cost- and time-effective manner [29]. This study aimed to design and model a candidate peptide-based vaccine against T. gondii infection using immunogenic epitopes from different tachyzoite-specific SRS proteins. Our approach integrates advanced immunoinformatics, structural modeling, and molecular simulations to predict and optimize the vaccine’s efficacy, antigenicity, and safety, paving the way for potential preclinical trials.
Methods
Retrieval of protein sequences
The FASTA sequence of five tachyzoite-specific SRS proteins including S8FBM7 (SRS20A), A0A125YP09 (SRS29B), S7W107 (SRS51), A0A125YJA5 (SRS52A) and A0A125YY85 (SRS57) were retrieved from the UniProtKB database to be submitted to various immunoinformatics related web-based tools.
Prediction and screening of B-cell epitopes
A multi-method approach was utilized to predict linear and conformational B-cell epitopes (BCEs) [30], involving five web servers including ElliPro, BepiPred v2.0, BCEPS, ABCpred and SVMTriP. Regarding Ellipro, the protein 3D model was submitted using default parameters. In BCEPS web server, a support vector machine (SVM) algorithm was exerted to yield linear B-cell epitopes. The threshold for BepiPred v2.0, BCEPS, ABCpred, and SVMTriP web servers were 0.5%, 0.5%, 0.75, and 0.2 respectively. Of note, B-cell epitopes predicted by the BepiPred v2.0 and BCEPS were of variable lengths, while those predicted by SVMTriP and ABCpred possessed fixed (n = 14) lengths. In the case of the SRS29B, B-cell epitopes were predicted through a PDB-derived template-based method. In this approach, alongside the predicted epitopes, the template crystal structure (PDB 1YNT) was utilized for more accurate B-cell epitope mapping (Fig. 2). The predicted epitopes shared by at least two prediction servers were then evaluated for antigenicity, allergenicity, water solubility and solubility using VaxiJen v2.0, AllergenFP v1.0, PepCalc and Toxinpred web servers, respectively. The most antigenic, non-allergenic, non-toxic, and soluble epitope from each protein sequence were selected for inclusion in the final vaccine construct.

The crystal structure (PDB 1ynt) of the monomeric form of T. gondii tachyzoite-derived SAG1 surface antigen complexed to a human Fab fragment. (a) The tachyzoite-derived SAG1 antigen is bound to a human Fab fragment. The antibody’s ligand binding site is highlighted. (b & c) This panel highlighted the SAG1 amino acid residues that are involved in H-bond interactions with the Fab. (d) The tachyzoite-specific SRS antigen SRS29B (A0A125YP09) is aligned with chains D and C of the Fab fragment. The alignment shows the Fab interacting rsidues of SRS29B (red coloured residues) are conserved and can be used as template for the validation of in silico predicted epitopes and so selection of the most B-cell immunodominant residues. (e) B-cell epitopes of the SAG1 are represented as CPK style and using BIOVIA Discovery Studio visualizer v21.1.0.20298
Prediction and screening of major histocompatibility complex (MHC)-binding epitopes
The prediction was performed using MHC-I and MHC-II binding tools of the Immune Epitope Database (IEDB), available at http://tools.iedb.org/mhci/ and http://tools.iedb.org/mhcii/, respectively. The prediction of 9-mer human MHC-I binders was done using IEDB recommended method 2020.09 (NetMHCpan EL 4.1) and the HLA allele reference set option. Moreover, 15-mer human MHC-II binders were predicted using IEDB recommended 2.22 method and selecting the full HLA reference set. Finally, those epitopes having lower percentile ranks (i.e., higher binding affinity) were selected for further screening [31,32,33]. In the following, MHC-I binders were screened in terms of antigenicity, allergenicity and toxicity, whereas MHC-II binders were evaluated regarding antigenicity, IFN-γ induction, and toxicity. Ultimately, two epitopes with strong affinity to MHC-I and MHC-II molecules (lowerst percentile rank) possessing good antigenicity/non-allergenicity (for MHC-I binders) or good antigenicity and IFN-γ induction (for MHC-binders) were selected from each protein for vaccine construction. The population coverage analysis of the predicted epitopes was done using Population coverage analysis tool in the IEDB web server.
Designing and assemblage of the multi-epitope vaccine construct
The final construct of the vaccine was achieved through the process of prediction, screening, and selection of potential immunogenic epitopes from five T. gondii SRS proteins, targeting both B- and T-cell responses. To facilitate the connection between B- and T-cell epitopes, two distinct peptide linkers, GPGPG and AAY, were utilized, respectively. Additionally, to enhance the activation of adaptive immunity, a synthetic T-helper peptide consisting of 13 amino acids, known as the Pan-DR epitope or PADRE sequence, was incorporated. This PADRE sequence was linked to a toll-like receptor 4 (TLR4) agonist called RS-09 peptide (APPHALS), employing a cleavable linker (PPGVS) at the N-terminus. The PADRE sequence was joined to the core vaccine construct through a cleavable linker called Cathepsin S (PMGLP) [34]. Furthermore, for the purpose of recombinant protein purification, a 6x histidine tag (6x His-Tag) was inserted at the C-terminus of the construct.
Physico-chemical properties of the vaccine construct
Primary physico-chemical features of the final vaccine model were predicted using the ExPASy ProtParam web tool, available at https://web.expasy.org/protparam/, comprising molecular weight (MW), speculated isoelectric pH, extinction coefficient, estimated half-life, instability index, aliphatic index and the grand average of hydropathicity (GRAVY) score [35].
Vaccine antigenicity, allergenicity, and solubility
In order to assess antigenicity, the vaccine sequence was submitted to two web servers, VaxiJen v2.0 (threshold: 0.5) and ANTIGENpro (http://scratch.proteomics.ics.uci.edu/). VaxiJen v2.0 performs based on an alignment-free method and auto cross-covariance (ACC) transformation of protein sequences into uniform vectors of principal amino acid properties. The prediction accuracy of the server is approximately 70–89% [36]. ANTIGENpro is a sequence-based prediction tool with 76% accuracy [37]. In the following, AllergenFP v1.0 (https://ddg-pharmfac.net/AllergenFP/) and AllerTOP v2.0 (https://www.ddg-pharmfac.net/AllerTOP/) were used for allergenicity prediction. AllergenFP v1.0 is a fingerprint descriptor-based approach without alignment reliance which identifies allergens from non-allergens [38]. The alignment-free server, AllerTOP v2.0, predicts based on the physico-chemical properties of a protein with an accuracy of 85.3% at best [39]. The vaccine solubility profile was, also, predicted using the Protein-Sol web server, available at https://protein-sol.manchester.ac.uk/ [40].
Structural modeling, refinement, and validation of the vaccine construct
The secondary structure of the vaccine construct was predicted using the NetSurfP-2.0 web tool [41]. This server predicts the surface accessibility, secondary structure, disorder, and phi/psi dihedral angles of amino acids in a given protein sequence. A single model, using a combination of Convolutional and Bi-Directional Long-Short Term Memory Neural Networks, predicts all structural features together [42]. In the following, the tertiary structure of the vaccine candidate was predicted using the Deep learning-based Iterative Threading ASSEmbly Refinement (D-I-TASSER) server [43]. The top 1 model was selected based on the estimated C-score, TM-score, and RMSD values for more analysis, i.e., structure refinement and validation.
Packing and re-establishing side chains for structural relaxations is a major procedure done by two time structure refinements, using the GalaxyRefine tool of the GalaxyWEB server [44]. The local/overall and stereochemistry quality of the refined model was evaluated using the ProSA-web and the structural validation tools developed by the UCLA SAVES v6.0 tools.
Prediction of vaccine antibody-interacting epitopes
Conformational B-cell epitopes, being essential regarding antigen-antibody interactions, were predicted by the ElliPro tool of the IEDB web server with a substantial AUC score of 0.732 and default settings of 6 Å max-distance and 0.5-min score [45]. Prediction and engineering of disulfide bonds were also performed via the DbD2 server in order to strengthen the total conformation and stability of the protein, if present. Those residues in the highly mobile region of the sequence will be mutated to cysteine, while only those residues having < 2.5 Kcal/mol energy and chi3 value between − 87 to + 97 can be qualified for disulfide engineering [46].
Molecular docking simulation: vaccine-TLR4
The intermolecular interactions and stability parameters of the TLR4-vaccine docked complex were evaluated using molecular docking and dynamic (MD) simulations, respectively. To simulate the vaccine’s interactions with an innate immune receptor, a protein-protein docking procedure was performed between the human TLR4 (PDB entity 3FXI) and the vaccine 3D model using the ZDOCK, a rigid-body docking online server [47]. The vaccine-TLR4 static interactions were visualized using BIOVIA Discovery Studio visualizer v21.1.0.20298.
Molecular dynamic (MD) simulations
Molecular dynamics (MD) simulation can provide valuable data about how the atoms in a system move over time [48, 49]. The folding and stability of three structures, including the human TLR4, the refined vaccine 3D model, and their docked complex were simulated using the MD simulation technique, as described previously [34, 50, 51]. In brief, the complex was imported to an MD simulation procedure using the GROMACS software (version 2022.4) and based on the GROMOS96 54a7 force field [52]. The simulations were conducted in a dodecahedron simulation box, while the SPC (simple point charge) model was used for the solvation of the required number of water molecules and also the neutralization with an appropriate number of counterions (i.e., Na and/or Cl). Subsequently, the system was checked for steric clashes and inappropriate geometry via the energy minimization (EM) technique with the steepest descent of 200,000 steps with a tolerance (Fmax) set to 1.00000e + 03.
