
Abstract
Background
Dysferlin is reduced in patients with limb girdle muscular dystrophy type 2B, Miyoshi myopathy, distal anterior compartment myopathy, and in certain Ethnic clusters.
Methods
We evaluated clinical and genetic patient data from three different Swiss Neuromuscular Centers.
Results
Thirteen patients from 6 non-related families were included. Age of onset was 18.8 ± 4.3 years. In all patients, diallelic disease-causing mutations were identified in the DYSF gene. Nine patients from 3 non-related families from Central Switzerland carried the identical homozygous mutation, c.3031 + 2T>C. A possible founder effect was confirmed by haplotype analysis. Three patients from two different families carried the heterozygous mutation, c.1064_1065delAA. Two novel mutations were identified (c.2869C>T (p.Gln957Stop), c.5928G>A (p.Trp1976Stop)).
Conclusions
Our study confirms the phenotypic heterogeneity associated with DYSF mutations. Two mutations (c.3031 + 2T>C, c.1064_1065delAA) appear common in Switzerland. Haplotype analysis performed on one case (c. 3031 + 2T>C) suggested a possible founder effect.
Similar content being viewed by others


Background
Dysferlin (DYSF) is a transmembrane protein linked to sarcolemmal repair mechanisms. Autosomal recessive mutations in the DYSF gene cause muscular dystrophies (MD): a limb girdle MD, the so-called LGMD 2B, with onset in the proximal lower limbs [1], and two distal MD, one initially affecting the gastrocnemius muscle during early adulthood, Miyoshi myopathy (MM) [2], and the other involving muscles of the anterior compartment (DMAT) [3]. Disease severity ranges from asymptomatic mutation carriers with elevated serum CK levels [4] and exercise intolerance [5] to severe functional disability [6]. Muscle weakness with pelvic girdle involvement on clinical examination distinguishes LGMD from MM. Other common phenotypes include a “proximo-distal” phenotype, characterized by simultaneous distal and proximal weakness onset [6]. Of note, a recent MRI study suggests that all patients have radiographic evidence of proximo-distal muscle involvement, regardless of the clinical phenotype [7].
The DYSF gene maps to chromosome 2p12-p14, contains 55 exons and is transcribed as an 8.5 Kb major transcript mainly expressed in skeletal and cardiac muscles. The protein product is a 230 kDa molecule involved in muscle fibre repair [8, 9]. More than 400 DYSF mutations have been described in the Leiden muscular dystrophy database (http://www.dmd.nl); however, a mutational hot spot has not been detected [10–12]. Ethnic clusters have been described in Jews from Libya [13] and the Caucasus region [14], as well as in the Italian [15] and Spanish [16] populations. Four mutations account for 60 % of all mutations in Japanese patients with MM [17]. Interestingly, the type of mutation does not correlate with phenotypic features. In fact, the same mutation has been found to be associated with a broad spectrum of inter—and intra-familial clinical phenotypes [18–22].
Within this range of clinical and genetic heterogeneity, we sought to determine frequently encountered DYSF gene mutations and phenotypes in the Swiss population and to uncover possible founder effects.
Methods
Subjects
The medical records of patients of Swiss origin with progressive muscle weakness, and DYSF mutations, who were admitted to Neuromuscular Centres in Basel, Lausanne and Zurich between 1989 and 2015 were anonymized and reviewed. All patients provided their written informed consent for genetic analysis. Clinical data collected included age and symptoms at onset, disease duration, family pedigrees, and muscle strength according to the Medical Research Council (MRC) scale. In addition, CK levels, muscle biopsy and mutational analysis results were evaluated. Electromyography, electrocardiography, echocardiography, pulmonary function and muscle MRI data were reviewed when available. The study was performed in accordance with the Declaration of Helsinki and approved by the local ethics committee (Kantonale Ethikkommission Zürich, KEK-ZH-Nr. 2015–0036).
Genetic analysis
Sequence analysis was performed using bidirectional fluorescent sequencing of all 55 exons of the DYSF gene, either in the Service de Cardiologie, Hôpital Cochin CHU Paris, France, or at the Institute of Human Genetics, University of Würzburg, Germany (Reference sequence: NM_003494.3). Multiplex ligation-dependent probe amplification (MLPA) was used in one case.
To evaluate the hypothesis that the DYSF mutation c.3031 + 2T>C is due to a founder effect, we performed haplotype analysis, as described previously, with 6 polymorphic microsatellite markers on chromosome 2p13.2 flanking the DYSF gene (D2S327, D2S2113, D2S292, D2S291, D21S2110 and D2S2111) [23].
