
Abstract
Background
Hemophagocytic lymphohistiocytosis (HLH) is a heterogeneous and potentially fatal disease that presents symptoms of persistent fever, splenomegaly and cytopenia. Primary HLH is identified as an autosomal recessive disorder with causative genes including HPLH1, PRF1, UNC13D, STX11 and STXBP2.
Case presentation
Here, we reported an 8-month-old female patient with compound heterozygosity in the UNC13D gene. The patient, who presented typical symptoms, was diagnosed with HLH based on HLH-2004 guidelines. High-throughput amplicon sequencing for the full-length exon, including a 5 bp padding region and 6 HLH-related genes, was performed to identify the pathogenic mutations in this patient. In all, 9 heterozygous variations were detected, namely, 7 nonpathogenic SNPs, one nonsense mutation (NM_199242.2:c.2206C > T, p.Gln736X), and one splicing mutation (NM_199242.2:c.2709 + 1G > A). These two mutations were considered pathogenic according to previous studies and functional prediction. A two-generation pedigree analysis based on Sanger sequencing was performed to confirm the result.
Conclusion
Compound heterozygosity in the UNC13D gene was identified in trans and considered a causative mutation in a female patient with HLH. The nonsense mutation (NM_199242.2:c.2206C > T, p.Gln736X) was novel in cases of HLH. Our data expand the spectrum of HLH-related mutations in China and demonstrate the potential of high-throughput amplicon sequencing in the diagnosis of HLH.
Similar content being viewed by others

Background
Hemophagocytic lymphohistiocytosis (HLH) is a heterogeneous and potentially fatal disease that presents with symptoms of persistent fever, splenomegaly and cytopenia. The first case was reported in 1952 by Farquhar et al. [1]; uncontrolled proliferation of activated macrophages and T lymphocytes are hallmarks of this syndrome [2]. Two types of HLH were described: a primary and a secondary form [3]. Primary HLH, also termed familial hemophagocytic lymphohistiocytosis (FHL), is an autosomal recessive genetic disorder with five major causative genes, namely, HPLH1, PRF1, UNC13D, STX11 and STXBP2 (Table 1) [4]. The incidence of FHL was estimated to approximately 1:100000 and 1:50000, respectively, according to two independent studies [5, 6]; however, worldwide, the incidence of FHL is still unclear. The secondary HLH, or acquired HLH, is mostly caused by the strong immunological activation of the immune system (e.g., severe infection of virus) rather than genetic mutations, but it may also develop during malignancies. It is difficult to distinguish these two types of HLH from each other, which necessitates an approach to differential diagnoses.
Here, we performed high-throughput amplicon sequencing to detect mutations in whole exons of 6 HLH-related genes for an 8-month-old female patient, who was diagnosed with HLH based on HLH-2004 guidelines [7]. As a result, 9 heterozygous variations, namely, 7 SNPs, one nonsense mutation (c.2206C > T) and one splicing mutation (c.2709 + 1G > A), in the UNC13D gene were detected. The nonsense mutation was reported in HLH for the first time. The compound heterozygous mutations in the UNC13D gene were considered causative mutations for the patient based on previous studies and functional predictions. A two-generation pedigree analysis showed that they were inherited from the parents. The result suggests great potential of high-throughput amplicon sequencing in the diagnosis of HLH.
Case presentation
This study was approved by the ethics committee on the use of human subjects at Wuhan Children’s Hospital. Informed consent from the patient’s parents was obtained before collecting blood samples. The patient and her parents are all Han Chinese from Hubei province of China.
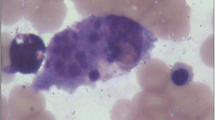
This 8-month-old female patient was admitted to Wuhan Children’s Hospital after 2 days of fever, with a temperature measured at 39.0 °C on regular examination. Blood analysis showed low platelets (PLT, 45 × 109/L) and low hemoglobin (Hb, 7.7 g/100 mL). Low NK cell activity (4.45%) and plasma albumin (18.2 g/L) were also observed. Moderate splenomegaly was revealed by ultrasound examination. The bone marrow examination suggested hemophagocytosis (Fig. 1), and no features of malignancy were observed. Since six of the eight criteria of HLH-2004 were fulfilled [7], this patient was diagnosed with HLH. To confirm the subtype of HLH and to investigate causative mutations in this patient, high-throughput amplicon sequencing and two-generation pedigree analysis were performed.

The bone marrow examination. Phagocytosis was clearly observed without any evidence of malignancy
Genomic DNA was purified from peripheral blood mononuclear cells (PBMCs) with a commercial kit purchased from TIANGEN Biotech (Beijing, China). Multiple PCR primers were designed for 6 genes using Ion AmpliSeq™ Designer (Table 1); the SH2D1A and XIAP genes were included for distinguishing X-linked lymphoproliferative syndrome (XLP), which has a strong resemblance to HLH [8]. All exons of these genes were covered by 198 amplicons, including 5 bp of padding region. High-throughput amplicon sequencing was performed using an ION S5XL genetic analyzer (Thermo Fisher Scientific, MA, USA). VariantCaller V1.0 was used to detect mutations from sequencing data.