Following the relaxation of the structure with EM, the NVT and the NPT ensembles (100.0 ps for each step) were performed. The equilibrated system was then subjected to MD simulations for 250 ns at a temperature of 300 K. Using the MD trajectory file, the following measures were analyzed: root-mean-square deviation (RMSD), root-mean-square fluctuation (RMSF), radius of gyration (Rg), hydrogen bond analysis, solvent accessible surface area (SASA) analysis, principal component analysis (PCA) and Gibbs free energy landscape (FEL). The visualization and interpretation of MD simulation results were conducted within the Spyder IDE v5.2.2 environment, utilizing Python v3.9.13 and using the NumPy package. The Matplotlib library v3.5.2 was utilized alongside its 3D plotting toolkits, specifically mpl_toolkits.mplot3d to generate the plots.
Vaccine immune response simulations
The immune profile of the injected vaccine candidate (without LPS) was evaluated using the C-ImmSim online server. A position-specific scoring matrix (PSSM) is applied by machine-learning techniques to predict likely immune interactions. The output of this server is provided based on the immunostimulatory activities in anatomical regions, comprising bone marrow, thymus, and lymph nodes. Default parameters such as random seed 12,345, simulation volume 10, simulation steps 100, and a time step of injection 1 were selected for this prediction [53].
Optimization of codon usage, in-silico cloning, and mRNA secondary structure
Due to the organism-specific differences in populations of cognate tRNAs, codon usage is also based on an organism-specific approach. Therefore, to improve the expression level of the designed peptide vaccine at the site of injection, codon, and mRNA secondary structures should be optimized. Here, we used the visual gene developer v1.9 software [54] and the RNAfold web server [55] to optimize the codon usage based on the Homo sapiens codon usage table and predict the mRNA secondary structure of the designed vaccine, respectively. The optimized DNA sequence of the vaccine construct was sub-cloned into the pET-22b(+) plasmid, using the SnapGene v5.2.4 software (from Insightful Science; available at www.snapgene.com).
Results
Screening and selection of B-cell epitopes
Multiple web servers with different ML-based techniques were utilized for the prediction of linear B-cell epitopes. The prediction thresholds for BepiPred, BCEPS and ABCpred were 0.5%, 0.5% and 0.75%, respectively. The outputs of four selected servers (BepiPred, BCEPS, ABCpred and SVMTriP) were compared and the non-allergen peptides having the best antigenicity and solubility values were selected. Only one B-cell epitope was chosen out of each SRS protein to design the final vaccine construct. Full details of the B-cell epitope prediction are provided in Tables S1-5. In the case of the SRS29B antigen (UniProtKB A0A125YP09), the final selected B-cell epitopes were identified after revalidation based on the experimental findings [56], and so this residue fragment (sequence: PKTALTEPPTLAYSPNRQICPAGTTSSCTSKAVTL) was selected as final B-cell epitope (Fig. 2).
Exploration of high-ranked human MHC binders
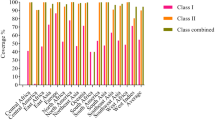
Top-ten high-ranked (lower percentile) human MHC binders were predicted through the IEDB web server and screened regarding antigenicity and allergenicity (MHC-I binders) as well as antigenicity and IFN-γ induction (MHC-II binders). Of each protein, two MHC-I and two MHC-II binding epitopes were selected for the final vaccine construction (Tables S6 and S7). Based on the population coverage analysis, T-cell epitope/HLA compositions predicted in the present study could cover 95.75% and 92.98% of the world population, regarding MHC-I and II alleles, respectively (Table 1).
Assemblage of the multi-epitope vaccine construct
In total, five immunodominant regions, including one B-cell, two MHC-I, and two MHC-II epitopes, were selected from each T. gondii SRS protein (Table 2) and further connected via specific peptide linkers. To promote the adjuvanticity and immune induction of the candidate vaccine, a TLR4 agonist (APPHALS) and PADRE sequence were added to the N-terminal of the sequence. Also, a C-end His-Tag was used for the upstream protein purification procedures [57]. The final complete sequence of the candidate vaccine had a length of 452 amino acids and so named “TgVax452” (Fig. 3a).

The amino acid sequence of the TgVax452, its three-dimensional (3D) model with various components, and 3D modeling validation plots. (a) Different components of TgVax452 as an amino acid sequence and three-dimensional (3D) model. (b) The residue-based ERRAT values of the 3D model are represented using the ERRAT method (top left). The ERRAT plot shows how well the predicted 3D model fits into its electron density map. The red-coloured regions of the ERRAT plot indicate areas with high errors that require further refinement. The initial model’s Ramachandran plot (top right). The verify3D’s 3D/1D plot (bottom left). The x-axis represents the residue number, while the y-axis represents the Verify3D score. The Verify3D score is a measure of how well the model fits into its environment, based on factors such as side-chain packing, backbone conformation, and solvent accessibility. (c) The consensus B-cell epitopes of TgVax452 are visualized using the Discovery Studio’s CPK (Corey-Pauling-Koltun) tool as an atom colouring scheme
Prediction of physico-chemical, antigenicity, allergenicity, and solubility characteristics
Based on ProtParam server output, the TgVax452 had a molecular weight of 44078.93 Dalton, relatively neutral speculated pI (6.8) and equal negatively-charged (Asp + Glu) [22] and positive-charged (Arg + Lys) [21] residues, extinction coefficients of 28,390 M-1 cm-1 (all pairs of Cys form cystines) and 27,390 M-1 cm-1 (all Cys are reduced). The estimated half-life in mammalian reticulocytes, yeast, and Escherichia coli was approximated to be 4.4, > 20 and > 10 h, respectively. The TgVax452 was estimated to be stable, as shown by an instability index of 33.20. Moreover, the aliphatic index and GRAVY score were computed as 70.75 and 0.194, respectively. A high antigenicity score was estimated for the vaccine model using VaxiJen (0.9639) and ANTIGENpro (0.882588) tools. Of note, the designed protein had no allergenic traits, as substantiated by AllergenFP and AllerTOP web servers. According to the Protein-Sol server, the predicted scaled solubility of the TgVax452 was calculated to be reasonable (0.498), in comparison with the population average for the experimental dataset (0.45).
Structural modeling of TgVax452
Based on the NetSurfP-2.0 server, coils and helices were the most abundant secondary structures in the final vaccine construct (Fig. S1). The D-I-TASSER estimates the modeling quality by measuring the structural similarity values (i.e., estimated TM-score and RMSD) between two structures, which are based on their correlation with C-score. A TM-score of more than 0.5 shows a model with correct topology, whereas a TM-score of less than 0.17 denotes a random structural similarity [58]. Also, the C-score is a confidence score, ranging from − 5 to 2, that estimates modeling quality by calculating the significance of threading template alignments and the convergence parameters of the structure assembly simulations. The C-score value close to 2.0 indicates a model with high confidence, and vice-versa [59]. Moreover, D-I-TASSER calculates the cluster density metric as the number of structure decoys per unit of space in the SPICKER cluster [58]. A higher density of clusters indicates that the structure occurs more frequently in the simulation trajectory, which means the model is better. In our study, the D-I-TASSER model quality metrics (i.e., expected TM-score and RMSD, C-score, number of decoys, and cluster density) for the top first predicted model were 0.46 ± 0.15, 11.3 ± 4.1, -1.18, 600 and 0.1035, respectively. This data implies that the quality of the modeled TgVax452 needs to be improved.
TgVax452’s 3D model refinement, validation and consensus B-cell epitopes
The quality measures of the TgVax452’s initial 3D model are shown in Fig. 3b. The initial model was refined two times using the GalaxyRefine web-server to optimize and energy-minimize the model’s hydrogen bonding network and atoms, respectively. Table 3 provides the quality validation metrics of the initially modeled TgVax452 and its refined formats. The ERRAT suite calculates the overall quality factor to quantify the 3D models’ quality. Herein, a high-resolution structure has higher overall quality factor values. According to the results obtained from the Verify3D, following the refinement, the percentage of residues having an averaged 3D-1D score more than equal to 0.2 has been increased. Based on the PROVE results, the total number of buried outlier atoms of the vaccine model decreased after the two times refinements. The ProSA and WHATCHECK stereochemical z-score values and PROCHECK’s Ramachandran plot relating to the vaccine’s psi/phi angles and backbone conformation were improved after the first and second refinements (Table 3). Altogether, the second refined model was selected for the following analysis. A consensus of the most immunodominant B-cell epitopes was selected based on a combination of online epitope predictors and experimental data (PDB 1YNT). Figure 3a and c illustrate the final B-cell epitopes that were selected for inclusion in the final construct of TgVax452.