Results
Clinical data
Thirteen patients were included in the study. Of those, 9 patients were from three non-related families from Central Switzerland (Canton of Schwyz). The clinical data of 10 patients from 6 different families are summarized in Tables 1 and 2. Three patients from central Switzerland (C-6, D-10, D-11) provided blood for haplotype analysis but refused clinical evaluation. Consanguinity was not reported and the parents of all patients were healthy. Mean disease onset was in late adolescence or early adulthood (18.7 ± 4.5 years). Early manifestations (Table 1) included leg pain, steppage gait, Gowers’ sign and difficulty running and jumping. Mean CK levels were 12500 ± 14400 IU/L (range 2200–46350, normal value for our laboratory < 167 U/l, Table 1).
The clinical phenotype could be determined in 10 patients (Table 2). In all patients, the facial muscles were spared. Four of the patients from Central Switzerland (C-4, C-5, C-7, F-13) presented without arm weakness a few years after the onset of symptoms (Table 2). All patients had distal lower limb flexor weakness and 2 (C-7, F-13) also showed distal lower limb extensor weakness. Three (C-4, C-5, C-7) patients had proximal lower limb weakness and therefore were classified as the “proximo-distal” (PD) phenotype [6]. Another patient (F-13) demonstrated isolated distal lower limb weakness (foot flexion > extension) and was classified as the MM phenotype. Three patients (C-4, C-5 and C-7) were followed for 11, 9 and 4 years, respectively (Table 2). All developed distal upper limb extensor weakness with 2 (DC-4, C-7) additionally showing distal upper limb flexor and proximal upper limb weakness. Two patients from central Switzerland (C-8, D-9) were examined 20 and 28 years after onset and demonstrated proximal and distal upper and lower limb flexor and extensor weakness (Table 2). Patient D-9 experienced a relatively “benign” course with muscle strength grade of at least 3 on the MRC scale (Table 2). One patient from the Canton Thurgau (E-12) presented 7 years after onset with proximal and distal lower limb flexor and extensor weakness, eventually developing distal flexor and extensor and proximal upper limb weakness (Table 2). A patient from the Canton Aargau (B-3) presented with severe distal lower limb flexor, proximal lower limb flexor and extensor, distal upper limb flexor and extensor and proximal upper limb weakness 15 years after onset. Two patients from the Canton Fribourg were initially examined 23 (A-1) and 17 (A-2) years after onset, respectively. Patient A-1 had generalized weakness, pronounced in the distal lower limb extensors, and over the course of 9 years developed proximal lower and upper limb palsies. Patient A-2 had generalized weakness, more pronounced in the distal lower limb flexors, that progressive minimally over 7 years. Interestingly, members of family C presented with a proximo-distal phenotype early in the course and members of family C and patient F-13 were notable for the lack of upper limb weakness during the early phases of the disease. Later in the course, all patients manifested a typical LGMD phenotype; however, the distribution of weakness was heterogeneous (Table 2). The DMAT phenotype was not observed in our cohort.
The use of mobility aids showed the following timeline: 2 patients required canes for walking assistance (A-1, A-2) at the age of 35 and 49 years, respectively (disease duration until first use of canes 12 and 21 years, respectively), and both were eventually wheelchair-bound (17 and 24 years after disease onset; 5 and 3 years after first cane use). Patient B-3, at age 29, 14 years after disease onset, used a wheelchair intermittently (B-3), and patient C-8 was wheelchair-bound, at age 43, 27 years after disease onset.
Cardiac, pulmonary, MRI and EMG data
Electrocardiography and ECG were normal in all 9 patients who underwent examination, the longest disease duration being 35 years (Table 1). Pulmonary function was normal in 6 out of 7 patients, with restrictive lung disease documented in patient A-1 (Table 1; initial pulmonary function testing performed after 29 years of disease duration). In 3 patients, muscle MRI showed fatty replacement of the lower limb muscles. In all three patients (A-2, E-12 and F-13), the short head of the biceps femoris and the sartorius and the gracilis muscles were symmetrically spared or appeared less affected than other muscles. In patients E-12 and F-13, the semitendinosus, the tibialis anterior and the extensor digitorum longus muscles also seemed less affected, while edema was suspected in selected lower limb muscles. In another patient (C-7), the pelvic and the lower limb muscles appeared normal. EMG was myopathic with short and small potentials in all 8 patients examined, with spontaneous activity found in 5 cases (Table 1).