As a result, 9 heterozygous variations were detected, namely, 5 synonymous mutations, one missense mutation, one noncoding region mutation, one nonsense mutation and one splicing mutation (Table 2). All mutations were considered to be benign according to the dbSNP database except for the nonsense mutation NM_199242.2:c.2206C > T and the splicing mutation NM_199242.2:c.2709 + 1G > A. The splicing mutation c.2709 + 1G > A has been reported in patients with HLH with compound heterozygous mutations in the UNC13D gene [9, 10], it was predicted to be broken wild-type donor site and most probably affecting splicing, according to the human splicing finder (HSF, version 3.1). However, the nonsense mutation c.2206C > T, which resulted in the introduction of a premature stop codon and produced truncated mRNA (p.Gln736X), was reported in HLH for the first time. The compound heterozygosity was considered to be causative in this case, based on functional alteration analysis and previous reports.
Both mutations were confirmed by Sanger sequencing, and a two-generation pedigree analysis was performed with the parents’ samples. The results showed these two mutations were inherited from the father and mother (Fig. 2), presenting a typical autosomal recessive mode of inheritance.

Two-generation pedigree analysis for two heterozygous mutations in the UNC13D gene. a Novel nonsense mutation (c.2206C > T) in UNC13D-exon24; b Reported splicing mutation (c.2709 + 1G > A) in UNC13D-exon28. The position of the mutation is marked with a red arrow. The results showed that the nonsense mutation was inherited from father and the splicing mutation was inherited from mother. Zygosity was indicated by letters of degenerate bases (Y: C/T, R: A/G)
Discussion and conclusions
In this case, an 8-month-old female patient was admitted to Wuhan Children’s Hospital and diagnosed with HLH based on clinical features, including persistent fever, moderate splenomegaly, low PLT and Hb, decreased NK-cell activity, and hemophagocytosis with no evidence of malignancy. Compound heterozygous mutations consisting of a nonsense mutation (c.2206C > T) and a splicing mutation (c.2709 + 1G > A) in the UNC13D gene were identified in this patient using high-throughput amplicon sequencing. A two-generation pedigree analysis based on Sanger sequencing confirmed the results and revealed a typical autosomal recessive inheritance mode in this family.
HLH is a heterogeneous disease with two major conditions and five subtypes; additionally, many other conditions can lead to the clinical picture of HLH, such as malignancies, rheumatoid disorders, and XLP [7]. Molecular diagnosis based on genetic tests provide a powerful tool for differential diagnoses of HLH, which is important for appropriate treatment. Bianca et al. reported an approach for the genetic screening of patients with HLH using targeted high-throughput sequencing of 12 related genes, which resulted in a diagnosis in 22 of 58 patients in the prospective cohort [11]. Further in-depth investigations of the phenotype-genotype relationship of HLH are required to develop better sequencing panels for diagnosis.
The UNC13D gene, which encodes a protein involved in the cytotoxic activity of T lymphocytes, was confirmed as a causative gene of familial HLH type 3. Pathogenic mutations in the UNC13D gene are very common in patients with HLH, as 17–19% of FHL patients from Turkey and Germany, 89% from Korea, and 30% from Japan were identified to have pathogenic mutations in the UNC13D gene [12,13,14]. Compound heterozygosity in the UNC13D gene was also reported, especially in the splicing site [9, 10]. However, there has been no genetic investigation on a substantial number of HLH patients in China. In this case, we identified the causative mutations of this patient in 24 h, using our customized panel which was designed automatically with online tool and purchased from Thermo Fisher Scientific (MA, USA). Average base coverage depth of the 55 kb target region was 1070-fold, 96.44% of total base was sequenced more than 100-fold. The concordance rate of amplicon sequencing and sanger sequencing is 100% in this case. The cost of this approach is about 1/5 to 1/10 of whole exome sequencing. The application of high-throughput amplicon sequencing would be helpful to diagnose genetic disease with an efficient and cost-saving way.
In conclusion, compound heterozygosity consisting of a nonsense mutation (c.2206C > T) and a splicing mutation (c.2709 + 1G > A) in the UNC13D gene was identified in a patient with HLH using high-throughput amplicon sequencing. Both mutations were confirmed by Sanger sequencing and were inherited from the parents. Our results expand the spectrum of UNC13D mutations in Chinese patients with HLH and demonstrate the potential of high-throughput amplicon sequencing in the diagnosis of heterogeneous genetic disorders.
Abbreviations
- FHL:
-
Familial hemophagocytic lymphohistiocytosis
- Hb:
-
Hemoglobin
- HLH:
-
Hemophagocytic lymphohistiocytosis
- PBMC:
-
Peripheral blood mononuclear cells
- PLT:
-
Platelets
- XLP:
-
X-linked lymphoproliferative syndrome
References
Bell RJ, Brafield AJ, Barnes ND, France NE. Familial Haemophagocytic Reticulosis. Arch Dis Child. 1952;27(136):519–25.