Molecular docking analysis: adjuvanticity profile of TgVax452
The designed vaccine contains the RS-09 peptide, which acts as a TLR4 agonist. TLR4 is known to effectively stimulate host innate immunity against T. gondii [60]. Herein, to identify the TLR4’s binding sites, the X-ray crystal structure of the human TLR4-human MD-2 in complex with E.coli LPS Ra (PDB ID: 3FXI; Fig. 4a) was retrieved from the Protein Data Bank (PDB) [61]. The additional molecules in the PDB structure were then removed (Fig. 4b) using UCSF Chimera v1.15 software [62]. The vaccine’s adjuvanticity was assessed via a protein-protein docking analysis between the human TLR4 (Fig. 4b), as a target immune receptor, and the modeled vaccine construct (Fig. 4c). Overall, the ZDOCK online program provided ten docked complexes, of which complex 1 was selected as the most optimal one to evaluate the vaccine-TLR4 interaction quality. As shown in Fig. 4d, three hydrophobic interactions and one hydrogen bond were observed in the docked complex. Details of the interacting residues and the length/type of interactions are provided in Table 4.

Protein-protein docking analysis between the vaccine construct and TLR4. (a) The antigen-interacting residues of the human TLR4 (green molecule) were identified based on the PDB ID 3fxi. (b) To prepare the TLR4 molecule for docking, the additional molecules were removed using the UCSF Chimera software. (c) The 3D-modeled vaccine is visualized using the UCSF Chimera software. (d) The docked complex of the vaccine-TLR4 is visualized on the right-hand panel. The 3D interacting interface of the vaccine model and the TLR4 is exhibited on the left-hand panel. The H-bond surfaces are selected to display the ligand H-bond interacting residues
Molecular dynamics (MD) simulations: evaluation of TgVax452’s adjuvanticity
The MD simulation of the complex (TgVax452:TLR4) was carried out for 250 ns, assessing the dynamics and thermodynamics based on temperature, pressure, and potential energy. As shown in Fig. 5a, the average temperature of the system is 299.771 K, with a small error estimate of 0.18 K, indicating a fairly constant temperature throughout the simulation with minimal fluctuations throughout the simulation.

Dynamics and thermodynamics of TgVax452:TLR4 complex. The variation plots in temperature (a), potential energy (b), and pressure (c) experienced by the TgVax452:TLR4 complex throughout the simulation. The plots were generated within the Spyder IDE 5.2.2 environment, running Python v3.9.13
Meanwhile, the system’s average potential energy was computed to be 3.3799 × 106 kJ/mol, accompanied by an error estimate of 57,000 kJ/mol (Fig. 5b). This energy signifies inter-particle interactions, where a highly negative potential energy implies strong attractive forces, while a positive value suggests repulsive interactions. The potential energy fluctuations were determined to be 161,732 kJ/mol, with a total drift of -374,381 kJ/mol, indicating a gradual decline in potential energy over the simulation period.
As shown in Fig. 5c, the average pressure of the system was 8.4512 bar, with a small error estimate of 5.7 bar. Pressure serves as an indicator of external forces exerted on the system, influenced by factors like temperature, volume, density, and intermolecular interactions [63].
In order to assess the stability of the complex, the root means square deviation (RMSD) and root-mean-square fluctuation (RMSF) were calculated for both TLR4 and TgVax452 structures. The RMSD values of the complex elements indicate that the TgVax452 and the TLR4 (Fig. 6a) have reached a stable condition during the simulation time process. At the same time, the RMSF plots highlighted the most fluctuated regions in the vaccine model and TLR4 (Fig. 6b). Notably, unlike TLR4, the vaccine model exhibited significant peaks at residues 171ANTAARVG178, 264QL265, 322RKI324, 373KVPQDNN379, 401PGDPS405, N411, 413GGP415, C441, and 449TCAC452, indicating highly fluctuated regions during interaction with TLR4. Remarkably, these highly fluctuating regions are not located within the RS09 peptide (1APPHALS7), which serves as a crucial component for facilitating effective interaction with the vaccine’s TLR4 agonist region.

Analysis of RMSD, RMSF, and radius of gyration for the TgVax452:TLR4 complex. (a) RMSD plots indicate the stability of the TgVax452 model and TLR4 structure over a 250 ns duration. (b) Residue-based local RMSF profiles are illustrated for the vaccine and TLR4 structures. The vaccine model displays some significant peaks above the threshold (average RMSF of 0.264), indicating regions of heightened fluctuation. (c) Radius of gyration (Rg) profiles of TLR4. (d) Radius of gyration (Rg) profiles of the TgVax452. The plots were generated within the Spyder IDE 5.2.2 environment, running Python v3.9.13
The Rg profiles provide insights into the compactness and structural fluctuations within the TgVax452:TLR4 complex. By using Python statistical libraries, such as pandas and scipy, we assessed the total Rg values across the X, Y, and Z dimensions. Through iterative analysis, we computed the variance between maximum and minimum values, conducting independent t-tests to evaluate statistical significance. The results revealed that, despite observable variations in Rg values of TLR4 (Fig. 6c) and TgVax452 (Fig. 6d), the differences between maximum and minimum values for Total_Rg(nm), Rg_X, Rg_Y, and Rg_Z did not reach statistical significance (p-value > 0.01).
Hydrogen bonds play a critical role in stabilizing intermolecular interactions. By quantifying the number of hydrogen bonds between the vaccine construct and TLR4, MD simulation allows for the characterization of the interaction strength and the fluctuations over time. Our analysis encompassed 25,000 frames of MD simulation data, employing a grid-search method with a 31 × 31 × 31 grid and a cutoff distance of 0.35 nm. Notably, within the TLR4 component, we observed an average of 462 hydrogen bonds per timeframe out of a potential 733,562 bonds (Fig. 7a), while within TgVax452, an average of 253 hydrogen bonds were detected out of a possible 309,650 bonds (Fig. 7b). The consistent presence of a relatively high number of hydrogen bonds, coupled with minimal fluctuations, underscores the robustness and stability of the interactions between the vaccine and TLR4 throughout the simulation. Specifically, both TLR4 and TgVax452 exhibited approximately 463 and 256 hydrogen bonds, respectively, with limited temporal variations, emphasizing the sustained formation of hydrogen bonds within the complex. The count of atom pairs within a 0.35 nm cutoff (Fig. 7c for TLR4 and Fig. 7d for TgVax452) highlights the close proximity and potential for interactions between atoms within the molecules. The assessment of a protein’s exposure to solvent is facilitated by computing its solvent-accessible surface area (SASA) [64]. In this study, SASA values for the TgVax452:TLR4 complex were computed using the gmx sasa module in GROMACS and plotted against 250,000 ps of simulation time (Fig. 7e for TLR4 and Fig. 7f for TgVax452).

Distribution of the hydrogen bond and solvent accessible surface area (SASA) curves related to the TgVax452 and TLR4. (a) the profile of the number of hydrogen bonds per 25,000 (ps) timeframe within TLR4, out of a potential 733,562 bonds. (b) the profile of the number of hydrogen bonds per timeframe (25,000 ps) within TgVax452, out of a potential 309,650 bonds. (c) Count of atom pairs within a 0.35 nm cutoff distance within TLR4, reflecting the proximity and potential for interactions. (d) Count of atom pairs within a 0.35 nm cutoff distance within TgVax452, indicating the spatial arrangement and potential interactions between atoms. (e) The SASA plot for TLR4. The curve indicates relatively constant fluctuations throughout the simulation, with a minor deviation observed within the initial 260 ps. (f) The SASA plot for TgVax452. After approximately 45 ns, the SASA curve for TgVax452 reaches an equilibrium state, reflecting a stabilized protein conformation. The plots were generated within the Spyder IDE 5.2.2 environment, running Python v3.9.13
This decrease in surface area may indicate the seclusion of hydrophobic residues or the development of secondary structures, resulting in a more condensed protein configuration. Subsequently, between 4 and 25 ns, the SASA curve stabilizes, maintaining a relatively constant level. This stabilization indicates that TLR4 has reached an equilibrium state. As depicted in Fig. 7e the SASA plot for TLR4 exhibits relatively constant fluctuations throughout the simulation, with a minor deviation observed within the initial 260 ps. Conversely, the SASA plot for TgVax452 (Fig. 7f) demonstrates a notable decrease from approximately 271 nm² to approximately 217 nm² within the first 45 ns of the MD simulation. This reduction in surface area may indicate the burial of hydrophobic residues or the formation of secondary structures, leading to a more compact protein conformation. Subsequently, between 45 and 250 ns, the SASA curve stabilizes, remaining relatively constant. This stabilization indicates that TLR4 has reached an equilibrium state.
Moreover, the principal component analysis (PCA) was conducted on the TgVax452:TLR4 complex to inspect atomic motions and assess stability in more detail. The eigenvalues were computed from the covariance matrix to discern the PCs, representing the eigenvectors extracted from the data derived from the MD simulations. As shown in Fig. 8a and d, the first few eigenvectors capture the majority of the variance in the data and are respectively retrieved from the TLR4 and TgVax452 structures, while subsequent eigenvectors explain progressively smaller amounts of variance. As shown in the eigenvalues of the covariance matrices, the first eigenvector (PC1) variance in the vaccine structure (11.6016 nm²) is higher than in the TLR4 structure (8.35805 nm²), suggesting that the vaccine structure might exhibit more extensive conformational changes or flexibility compared to TLR4. Figure 8b and e show the distribution of conformations or states sampled during the MD simulations’ trajectory projection onto the first two eigenvectors obtained from the covariance matrix of Cα atoms respectively for the TLR4 and TgVax452 molecules. Analysis of the 2D and 3D projections of the TLR4 trajectory reveals closely clustered data points, indicative of low-energy and stable states that are energetically favourable for the system (Fig. 8b and c). In contrast, examination of the vaccine trajectory shows distinct clusters of points distributed across varied regions (Fig. 8e and f), suggesting significant conformational flexibility or structural diversity within TgVax452. These clusters imply the exploration of a broad spectrum of structural configurations, including higher-energy or transiently stable states that the vaccine intermittently samples throughout the simulation period.