Pathological features and protein studies
A total of 5 surgical muscle biopsies were available (Table 3) for evaluation. In 2 patients, biopsies were not available because they were taken in 1986 and 1977. In 6 patients, the genetic diagnosis was made without prior biopsy. The mean age at biopsy was 28.25 ± 13.7 years, while the mean disease duration at biopsy was 8.3 ± 9.8 years. Samples were removed from the trapezius, quadriceps and gracilis muscles. All patients exhibited dystrophic changes with an increased spectrum of myofiber diameter and variable fibrosis. In one case with a LGMD phenotype (B-3), striking inflammatory changes were observed within the endomysium. Western blots (WB) and/or immunohistochemistry (IHC) were performed in 6 samples (WB only in 2 samples, IHC only in 3 samples, WB and IHC in a single sample). On WB, dysferlin was totally absent in all samples tested, whereas by IHC, dysferlin was judged absent in 2 cases and reduced in two.
Genetic studies
Genetic data are summarized in Table 4. In 13 patients, 2 disease-causing mutations on separate alleles were identified. Nine patients from 3 non-related families from Central Switzerland (Canton Schwyz) carried the identical splice site mutation, c.3031 + 2T>C, 7 were homozygous (Table 4). Three patients from 2 different families from the Cantons of Fribourg and Aargau carried a heterozygous frameshift mutation c.1064_1065delAA, and 2 members from family A, harbored the heterozygous nonsense mutation c.2217C>A (Table 4).
Two novel mutations were identified in this patient set. Patient C-6, who was compound heterozygous for the c.3031 + 2T >C mutation on one allele, showed a novel mutation, c.2869C>T on the other allele. Patient E-12 carried the novel homozygous c.5928G > A mutation.
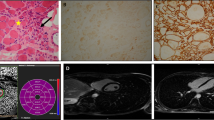
Haplotype analyses using markers flanking the DYSF gene revealed homozygosity for at least one nearest marker (D2S291) in all patients from the two apparently unrelated C and D families carrying the homozygous c.3031 + 2T>C mutation, suggesting a possible founder mutation (See Fig. 1). In patient C-7, carrying a compound heterozygous DYSF mutation (c.3031 + 2 T >C and c.2869C>T), marker D2S291 shows a heterozygous genotype (Fig. 1).

Haplotype Analysis. Haplotype analysis using six microsatellite markers flanking the DYSF gene in families C and D. The DYSF gene is located between markers D2S292 and D2S291. In all patients carrying the homozygous c.3031 + 2T>C mutation, the adjacent marker D2S291 also shows a homozygous genotype (allele 178) suggesting that this mutation may be a founder mutation in these two families. Accordingly, in patient C-6 carrying a compound heterozygous DYSF mutation (c.3031 + 2T>C and c.2869C>T), the marker D2S291 shows a heterozygous genotype (278–290)
Discussion
Evaluation of the present cohort of 6 families brings new genetic information about dysferlinopathy in Switzerland, and novel data regarding a potential founder effect in Central Switzerland. The fact that unrelated families with identical mutations come from the same region indicates that these mutations are common, and indeed, haplotype analysis of 2 families carrying the mutation c.3031 + 2T>C suggests a possible founder effect for this mutation in Switzerland. The limitations of this study are twofold: the analysis is limited by the small cohort size and 3 out of 13 patients had not been examined clinically.
From a clinical point of view, the onset age in early adulthood confirms previous findings [6, 7, 24]. In addition, late-onset [25] or congenital forms were not encountered [26]. Three out of 4 patients from Central Switzerland presented with the PD phenotype, while another patient with the same mutation was considered to represent the MM phenotype. Upper limb weakness was not a presenting feature in the current cohort. Some reports suggest that the proximo-distal phenotype occurs in up to one third of dysferlinopathy cases [6]. Of note, 9 out of 10 patients developed distal upper limb weakness later in the disease course, which has been frequently reported [27–29]. Muscle MRI is known to reveal fatty replacement of distal and proximal lower limb muscles in the early stages of dysferlinopathy, including patients with clinically isolated proximal or distal involvement [7, 30]. However, an adolescent patient with a MM phenotype (C-7) showed a normal pelvic and whole lower limb muscle MRI, similarly to a patient described with isolated hyperCKemia [31].