Filipovich AH. Hemophagocytic lymphohistiocytosis (HLH) and related disorders. Hematology. 2009;2009(1):127–31.
Janka GE. Familial and acquired hemophagocytic lymphohistiocytosis. Annu Rev Med. 2012;63(1):233–46.
Mehta RS, Smith RE. Hemophagocytic lymphohistiocytosis (HLH): a review of literature. Med Oncol. 2013;30(4):740.
Henter J, Elinder G, Soder O, Ost A. Incidence in Sweden and clinical features of familial Hemophagocytic Lymphohistiocytosis. Acta Paediatr. 1991;80(4):428–35.
Niece JA, Rogers ZR, Ahmad N, Langevin AR, Mcclain KL. Hemophagocytic lymphohistiocytosis in Texas: observations on ethnicity and race. Pediatr Blood Cancer. 2010;54(3):424–8.
Henter J, Horne A, Egeler RM, Filipovich AH, Imashuku S, Ladisch S, Mcclain K, Webb D, Winiarski J, Janka G. HLH-2004: diagnostic and therapeutic guidelines for hemophagocytic lymphohistiocytosis. Pediatr Blood Cancer. 2007;48(2):124–31.
Arico M, Imashuku S, Clementi R, Hibi S, Teramura T, Danesino C, Haber DA, Nichols KE. Hemophagocytic lymphohistiocytosis due to germline mutations in SH2D1A, the X-linked lymphoproliferative disease gene. Blood. 2001;97(4):1131–3.
Zhizhuo H, Junmei X, Yuelin S, Qiang Q, Chunyan L, Zhengde X, Kunling S. Screening the PRF1, UNC13D, STX11, SH2D1A, XIAP, and ITK gene mutations in Chinese children with Epstein–Barr virus-associated hemophagocytic lymphohistiocytosis. Pediatr Blood Cancer. 2012;58(3):410–4.
Liu D, Hu X, Jiang X, Gao B, Wan C, Chen C. Characterization of a novel splicing mutation in UNC13D gene through amplicon sequencing: a case report on HLH. BMC Med Genet. 2017;18(1):135.
Tesi B, Lagerstedt-Robinson K, Chiang SC, Ben Bdira E, Abboud M, Belen B, Devecioglu O, Fadoo Z, Yeoh AE, Erichsen HC, et al. Targeted high-throughput sequencing for genetic diagnostics of hemophagocytic lymphohistiocytosis. Genome Med. 2015;7:130.
Stadt UZ, Beutel K, Kolberg S, Schneppenheim R, Kabisch H, Janka G, Hennies HC. Mutation spectrum in children with primary hemophagocytic lymphohistiocytosis: molecular and functional analyses of PRF1, UNC13D, STX11, and RAB27A†. Hum Mutat. 2006;27(1):62–8.
Yoon HS, Kim H, Yoo K, Sung K, Koo H, Kang HJ, Shin HY, Ahn HS, Kim J, Lim Y. UNC13D is the predominant causative gene with recurrent splicing mutations in Korean patients with familial hemophagocytic lymphohistiocytosis. Haematologica. 2010;95(4):622–6.
Yamamoto K, Ishii E, Sako M, Ohga S, Furuno K, Suzuki N, Ueda I, Imayoshi M, Yamamoto S, Morimoto A. Identification of novel MUNC13-4 mutations in familial haemophagocytic lymphohistiocytosis and functional analysis of MUNC13-4-deficient cytotoxic T lymphocytes. J Med Genet. 2004;41(10):763–7.
Acknowledgments
The authors appreciate the participation of the patient’s family in this clinical study. This work was supported by the Wuhan Children’s Hospital and Taihe Hospital. We are grateful to Cheng Wan from Peking University for helping with the manuscript and providing meaningful discussion.
Funding
Key Research Projects of Henan Provincial Department of Education. ID: 17A320051.
Availability of data and materials
The sequencing data were deposited into the NCBI Short Read Archive under accession number SRP124825.
Author information
Authors and Affiliations
Contributions
XH, DL and XJ performed the experiments. BG carried out the blood analysis and analyzed the data. XH and DL analyzed the data and wrote the manuscript. XJ revised the manuscript. BG and CC designed this study and edited the manuscript. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
This study was approved according to the guidelines of the Committee on the Use of Human Subjects in Wuhan Children’s Hospital.
Consent for publication
Written informed consent for the publication of the patient’s medical data was obtained from the parents before collecting samples.A copy of the written consent is available for review by the editor of this journal.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Hu, X., Liu, D., Jiang, X. et al. Identification of a novel nonsense mutation in the UNC13D gene from a patient with hemophagocytic lymphohistiocytosis: a case report. BMC Med Genet 19, 82 (2018). https://doi.org/10.1186/s12881-018-0600-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12881-018-0600-2