Principal component analysis (PCA) and free energy landscape (FEL) analysis of TgVax452:TLR4 complex in 2D and 3D graphical presentation. (a) Variance of eigenvectors computed from the covariance matrix of the TLR4 structure. (b) Distribution of conformations or states sampled during MD simulations’ trajectory projection onto the first two eigenvectors for TLR4 (c) FEL plot of TLR4 in 2D (left) and 3D (right) graphical presentation corresponding to energy minimized structure. (d) Variance of eigenvectors computed from the covariance matrix of TgVax452 structure. (e) Projection of the motion for the vaccine along the PC1 and PC2. (f) FEL plot of the TgVax452 in 2D (left) and 3D (right) graphical formats corresponding to energy-minimized structure. The plots were generated within the Spyder IDE 5.2.2 environment, utilizing the Matplotlib library of Python v3.9.13
Simulation of the specific vaccine immune responses
After vaccine injection, a rise in IgM was concomitant with a significantly high rate of specific IgM + IgG responses, reaching a peak 10–15 days post-vaccination (PV). Also, after about 5 days PV B-cell population activated, B-memory cells increased and plasma B lymphocytes reached a peak regarding IgM and IgM + IgG. Memory CD4+ T-helper lymphocytes began to increase and reached a plateau around 7 days PV. Also, substantial CD8+ T-cytotoxic lymphocytes were elicited and a low but persistent number of dendritic cells (DCs) were active for over a month PV. Macrophages followed a manner similar to DCs and a remarkably high rate of IFN-γ (over 400000 ng/ml) was provoked PV (Fig. 9).

In silico immune simulation profile of the multi-epitope vaccine construct using C-ImmSim server. A single dose of vaccine injection demonstrated significant rises in IgG + IgM antibodies, followed by IFN-γ upsurge (over 400000 ng/ml), substantial increase in CTL, memory HTL and memory B-cells
Optimization of codon usage, in silico cloning, and mRNA secondary structure analysis
The vaccine’s amino acid sequence was reversely translated into nucleotide sequence based on the Escherichia coli K12 codon usage and the w-table file obtained from http://www.kazusa.or.jp/codon. The G/C content, codon adaptation index (CAI), effective number of codons (Nc), and tRNA adaptation index (tAI) of the codon-optimized vaccine construct were calculated 63.57%, 1.00, 35.667, and 0.386, respectively. The CAI is a normalized score, so values close to 1.0 mean that the most frequent codons are used in a codon-optimized gene [65]. The Nc measures the overall level of codon usage bias of a nucleotide sequence across all codon families. It ranges from 20 (extremely biased genes) to 61 (no bias in codon usage) [66]. The tAI, an index for measuring translation efficiency, is based on the tRNA copy number, therefore it computes a weight (from 0 to 1) for the tAI codon-anticodon interaction. A gene with higher tAI values is more suited to the tRNA pool [67]. By using the optimal codons (Fig. 10a), it can be expected that the transcribed mRNA may have an increased functional half-life and so be translated more efficiently in the bacterial expression system. The codon-optimized vaccine construct was subcloned between XbaI and XhoI restriction sites of the pET-22b(+) vector using SnapGene v5.2.4 software (Fig. 10b). The transcribed mRNA sequence of the vaccine construct is shown in Fig. 10c.

The codon-optimized DNA sequence of the vaccine construct, its DNA-to-RNA transcribed format, subcloning into the pET-22b(+) vector, and mRNA secondary structure. (a) The protein-to-DNA back-translated and codon-optimized sequence of the vaccine construct. (b) The DNA sequence of the vaccine is subcloned between XhoI and XbaI restriction sites of the pET-22b(+) expression vector. (c) The DNA-to-RNA transcribed sequence of the vaccine. The mRNA secondary structure of the vaccine is mapped as positional entropy and base-pairing possibilities. (d) The minimum free energy (MFE) secondary structure is shown as positional entropy probability. (e) The MFE secondary structure is shown as a positional base-pairing probability
The secondary structure of the mRNA molecule can impact the efficiency of translation initiation, as a complex secondary structure can hinder ribosome binding to the ribosome binding site (RBS). Therefore, analyzing the secondary structure of the mRNA molecule can be an effective strategy to optimize translation initiation [68]. The optimal mRNA secondary structure of the codon-optimized vaccine sequence, along with its positional base-pair and entropy probabilities, with a minimum free energy (MFE) of -660.60 kcal/mol is represented in Fig. 10d and e, respectively. The Gibbs free energy of the thermodynamic ensemble, which measures the stability of the secondary structure, was found to be -682.04 kcal/mol. The frequency of the MFE structure in the ensemble, which indicates how often the MFE structure is predicted to occur among all possible secondary structures, was found to be 0.00%. This suggests that the MFE structure is unique and stable. The ensemble diversity was measured to be 240.58, which is relatively high due to the large size of the mRNA sequence. The structural diversity within the Gibbs thermodynamic ensemble is calculated based on the average base-pair distance between all the structures.
Discussion
The ubiquitous protozoan, T. gondii, possesses a complicated life cycle posing a significant burden in different groups of animals and humans, hence a One-Health approach should accompany control measures [69]. Despite years of research, an effective vaccine for clinical use in humans is still lacking [70]. Thus far, most vaccination studies have been focused on the well-known antigenic repertoire of T. gondii, comprising SAGs, GRAs, ROPs, and MICs for vaccine design and immunization purposes. In addition, various proteins such as apical membrane antigen-1 (AMA-1), microneme-2 associated protein (M2AP), apicoplast proteins, rhoptry neck proteins (RONs), cyst matrix antigen (MAG), etc. have been shown to possess good antigenic features as potent vaccine candidates [21]. A set of vaccine platforms (e.g., DNA, vectored, protein/subunit, live-attenuated, and carbohydrate-based) have been utilized with different efficacies and a general consensus seems to be higher efficacy of multiple antigen-expressing vaccines than single antigenic ones [71]. In view of this, the meticulous selection of highly immunogenic antigens and/or epitopes to be combined and constructed in multi-epitope vaccines is an extremely critical step in vaccine design strategy [72].
The reverse vaccinology approach was first introduced by Rino Rappuoli [73], indicating the applicability of deposited data of a given organism in genetic and protein databases towards vaccine design, instead of the organism itself. Since then, unimagined dimensions emerged in the field of vaccine design using cutting-edge machine-learning technologies for the prediction of immunogenic epitopes and evaluation of the multi-epitope vaccines regarding stability, safety, and efficacy [74]. This impressive functionality in vaccine design offered by in silico methods is considerably time- and cost-effective, comparable to conventional vaccine development, leading to rapid, large-scale production of vaccines that benefit global health [29, 75].
In this study, for the first time different tachyzoite-specific Toxoplasma SRS proteins were targeted, due to their surface localization and immunogenicity [25], and deeply analyzed for multi-epitope vaccine design against toxoplasmosis using a set of comprehensive computational methods. Initially, sequential characterization of the selected proteins (SRS29B, SRS20A, SRS51, SRS52A, and SRS57) was performed through the UniProt database, and the text-based FASTA sequences were saved for further analyses. To predict epitopes within the selected proteins, a stringent filtering process was employed. This involve identifying overlapping B-cell epitopes using various online servers, including BepiPred, BCEPS, ABCpred, and SVMTriP. Additionally, the IEDB HLA reference set was consulted to predict human MHC epitopes. The recommended method was used for this, as it is recognized as the most reliable and is regularly updated. Once all epitopes were carefully predicted and screened, one B-cell, two MHC-I, and two MHC-II epitopes were chosen from each SRS protein, to be compiled in the final vaccine construct. Actually, for B-cells, those epitopes with the highest antigenicity, good solubility, and without allergenicity were selected, while MHC-binders were screened for antigenicity, allergenicity, and IFN-γ induction. Of note, predicted MHC-binders showed very high population coverage percentage regarding both MHC-I (95.75%) and MHC-II alleles (92.98%). Our final multi-epitope vaccine construct was built on this basis, with the application of linkers among B-cell (GPGPG), HTL (GPGPG), and CTL (AAY) epitopes. Linkers are indispensable components of recombinant proteins and multi-domain peptides, which preserve biological activity and inter-domain interactions [76]. The “GPGPG” peptide was first reported as a functional inducer of T-lymphocytes responses in vivo by Livingston and colleagues. It also prevents the formation of junctional epitopes [77]. Moreover, “AAY” linkers, mostly used to join CTL epitopes, were embedded as the proteasome cleavage site in mammalian cells [78]. The addition of adjuvants to the vaccine sequence would rectify the problem of low immunogenicity of peptide-based vaccines. Since many effective adjuvants are not available in amino acid sequences, options for intramolecular adjuvants are scarce [79]. With the emphasis on the in vivo cleavable linker, “PPGVS”, surrounded by intramolecular adjuvant (APPHALS) and PADRE sequence, high levels of innate TLR-dependent immunity by adjuvant as well as adaptive immune responses by PADRE are anticipated upon vaccination. The possible synergy between APPHALS and PADRE is expected to enhance both the breadth and depth of the immune response, providing a dual mechanism of action that is more effective than using a single adjuvant alone [80]. By integrating these adjuvants into the vaccine construct, we aim to achieve a balance of safety, immunogenicity, and efficacy, potentially overcoming the limitations faced by conventional peptide-based vaccines.