Interestingly, we found clinical phenotype variability within families and within unrelated carriers with similar mutations. Such variability has already been reported in Libyan Jews [13] and was found to be particularly relevant in a 6-generation, highly consanguineous family originating from an isolated village in Dagestan [32]. This observation may indicate that to date, unknown genetic or environmental modifiers play a role in influencing the clinical phenotypes of the mutated DYSF gene [22]. Accordingly, it would be interesting to study micro-RNAs known to be involved in many biological processes, including epigenetic changes. Other cardinal features of our cohort were very high CK levels, as previously described [7], the absence of cardiac impairment (nevertheless encountered in few patients [6, 25, 33, 34] and present in a knock out mouse model [35]), and the risk of developing respiratory failure late in the disease course [36, 37], as observed in one of our patients.
EMG showed myopathic changes and pathologic spontaneous activity. In one case, the biopsy revealed striking endomysial inflammation, in line with frequently observed inflammatory changes in dysferlinopathy and other muscular dystrophies [18, 38]. Immunosuppressive treatment, however, fails to improve muscle weakness [39]. It has been postulated that dysferlin may be linked to membrane repair and/or inflammatory activation [40], and in a few cases, splice site mutations disrupting dysferlin are known to produce inflammation [18]. Conversely, most of our patients with splice mutations showed no evidence of inflammation on muscle biopsy, indicating the lack of clear correlation between inflammation and genotype. On Western blot (WB) 3 cases showed a consistent reduction in dysferlin, as has been previously described [41].
In dysferlinopathies, genotype is not predictive of disease severity, and genotype-phenotype correlations are not clear-cut [15, 42]. However, correlation has been found between certain geographical areas and the age of onset, notably in Japanese and Italian MM patients [17], who develop symptoms earlier than patients with LGMD 2B [10]. The most severely affected patients in our study (Patients A-1 and A-2, described previously [12]) carried the truncating mutation c.2217C>A (p.Tyr739X) with a disease duration of 17 and 24 years, respectively, before the patients became wheelchair-bound. One of the older patients (C-8), carrying the c.3031 + 2T>C homozygous mutation, became wheelchair bound after a disease duration of 27 years, and patient D-9 showed relatively mild impairment 20 years after onset. However, in all other patients carrying this mutation, disease duration was shorter; thus precluding longitudinal comparisons. A longer follow-up is necessary to assess the onset and time-course of the various disabling symptoms.
Conclusion
Our study confirms the broad phenotypic heterogeneity, often accompanied by markedly elevated CK levels, associated with DYSF mutations in the Swiss population. Due to a potential founder effect in the past, two mutations appear to be endemic in central Switzerlans. These findings are important for genetic counselling and should facilitate targeted molecular diagnosis of dysferlin deficiency in patients of Swiss origin.
References
Bashir R, Britton S, Strachan T, Keers S, Vafiadaki E, Lako M, et al. A gene related to Caenorhabditis elegans spermatogenesis factor fer-1 is mutated in limb-girdle muscular dystrophy type 2B. Nat Genet. 1998;20(1):37–42.
Liu J, Aoki M, Illa I, Wu C, Fardeau M, Angelini C, et al. Dysferlin, a novel skeletal muscle gene, is mutated in Miyoshi myopathy and limb girdle muscular dystrophy. Nat Genet. 1998;20(1):31–6.
Illa I, Serrano-Munuera C, Gallardo E, Lasa A, Rojas-Garcia R, Palmer J, et al. Distal anterior compartment myopathy: a dysferlin mutation causing a new muscular dystrophy phenotype. Ann Neurol. 2001;49(1):130–4.
Fanin M, Nascimbeni AC, Aurino S, Tasca E, Pegoraro E, Nigro V, et al. Frequency of LGMD gene mutations in Italian patients with distinct clinical phenotypes. Neurology. 2009;72(16):1432–5.
Xi J, Blandin G, Lu J, Luo S, Zhu W, Beroud C, et al. Clinical heterogeneity and a high proportion of novel mutations in a Chinese cohort of patients with dysferlinopathy. Neurol India. 2014;62(6):635–9.
Nguyen K, Bassez G, Krahn M, Bernard R, Laforet P, Labelle V, et al. Phenotypic study in 40 patients with dysferlin gene mutations: high frequency of atypical phenotypes. Arch Neurol. 2007;64(8):1176–82.
Paradas C, Llauger J, Diaz-Manera J, Rojas-Garcia R, De Luna N, Iturriaga C, et al. Redefining dysferlinopathy phenotypes based on clinical findings and muscle imaging studies. Neurology. 2010;75(4):316–23.