In the next step, several online servers were used to gather valuable information about vaccine protein function and structure, peptide folding, and accurate molecular dynamic simulations using deep machine-learning algorithms. Based on the ProtParam server, the vaccine should be stable to proceed, as evidenced in this study with an instability index of 33.20, in line with other previous studies on T. gondii multi-epitope vaccine constructs [81,82,83]. Also, the GRAVY score is important regarding vaccine interaction with the surrounding water milieu and may need to adjust treatment to prevent suboptimal performances. A lower GRAVY score indicates higher solubility and hydrophilicity of the molecule [84]. Here, we predicted a hydrophobic (GRAVY: 0.194) and thermal stable (aliphatic index: 70.75) vaccine molecule. The result of solubility was somehow in line with the GRAVY score calculation, showing borderline solubility (0.498). A positive GRAVY score (hydrophobic protein), similar to the score (0.208) obtained by the construct designed by Asghari and colleagues [70], should be considered only in case of protein vaccine production during protein purification experiment by modifying salt gradient concentration, while it may not be implicated in other vaccine formulations (e.g., DNA vaccines). We excluded the allergenic epitopes and picked antigenic ones during the epitope selection process; consequently, our designed multimeric vaccine was shown to be antigenic and non-allergenic in nature.
The multi-epitope vaccine model was further subjected to structural analysis and molecular stability interaction with human TLR4. Secondary structure analysis showed the predominance of exposed residues and disordered regions in the vaccine sequence, which facilitate antigen-antibody interactions. Moreover, random coils, as surface-located protruding structures, were our frequent in the vaccine model; such mobile fragments provide robust insights into epitope identification [85]. The 3D model was predicted through D-I-TASSER homology modeling, while the quality metrics of the selected model, including C-score (-1.18), TM-score, and RMSD (0.46 ± 0.15, 11.3 ± 4.1), number of decoys (600) and cluster density (0.1035), made us perform multiple refinements and subsequent validations, in order to getting it closer to the native structure. This model was subjected to a two-step structural optimization and refinement by GalaxyRefine online server, with further validations using online tools provided by the ProSA-web and SAVES 6.0 web servers. The results showed improvements in some parameters such as Ramachandran plot and Z-score. Although, the cell-mediated immune responses are highly important for parasite clearance, B-cell induction, maturation and the production of neutralizing antibodies are, also, beneficial to combat the infection [86]. Thus, ElliPro tool of the IEDB server was used to predict conformational B-cell epitopes, showing seven epitopic regions with 69, 25, 49, 91, 3, 3 and 15 residues in length and respective scores of 0.778, 0.727, 0.715, 0.64, 0.571, 0.57 and 0.541, respectively.
Proper sensing of self from non-self molecules requires a finely-tuned system of molecular pattern recognition to promptly identify the invasive microbes like T. gondii [87]. In this sense, toll-like receptors (TLRs) play a significant role in reconition of pathogen-associated of molecular patterns (PAMPs) [88]. Induction of innate immune responses in the murine small intestine affected by T. gondii infection was shown to be significantly associated with TLR4 [89]. Further evidence showed that Toxoplasma surface GPIs induces immune cells activation via a MYD88-dependent manner [90]. This led us to perform a protein-protein docking between the finally designed vaccine model (as ligand) and human TLR4 (as receptor) using ZDOCK online program. Of 10 docked complexes provided by this server, the first one was evaluated to be optimal and was shown to possess seven tight hydrogen bonds and five hydrophobic interactions. This robustly-docked vaccine-TLR4 complex was further subjected to MD simulation to accurately predict the stability of the vaccine-receptor construct. The RMSD plot indicated that the vaccine-TLR4 have reached a stable condition during MD simulation. Also, RMSF analysis demonstrated relatively low fluctuations between both molecules, while it increased in the last residues. Further PCA analysis showed higher density of the vaccine model than TLR4, being consistent with higher RMSD values. In line with the residue fluctuation extent for the examined complex, the FEL plots of the TLR4 showed broader energy minima than the vaccine model.
The codon usage of the vaccine model was optimized for a higher efficiency in the bacterial expression system, so that a GC content of 63.57%, CAI of 1.00, effective number of codons of 35.667 and tAI of 0.386 were predicted. Since a GC content between 30 and 70% and a CAI value of over 0.8 are of great interest for better expression, the results of this section were shown to be satisfactory. The mRNA’s free energy and entropy profile particulary for a short sequence at the beginning of the mRNA are critical for the efficiency of translation initiation. The resides that have a low entropy probability can make correct Watson-Crick (GC or AU) and Wobble (GU) base-pairing [91]. In the present study, we used the RNAfold web server for mRNA secondary structure prediction. The total MFE of the codon-optimized vaccine model, its positional base-pair and entropy probabilities was calculated to be -660.60 kcal/mol. According to the large negative values of the Gibbs free energy in the thermodynamic ensemble (682.04 kcal/mol), the transcribed mRNA can likely produce a secondary structure with thermodynamic stability in the host expression system. Upon sensing T. gondii-associated antigens, that are mostly introduced by oral ingestion of oocysts, via innate receptors (e.g., TLR4), the activation of innate lymphoid cells (ILCs) groups occurs, including natural killers (NKs) and groups 2 ILCs which produce Th1 (IFN-γ, TNF-α) and Th2 (IL-4, IL-5, IL-9, IL-13) cytokines, respectively, leading to subsequent specific immune responses. The release of IL-12 by typical and plasmacytoid dendritic cells as well as macrophages leads to the activation of NKs, CD4+ T cells, and CD8+ T cells, promoting cytotoxic effects and the generation of significant levels of IFN-γ as the most imoportant molecule in tachyzoite clearance and parasite elimination [92]. Moreover, specific antibodies are crucial for complement-mediated lysis of tachyzoites and hampering the parasite multiplication [92]. Based on the C-ImmSim server output, a single dose of vaccine injection demonstrated significant rises in IgG + IgM antibodies, followed by IFN-γ upsurge (over 400000 ng/ml), substantial increase in CTL, memory HTL and memory B-cells.
There exist several studies focusing on the multi-epitope vaccine construction using different antigenic fragments of T. gondii to prevent toxoplasmosis. Asghari et al. (2021) designed a novel vaccine candidate using B- and T-cell epitopes derived from T. gondii SAG1 and apicoplast proteins (S2, S5, and L11); they selected T-cell epitopes upon two MCH-I alleles (HLA-A*03:01, HLA-A*23:01) and a single MHC-II allele (HLA-DRB1*04:01). Moreover, a multi-method (BCPREDS, ABCpred, SVMTriP) approach was done for B-cell epitope prediction, similar to our study. Various components connected together using AAY and GPGPG linkers. It was a soluble, 291-residue candidate with a MW of 31.46 kDa and good antigenicity (0.8324). The docked vaccine-human TLR4 complex showed a binding energy score of -893.6 [70]. Forouharmehr (2021) predicted B-cell epitopes (ABCpred, Bcepred, IEDB servers) and T-cell epitopes (SYFPEITHI, NetCTL, IEDB servers) out of 10 Toxoplasma proteins (BiP, GRA1, GRA2, GRA5, MIC8, MIC13, ROP2, P30, PI1, SOD), and selected one B-cell, one CTL and one HTL epitopes out of each protein upon antigenicity evaluation. Internal linkers were (KP)2 and EAAAK was used to connect B-cell epitopes to CTL and HTL epitopes. Notably, mycobacterial heparin-binding hemagglutinin (HBHA) as adjuvant was used. The designed candidate had 730 residues with a MW of 77.67 kDa, pI of 9.32, moderate thermotolerance (54.12) and hydrophilicity (-0.906). Docking performed with TLR4/MD2 using HawkDock server showed that HBHA domain interacted with the receptor (best model energy: 6223.43), with the free binding energy of -43.74 kcal/mol [93]. In a study by Hammed-Akanmu et al. (2022), BCPREDS server was used to predict B-cell epitopes out of Toxoplasma MIC3, GRA7, and ROP2. Also, predicted CTL (NetCTL 1.2) and HTL (IEDB mhcii) epitopes were screened regarding antigenicity, allergenicity, toxicity and immunogenicity (MHC-I) or IFN-γ induction (MHC-II). HTL epitopes were predicted based on HLA-DQA1*05:01, HLA-DQB1*02:01, and HLADRB1*03:01 alleles. Linkers were AAY (CTL epitopes) and KK (HTL and B-cell epitopes) and 50 S ribosomal protein L7/L12 was used as adjuvant. The proposed candidate had a MW of 51 kDa with 469 residues in length, a pI of 5.46, with good antigenicity (0.6182), hydrophilicity (-0.485), and solubility. Docking with TLR4 showed better results than with TLR2, demonstrating a global energy of 3.87 [94]. In a recent vaccine design study by Dalir-Ghaffari and Rahimi (2024), 12 B-cell epitopes (ABCpred server) and 24 T-cell epitopes (IEDB; HLA reference set) derived from calcium-dependent protein kinases (CDPKs) 1, 2, 3, and 5 were selected for multi-epitope vaccine construction using AAY and KK linkers, and 50 S ribosomal protein L7/L12 as adjuvant. This polytope contained 680 residues with 74.66 MW, an alkaline nature (pI: 9.21), high antigenic index (0.9099), high thermotolerance (75.51), and negative GRAVY (-0.467). The authors refined the 3D structure, while no docking was performed [95]. In comparison with above studies, we selected surface-localized, immunogenic SRS proteins expressed in tachyzoites and gathered B-cell (multi-method approach) and T-cell epitopes (IEDB HLA reference set) with high population coverage and extensive screening, and those selected were connected with regular (AAY, GPGPG) and specific linkers (PPGVS, PMGLP). Our designed candidate was a stable (33.20) protein with a 44.07 kDa MW and high antigenicity (0.9639). The vaccine candidate could effectively bound with TLR4 with relatively low fluctuations and the elicited immune responses were equal (IFN-γ) and even higher (specific antibodies) than those predicted in Hammed-Akanmu et al. (2022) study. There are growing experimental evidences that Toxoplasma SRS-related proteins are among robust immunogenic proteins which can substantially elicit specific immunity in animal modes of toxoplasmosis [96,97,98]. As a final note, all predictions in the current study are merely computer-based estimations that must be validated through wet experiments.
It is noteworthy that computer-based studies may have some limitations and the following considerations should be taken into account in future studies: i) experimental data could enhance the reliability of the obtained protein sequence and its 3D model prediction; ii) the accuracy of predictions (e.g., antigenicity, allergenicity, epitope mapping) depends on the quality of algorithms and databases used; iii) the immunogenicity of in silico predicted epitopes should be validated using proper tools such as the enzyme-linked immunosorbent assay (ELISA), enzyme-linked immunosorbent spot (ELISpot), lymphocyte proliferation assay (LPA), etc.; iv) experimental validation with diverse HLA types should be considered for HLA-binding epitopes predicted in the current study; and v) the clinical relevance (i.e., vaccine development) of the proposed vaccine candidate should be further clarified both in vitro and in vivo.
Conclusion
Rational designing and evaluation of a vaccine candidate demand the utilization of stringent Immunoinformatics algorithms for the prediction of immunodominant epitopes and other vaccine-related parameters. However, the main challenge in vaccine development against T. gondii lies in the fact that its strains are highly diverse, so may have different virulence factors, antigenic profiles, and immune evasion mechanisms. Therefore, broad protection and effectiveness of anti-T. gondii vaccines mainly depend on (i) using other strains of different genotypes, (ii) identification of conserved antigens (or epitopes) that are present across all strains and can elicit robust immune responses, (iii) identifying more prevalent or more virulent T. gondii strains to develop vaccines that are tailored to specific geographic locations or demographics (adapted the approach of personalized vaccinology), and (iv) avoiding antigens that may induce immune evasion mechanisms or exacerbate disease symptoms. Almost all these parameters can be measured through computational vaccinology. In this in silico study, the most immunodominant and safe B- and T-cell epitopes of several T. gondii tachyzoite-specific SRS antigens were used to increase the coverage of the designed vaccine candidate. In addition, some critical items (i.e., safety, antigenicity, allergenicity, vaccine adjuvanticity, immune response profile, codon usage in a bacterial expression system, and mRNA secondary structure) were checked in the computer environment. Plus, molecular docking and dynamic simulations were used to provide rewarding insights at an atomic level into the interactions between the vaccine candidate and human TLR4. These are preliminary data which are helpful in predicting the efficacy and safety of a vaccine candidate. However, moving forward, further experimental validation and preclinical studies are warranted to assess the efficacy and safety of the multi-epitope vaccine in animal models and eventually in human populations. Today, there is an increasing demand for machine learning algorithms and computational simulations in vaccine production companies not only to optimize and improve the vaccine development process but also potentially lead to more efficient and effective manufacturing. Nevertheless, such computer-aided studies meet some pitfalls such as a lack of biochemical and safety assessment in laboratory animal models challenged with different acute T. gondii strains. Thus, further interpretation of the vaccine efficacy demands subsequent experimental studies.
Data availability
The datasets analysed during the current study are available in the UniProtKB repository, with accession numbers of S8FBM7 (SRS20A), A0A125YP09 (SRS29B), S7W107 (SRS51), A0A125YJA5 (SRS52A), and A0A125YY85 (SRS57).
Change history
13 September 2024
A Correction to this paper has been published: https://doi.org/10.1186/s12879-024-09881-1
References
Dubey JP. Outbreaks of clinical toxoplasmosis in humans: five decades of personal experience, perspectives and lessons learned. Parasites Vectors. 2021;14(1):1–12.
Amouei A, Sarvi S, Sharif M, Aghayan SA, Javidnia J, Mizani A, et al. A systematic review of Toxoplasma gondii genotypes and feline: geographical distribution trends. Transbound Emerg Dis. 2020;67(1):46–64.
Fallahi S, Rostami A, Shiadeh MN, Behniafar H, Paktinat S. An updated literature review on maternal-fetal and reproductive disorders of Toxoplasma Gondii infection. J Gynecol Obstet Hum Reprod. 2018;47(3):133–40.
Foroutan-Rad M, Majidiani H, Dalvand S, Daryani A, Kooti W, Saki J, et al. Toxoplasmosis in blood donors: a systematic review and meta-analysis. Transfus Med Rev. 2016;30(3):116–22.
Dard C, Marty P, Brenier-Pinchart M-P, Garnaud C, Fricker-Hidalgo H, Pelloux H, et al. Management of toxoplasmosis in transplant recipients: an update. Expert Rev Anti-Infective Therapy. 2018;16(6):447–60.
Pereira KS, Franco RM, Leal DA. Transmission of toxoplasmosis (Toxoplasma Gondii) by foods. Adv Food Nutr Res. 2010;60:1–19.
Daryani A, Sarvi S, Aarabi M, Mizani A, Ahmadpour E, Shokri A, et al. Seroprevalence of Toxoplasma Gondii in the Iranian general population: a systematic review and meta-analysis. Acta Trop. 2014;137:185–94.
Moncada PA, Montoya JG. Toxoplasmosis in the fetus and newborn: an update on prevalence, diagnosis and treatment. Expert Rev anti-infective Therapy. 2012;10(7):815–28.
Wang Z-D, Liu H-H, Ma Z-X, Ma H-Y, Li Z-Y, Yang Z-B, et al. Toxoplasma Gondii infection in immunocompromised patients: a systematic review and meta-analysis. Front Microbiol. 2017;8:389.
Stelzer S, Basso W, Silván JB, Ortega-Mora LM, Maksimov P, Gethmann J, et al. Toxoplasma Gondii infection and toxoplasmosis in farm animals: risk factors and economic impact. Food Waterborne Parasitol. 2019;15:e00037.
Jafarpour Azami S, Mohammad Rahimi H, Mirjalali H, Zali MR. Unravelling Toxoplasma treatment: conventional drugs toward nanomedicine. World J Microbiol Biotechnol. 2021;37(3):1–9.
Sharif M, Sarvi S, Pagheh AS, Asfaram S, Rahimi MT, Mehrzadi S, et al. The efficacy of herbal medicines against Toxoplasma Gondii during the last 3 decades: a systematic review. Can J Physiol Pharmacol. 2016;94(12):1237–48.
Dunay IR, Gajurel K, Dhakal R, Liesenfeld O, Montoya JG. Treatment of toxoplasmosis: historical perspective, animal models, and current clinical practice. Clin Microbiol Rev. 2018;31(4):e00057–17.
Elmore SA, Jones JL, Conrad PA, Patton S, Lindsay DS, Dubey J. Toxoplasma Gondii: epidemiology, feline clinical aspects, and prevention. Trends Parasitol. 2010;26(4):190–6.
Wang J-L, Zhang N-Z, Li T-T, He J-J, Elsheikha HM. Zhu X-QJTip. Advances in the development of anti-toxoplasma gondii vaccines: challenges, opportunities, and perspectives. 2019;35(3):239–53.
Li Y, Zhou H. Moving towards improved vaccines for Toxoplasma Gondii. Expert Opin Biol Ther. 2018;18(3):273–80.
Hiszczyńska-Sawicka E, Gatkowska JM, Grzybowski MM, Długońska H. Veterinary vaccines against toxoplasmosis. Parasitology. 2014;141(11):1365–78.
Munoz M, Liesenfeld O, Heimesaat MM. Immunology of Toxoplasma Gondii. Immunol Rev. 2011;240(1):269–85.
Boothroyd JC. Toxoplasma Gondii: 25 years and 25 major advances for the field. Int J Parasitol. 2009;39(8):935–46.
Daryani A, Kalani H, Sharif M, Ziaei H, Sarvi S, Ahmadpour E. Toxoplasma Gondii: a review of excretory secretory antigens. J Mazandaran Univ Med Sci. 2013;22(2):220–32.
Mamaghani AJ, Fathollahi A, Arab-Mazar Z, Kohansal K, Fathollahi M, Spotin A, et al. Toxoplasma Gondii vaccine candidates: a concise review. Ir J Med Sci (1971-). 2023;192(1):231–61.
Zhang X, Yuan H, Mahmmod YS, Yang Z, Zhao M, Song Y, et al. Insight into the current Toxoplasma gondii DNA vaccine: a review article. Expert Rev Vaccines. 2023;22(1):66–89.