Lennon NJ, Kho A, Bacskai BJ, Perlmutter SL, Hyman BT, Brown RH. Dysferlin interacts with Annexins A1 and A2 and mediates sarcolemmal wound-healing. J Biol Chem. 2003;278(50):50466–73.
Bansal D, Miyake K, Vogel SS, Groh S, Chen CC, Williamson R, et al. Defective membrane repair in dysferlin-deficient muscular dystrophy. Nature. 2003;423(6936):168–72.
Cagliani R, Magri F, Toscano A, Merlini L, Fortunato F, Lamperti C, et al. Mutation finding in patients with dysferlin deficiency and role of the dysferlin interacting proteins annexin A1 and A2 in muscular dystrophies. Hum Mutat. 2005;26(3):283.
Nguyen K, Bassez G, Bernard R, Krahn M, Labelle V, Figarella-Branger D, et al. Dysferlin mutations in LGMD2B, Miyoshi myopathy, and atypical dysferlinopathies. Hum Mutat. 2005;26(2):165.
Krahn M, Beroud C, Labelle V, Nguyen K, Bernard R, Bassez G, et al. Analysis of the DYSF mutational spectrum in a large cohort of patients. Hum Mutat. 2009;30(2):E345–375.
Argov Z, Sadeh M, Mazor K, Soffer D, Kahana E, Eisenberg I, et al. Muscular dystrophy due to dysferlin deficiency in Libyan Jews. Clinical and genetic features. Brain. 2000;123(Pt 6):1229–37.
Leshinsky-Silver E, Argov Z, Rozenboim L, Cohen S, Tzofi Z, Cohen Y, et al. Dysferlinopathy in the Jews of the Caucasus: a frequent mutation in the dysferlin gene. Neuromuscul Disord. 2007;17(11–12):950–4.
Cagliani R, Fortunato F, Giorda R, Rodolico C, Bonaglia MC, Sironi M, et al. Molecular analysis of LGMD-2B and MM patients: identification of novel DYSF mutations and possible founder effect in the Italian population. Neuromuscul Disord. 2003;13(10):788–95.
Vilchez JJ, Gallano P, Gallardo E, Lasa A, Rojas-Garcia R, Freixas A, et al. Identification of a novel founder mutation in the DYSF gene causing clinical variability in the Spanish population. Arch Neurol. 2005;62(8):1256–9.
Takahashi T, Aoki M, Tateyama M, Kondo E, Mizuno T, Onodera Y, et al. Dysferlin mutations in Japanese Miyoshi myopathy: relationship to phenotype. Neurology. 2003;60(11):1799–804.
McNally EM, Ly CT, Rosenmann H, Mitrani Rosenbaum S, Jiang W, Anderson LV, et al. Splicing mutation in dysferlin produces limb-girdle muscular dystrophy with inflammation. Am J Med Genet. 2000;91(4):305–12.
Aoki M, Liu J, Richard I, Bashir R, Britton S, Keers SM, et al. Genomic organization of the dysferlin gene and novel mutations in Miyoshi myopathy. Neurology. 2001;57(2):271–8.
Nakagawa M, Matsuzaki T, Suehara M, Kanzato N, Takashima H, Higuchi I, et al. Phenotypic variation in a large Japanese family with Miyoshi myopathy with nonsense mutation in exon 19 of dysferlin gene. J Neurol Sci. 2001;184(1):15–9.
Ueyama H, Kumamoto T, Nagao S, Masuda T, Horinouchi H, Fujimoto S, et al. A new dysferlin gene mutation in two Japanese families with limb-girdle muscular dystrophy 2B and Miyoshi myopathy. Neuromuscul Disord. 2001;11(2):139–45.
Weiler T, Bashir R, Anderson LV, Davison K, Moss JA, Britton S, et al. Identical mutation in patients with limb girdle muscular dystrophy type 2B or Miyoshi myopathy suggests a role for modifier gene(s). Hum Mol Genet. 1999;8(5):871–7.
von der Hagen M, Schallner J, Kaindl AM, Koehler K, Mitzscherling P, Abicht A, et al. Facing the genetic heterogeneity in neuromuscular disorders: linkage analysis as an economic diagnostic approach towards the molecular diagnosis. Neuromuscul Disord. 2006;16(1):4–13.
Nalini A, Gayathri N. Dysferlinopathy: a clinical and histopathological study of 28 patients from India. Neurol India. 2008;56(3):379–85. discussion 386–377.