Zhang Y, Li D, Lu S, Zheng B. Toxoplasmosis vaccines: what we have and where to go? npj Vaccines. 2022;7(1):131.
Jung C, Lee CY-F, Grigg ME. The SRS superfamily of Toxoplasma surface proteins. Int J Parasitol. 2004;34(3):285–96.
Theisen TC, Boothroyd JC. Transcriptional signatures of clonally derived Toxoplasma tachyzoites reveal novel insights into the expression of a family of surface proteins. PLoS ONE. 2022;17(2):e0262374.
Salemi A, Pourseif MM, Omidi Y. Next-generation vaccines and the impacts of state-of-the-art in-silico technologies. Biologicals. 2021;69:83–5.
Kelly DF, Rappuoli R. Reverse vaccinology and vaccines for serogroup B Neisseria meningitidis. Adv Exp Med Biol. 2005;568:217–23.
Pourseif MM, Moghaddam G, Nematollahi A, Khordadmehr M, Naghili B, Dehghani J, et al. Vaccination with rEGVac elicits immunoprotection against different stages of Echinococcus granulosus life cycle: a pilot study. Acta Trop. 2021;218:105883.
Omidi Y, Pourseif MM, Omidian H, Barar J. Nanoscale vaccines: Design, delivery, and applications. In: Sougata Jana SJ, editor. Nanoengineering of Biomaterials. Volume I. Wiley Online Library; 2022. pp. 437–68.
Pourseif MM, Yousefpour M, Aminianfar M, Moghaddam G, Nematollahi A. A multi-method and structure-based in silico vaccine designing against Echinococcus granulosus through investigating enolase protein. Bioimpacts. 2019;9(3):131–44.
Vita R, Mahajan S, Overton JA, Dhanda SK, Martini S, Cantrell JR, et al. The immune epitope database (IEDB): 2018 update. Nucleic Acids Res. 2019;47(D1):D339–43.
Mahapatra SR, Dey J, Raj TK, Misra N, Suar M. Designing a Next-Generation Multiepitope-based vaccine against Staphylococcus aureus using reverse vaccinology approaches. Pathogens. 2023;12(3):376.
Dey J, Mahapatra SR, Singh PK, Prabhuswamimath SC, Misra N, Suar M. Designing of multi-epitope peptide vaccine against Acinetobacter baumannii through combined immunoinformatics and protein interaction–based approaches. Immunol Res. 2023;71(4):639–62.
Pourseif MM, Parvizpour S, Jafari B, Dehghani J, Naghili B, Omidi Y. A domain-based vaccine construct against SARS-CoV-2, the causative agent of COVID-19 pandemic: development of self-amplifying mRNA and peptide vaccines. Bioimpacts. 2021;11(1):65–84.
Gasteiger E, Hoogland C, Gattiker A, Wilkins MR, Appel RD, Bairoch A. Protein identification and analysis tools on the ExPASy server. Proteom Protocols Handb. 2005:571–607.
Doytchinova IA, Flower DR. VaxiJen: a server for prediction of protective antigens, tumour antigens and subunit vaccines. BMC Bioinformatics. 2007;8(1):1–7.
Magnan CN, Zeller M, Kayala MA, Vigil A, Randall A, Felgner PL, et al. High-throughput prediction of protein antigenicity using protein microarray data. Bioinformatics. 2010;26(23):2936–43.
Dimitrov I, Naneva L, Doytchinova I, Bangov I. AllergenFP: allergenicity prediction by descriptor fingerprints. Bioinformatics. 2014;30(6):846–51.
Dimitrov I, Bangov I, Flower DR, Doytchinova I. AllerTOP v. 2—a server for in silico prediction of allergens. J Mol Model. 2014;20(6):1–6.
Hebditch M, Carballo-Amador MA, Charonis S, Curtis R, Warwicker J. Protein–Sol: a web tool for predicting protein solubility from sequence. Bioinformatics. 2017;33(19):3098–100.
Klausen MS, Jespersen MC, Nielsen H, Jensen KK, Jurtz VI, Sonderby CK, et al. NetSurfP-2.0: improved prediction of protein structural features by integrated deep learning. Proteins. 2019;87(6):520–7.
Klausen MS, Jespersen MC, Nielsen H, Jensen KK, Jurtz VI, Soenderby CK et al. NetSurfP-2.0: Improved prediction of protein structural features by integrated deep learning. Proteins: Structure, Function, Bioinformatics. 2019;87(6):520-7.
Li Y, Zhang C, Feng C, Pearce R, Lydia Freddolino P, Zhang Y. Integrating end-to-end learning with deep geometrical potentials for ab initio RNA structure prediction. Nat Commun. 2023;14(1):5745.
Lee GR, Heo L, Seok C. Effective protein model structure refinement by loop modeling and overall relaxation. Proteins. 2016;84(Suppl 1):293–301.
Ponomarenko J, Bui H-H, Li W, Fusseder N, Bourne PE, Sette A, et al. ElliPro: a new structure-based tool for the prediction of antibody epitopes. BMC Bioinformatics. 2008;9(1):1–8.
Craig DB, Dombkowski AA. Disulfide by Design 2.0: a web-based tool for disulfide engineering in proteins. BMC Bioinformatics. 2013;14(1):1–7.
Pierce BG, Wiehe K, Hwang H, Kim BH, Vreven T, Weng Z. ZDOCK server: interactive docking prediction of protein-protein complexes and symmetric multimers. Bioinformatics. 2014;30(12):1771–3.
Hollingsworth SA, Dror RO. Molecular Dynamics Simulation for all. Neuron. 2018;99(6):1129–43.
Moin AT, Singh G, Ahmed N, Saiara SA, Timofeev VI, Ahsan Faruqui N, et al. Computational designing of a novel subunit vaccine for human cytomegalovirus by employing the immunoinformatics framework. J Biomol Struct Dyn. 2023;41(3):833–55.
Moin AT, Ullah MA, Patil RB, Faruqui NA, Araf Y, Das S, et al. A computational approach to design a polyvalent vaccine against human respiratory syncytial virus. Sci Rep. 2023;13(1):9702.
Araf Y, Moin AT, Timofeev VI, Faruqui NA, Saiara SA, Ahmed N et al. Immunoinformatic Design of a multivalent peptide vaccine against mucormycosis: Targeting FTR1 protein of major causative Fungi. Front Immunol. 2022;13.
Schmid N, Eichenberger AP, Choutko A, Riniker S, Winger M, Mark AE, et al. Definition and testing of the GROMOS force-field versions 54A7 and 54B7. Eur Biophys J. 2011;40(7):843–56.
Rapin N, Lund O, Castiglione F. Immune system simulation online. Bioinformatics. 2011;27(14):2013–4.
Jung SK, McDonald K. Visual gene developer: a fully programmable bioinformatics software for synthetic gene optimization. BMC Bioinformatics. 2011;12:340.
Gruber AR, Lorenz R, Bernhart SH, Neubock R, Hofacker IL. The Vienna RNA websuite. Nucleic Acids Res. 2008;36(Web Server issue):W70–4.
Graille M, Stura EA, Bossus M, Muller BH, Letourneur O, Battail-Poirot N, et al. Crystal structure of the complex between the monomeric form of Toxoplasma Gondii surface antigen 1 (SAG1) and a monoclonal antibody that mimics the human immune response. J Mol Biol. 2005;354(2):447–58.
Pourseif MM, Masoudi-Sobhanzadeh Y, Azari E, Parvizpour S, Barar J, Ansari R, et al. Self-amplifying mRNA vaccines: Mode of action, design, development and optimization. Drug Discov Today. 2022;27(11):103341.
Yang J, Yan R, Roy A, Xu D, Poisson J, Zhang Y. The I-TASSER suite: protein structure and function prediction. Nat Methods. 2015;12(1):7–8.
Roy A, Kucukural A, Zhang Y. I-TASSER: a unified platform for automated protein structure and function prediction. Nat Protoc. 2010;5(4):725–38.
Denkers EY. Toll-like receptor initiated host defense against Toxoplasma Gondii. J Biomed Biotechnol. 2010;2010:737125.
Park BS, Song DH, Kim HM, Choi BS, Lee H, Lee JO. The structural basis of lipopolysaccharide recognition by the TLR4-MD-2 complex. Nature. 2009;458(7242):1191–5.
Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, et al. UCSF Chimera–a visualization system for exploratory research and analysis. J Comput Chem. 2004;25(13):1605–12.
Pawlus S, Grzybowski A, Kolodziej S, Wikarek M, Dzida M, Goralski P, et al. Density scaling based detection of thermodynamic regions of Complex Intermolecular interactions characterizing Supramolecular structures. Sci Rep. 2020;10(1):9316.
Zhang D, Lazim R. Application of conventional molecular dynamics simulation in evaluating the stability of apomyoglobin in urea solution. Sci Rep. 2017;7:44651.
Bahiri-Elitzur S, Tuller T. Codon-based indices for modeling gene expression and transcript evolution. Comput Struct Biotechnol J. 2021;19:2646–63.
Fuglsang A. The ‘effective number of codons’ revisited. Biochem Biophys Res Commun. 2004;317(3):957–64.
Miller JB, Brase LR, Ridge PG. ExtRamp: a novel algorithm for extracting the ramp sequence based on the tRNA adaptation index or relative codon adaptiveness. Nucleic Acids Res. 2019;47(3):1123–31.
Chen YL, Wen JD. Translation initiation site of mRNA is selected through dynamic interaction with the ribosome. Proc Natl Acad Sci U S A. 2022;119(22):e2118099119.
Innes EA, Hamilton C, Garcia JL, Chryssafidis A, Smith D. A one health approach to vaccines against Toxoplasma Gondii. Food Waterborne Parasitol. 2019;15:e00053.
Asghari A, Shamsinia S, Nourmohammadi H, Majidiani H, Fatollahzadeh M, Nemati T, et al. Development of a chimeric vaccine candidate based on Toxoplasma Gondii major surface antigen 1 and apicoplast proteins using comprehensive immunoinformatics approaches. Eur J Pharm Sci. 2021;162:105837.
Chu K-B, Quan F-S. Advances in Toxoplasma gondii vaccines: current strategies and challenges for vaccine development. Vaccines. 2021;9(5):413.
Pourseif MM, Moghaddam G, Daghighkia H, Nematollahi A, Omidi Y. A novel B- and helper T-cell epitopes-based prophylactic vaccine against Echinococcus Granulosus. Bioimpacts. 2018;8(1):39–52.
Rappuoli R. Reverse vaccinology. Curr Opin Microbiol. 2000;3(5):445–50.
Martinelli DD. Silico vaccine design: a tutorial in immunoinformatics. Healthc Analytics. 2022;2:100044.
Ysrafil Y, Sapiun Z, Astuti I, Anasiru MA, Slamet NS, Hartati H, et al. Designing multi-epitope based peptide vaccine candidates against SARS-CoV-2 using immunoinformatics approach. Bioimpacts. 2022;12(4):359–70.
Chen X, Zaro JL, Shen W-C. Fusion protein linkers: property, design and functionality. Adv Drug Deliv Rev. 2013;65(10):1357–69.
Livingston B, Crimi C, Newman M, Higashimoto Y, Appella E, Sidney J, et al. A rational strategy to design multiepitope immunogens based on multiple th lymphocyte epitopes. J Immunol. 2002;168(11):5499–506.
Yang Y, Sun W, Guo J, Zhao G, Sun S, Yu H, et al. In silico design of a DNA-based HIV-1 multi-epitope vaccine for Chinese populations. Hum Vaccines Immunotherapeutics. 2015;11(3):795–805.
Lei Y, Zhao F, Shao J, Li Y, Li S, Chang H, et al. Application of built-in adjuvants for epitope-based vaccines. PeerJ. 2019;6:e6185.
Zhao T, Cai Y, Jiang Y, He X, Wei Y, Yu Y, et al. Vaccine adjuvants: mechanisms and platforms. Signal Transduct Target Ther. 2023;8(1):283.
Khodadadi M, Ghaffarifar F, Dalimi A, Ahmadpour E. Immunogenicity of in-silico designed multi-epitope DNA vaccine encoding SAG1, SAG3 and SAG5 of Toxoplasma Gondii adjuvanted with CpG-ODN against acute toxoplasmosis in BALB/c mice. Acta Trop. 2021;216:105836.
Onile OS, Ojo GJ, Oyeyemi BF, Agbowuro GO, Fadahunsi AI. Development of multiepitope subunit protein vaccines against Toxoplasma gondii using an immunoinformatics approach. NAR Genomics Bioinf. 2020;2(3):lqaa048.
Ahmed N, Rani NA, Robin TB, Shovo MI, Prome AA, Sultana S et al. Designing a Multi-Epitope Subunit Vaccine Against Toxoplasma Gondii Through Reverse Vaccinology Approach. Available at SSRN 4733701.
Kar T, Narsaria U, Basak S, Deb D, Castiglione F, Mueller DM, et al. A candidate multi-epitope vaccine against SARS-CoV-2. Sci Rep. 2020;10(1):1–24.
Wang Y. Bioinformatics analysis of NetF proteins for designing a multi-epitope vaccine against Clostridium perfringens infection. Infect Genet Evol. 2020;85:104461.
Mévélec M-N, Lakhrif Z, Dimier-Poisson I. Key limitations and new insights into the Toxoplasma Gondii parasite stage switching for future vaccine development in human, livestock, and cats. Front Cell Infect Microbiol. 2020:697.
Moresco EMY, LaVine D, Beutler B. Toll-like receptors. Curr Biol. 2011;21(13):R488–93.
Zare-Bidaki M, Hakimi H, Abdollahi SH, Zainodini N, Arababadi MK, Kennedy D. TLR4 in toxoplasmosis; friends or foe? Microb Pathog. 2014;69:28–32.
Furuta T, Kikuchi T, Akira S, Watanabe N, Yoshikawa Y. Roles of the small intestine for induction of toll-like receptor 4-mediated innate resistance in naturally acquired murine toxoplasmosis. Int Immunol. 2006;18(12):1655–62.
Debierre-Grockiego F, Campos MA, Azzouz N, Schmidt J, Bieker U, Resende MG, et al. Activation of TLR2 and TLR4 by glycosylphosphatidylinositols derived from Toxoplasma Gondii. J Immunol. 2007;179(2):1129–37.
Garcia-Martin JA, Clote P. RNA thermodynamic structural entropy. PLoS ONE. 2015;10(11):e0137859.
Sasai M, Pradipta A, Yamamoto M. Host immune responses to Toxoplasma Gondii. Int Immunol. 2018;30(3):113–9.
Forouharmehr A. Engineering an efficient poly-epitope vaccine against Toxoplasma gondii infection: a computational vaccinology study. Microb Pathog. 2021;152:104646.
Hammed-Akanmu M, Mim M, Osman AY, Sheikh AM, Behmard E, Rabaan AA, et al. Designing a multi-epitope vaccine against Toxoplasma Gondii: an immunoinformatics approach. Vaccines. 2022;10(9):1389.
Ghaffari AD, Rahimi F. Immunoinformatics studies and design of a novel multi-epitope peptide vaccine against Toxoplasma Gondii based on calcium-dependent protein kinases antigens through an in-silico analysis. Clin Experimental Vaccine Res. 2024;13(2):146.
Gül C, Gül A, Karakavuk T, Erkunt Alak S, Karakavuk M, Can H et al. A novel DNA vaccine encoding the SRS13 protein administered by electroporation confers protection against chronic toxoplasmosis. Vaccine. 2024.
Guo X-D, Zhou C-X, Cui L-L, Qiu H-J, Wang Y-L, Fu M, et al. Evaluation of protective immunity induced by a DNA vaccine encoding SAG2 and SRS2 against Toxoplasma gondii infection in mice. Acta Trop. 2024;257:107302.
Yan A, Tian J, Ye J, Gao C, Ye L, Zhang D, et al. Construction of Toxoplasma Gondii SRS29C nucleic acid vaccine and comparative immunoprotective study of an SRS29C and SAG1 combination. Mol Biochem Parasitol. 2024;259:111630.
Acknowledgements
The authors of the present study would like to apprecaite the Deputy of Research and Technology, Neyshabur University of Medical Sciences, Neyshabur, for their kind support.
Funding
This research did not receive any specific grant from funding agencies in the public, commercial, or not-for-profit sectors.
Author information
Authors and Affiliations
Contributions
HM & MMP: Conceptualized the main research idea; performed molecular docking and dynamic simulations, visualizations and interpretations; immunoinformatics analyses; supervised the research procedure and reviewed the final version of the manuscript. BK, MRS, and AN: Curated the research data and drafted the initial version of the manuscript; All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Ethical approval
The present study was approved by the ethic committee of the Neyshabur University of Medical Sciences (Code No.: IR.NUMS.REC.1400.050).
Consent for publication
Not applicable.
Consent to participate
Not applicable.
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
The original online version of this article was revised: Following publication of the original article, we have been notified that due to a misunderstanding in proof corrections, there are some corrections needed in the affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Supplementary Material 1
: Table S1. Linear B-cell epitopes of T. gondii SRS20A predicted using the four different web-servers
Supplementary Material 2
: Table S2. Linear B-cell epitopes of T. gondii SRS29B predicted using the four different web-servers
Supplementary Material 3
: Table S3. Linear B-cell epitopes of T. gondii SRS51 predicted using the four different web-servers
Supplementary Material 4
: Table S4. Linear B-cell epitopes of T. gondii SRS52A predicted using the four different web-servers
Supplementary Material 5
: Table S5. Linear B-cell epitopes of T. gondii SRS57 predicted using the four different web-servers
Supplementary Material 6
: Table S6. Predicted MHC-I binding peptides of the tachyzoite-specific SRS proteins using frequently-occurring HLA alleles of the IEDB Recommended method
Supplementary Material 7
: Table S7. Predicted MHC-II binding epitopes of the selected tachyzoite-specific SRS proteins using frequently-occurring HLA alleles of the IEDB Recommended method
Supplementary Material 8
: Fig. S1. Secondary structure prediction of the final multi-epitope vaccine construct using NetSurfP-2.0 web server
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Majidiani, H., Pourseif, M.M., Kordi, B. et al. TgVax452, an epitope-based candidate vaccine targeting Toxoplasma gondii tachyzoite-specific SAG1-related sequence (SRS) proteins: immunoinformatics, structural simulations and experimental evidence-based approaches. BMC Infect Dis 24, 886 (2024). https://doi.org/10.1186/s12879-024-09807-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12879-024-09807-x