Klinge L, Aboumousa A, Eagle M, Hudson J, Sarkozy A, Vita G, et al. New aspects on patients affected by dysferlin deficient muscular dystrophy. J Neurol Neurosurg Psychiatry. 2010;81(9):946–53.
Paradas C, Gonzalez-Quereda L, De Luna N, Gallardo E, Garcia-Consuegra I, Gomez H, et al. A new phenotype of dysferlinopathy with congenital onset. Neuromuscul Disord. 2009;19(1):21–5.
Linssen WH, Notermans NC, Van der Graaf Y, Wokke JH, Van Doorn PA, Howeler CJ, et al. Miyoshi-type distal muscular dystrophy. Clinical spectrum in 24 Dutch patients. Brain. 1997;120(Pt 11):1989–96.
Rosales XQ, Gastier-Foster JM, Lewis S, Vinod M, Thrush DL, Astbury C, et al. Novel diagnostic features of dysferlinopathies. Muscle Nerve. 2010;42(1):14–21.
Woudt L, Di Capua GA, Krahn M, Castiglioni C, Hughes R, Campero M, Trangulao A, Gonzalez-Hormazabal P, Godoy-Herrera R, Levy N et al.: Toward an objective measure of functional disability in dysferlinopathy. Muscle Nerve 2015 doi:10.1002/mus.24685
Fischer D, Walter MC, Kesper K, Petersen JA, Aurino S, Nigro V, et al. Diagnostic value of muscle MRI in differentiating LGMD2I from other LGMDs. J Neurol. 2005;252(5):538–47.
Kesper K, Kornblum C, Reimann J, Lutterbey G, Schroder R, Wattjes MP. Pattern of skeletal muscle involvement in primary dysferlinopathies: a whole-body 3.0-T magnetic resonance imaging study. Acta Neurol Scand. 2009;120(2):111–8.
Illarioshkin SN, Ivanova-Smolenskaya IA, Tanaka H, Vereshchagin NV, Markova ED, Poleshchuk VV, et al. Clinical and molecular analysis of a large family with three distinct phenotypes of progressive muscular dystrophy. Brain. 1996;119(Pt 6):1895–909.
Wenzel K, Geier C, Qadri F, Hubner N, Schulz H, Erdmann B, et al. Dysfunction of dysferlin-deficient hearts. J Mol Med. 2007;85(11):1203–14.
Ueyama H, Kumamoto T, Horinouchi H, Fujimoto S, Aono H, Tsuda T. Clinical heterogeneity in dysferlinopathy. Intern Med. 2002;41(7):532–6.
Han R, Bansal D, Miyake K, Muniz VP, Weiss RM, McNeil PL, et al. Dysferlin-mediated membrane repair protects the heart from stress-induced left ventricular injury. J Clin Invest. 2007;117(7):1805–13.
Takahashi T, Aoki M, Suzuki N, Tateyama M, Yaginuma C, Sato H, et al. Clinical features and a mutation with late onset of limb girdle muscular dystrophy 2B. J Neurol Neurosurg Psychiatry. 2013;84(4):433–40.
Mahjneh I, Marconi G, Bushby K, Anderson LV, Tolvanen-Mahjneh H, Somer H. Dysferlinopathy (LGMD2B): a 23-year follow-up study of 10 patients homozygous for the same frameshifting dysferlin mutations. Neuromuscul Disord. 2001;11(1):20–6.
Fanin M, Angelini C. Muscle pathology in dysferlin deficiency. Neuropathol Appl Neurobiol. 2002;28(6):461–70.
Walter MC, Reilich P, Thiele S, Schessl J, Schreiber H, Reiners K, et al. Treatment of dysferlinopathy with deflazacort: a double-blind, placebo-controlled clinical trial. Orphanet J Rare Dis. 2013;8:26.
Han R. Muscle membrane repair and inflammatory attack in dysferlinopathy. Skelet Muscle. 2011;1(1):10.
Cacciottolo M, Numitone G, Aurino S, Caserta IR, Fanin M, Politano L, et al. Muscular dystrophy with marked Dysferlin deficiency is consistently caused by primary dysferlin gene mutations. Eur J Hum Genet. 2011;19(9):974–80.
Kawabe K, Goto K, Nishino I, Angelini C, Hayashi YK. Dysferlin mutation analysis in a group of Italian patients with limb-girdle muscular dystrophy and Miyoshi myopathy. Eur J Neurol. 2004;11(10):657–61.
Acknowledgments
We thank P. Mitzscherling for expert technical assistance.
MvdH, AH and WK are members of the German network on muscular dystrophies (MD-NET, 01GM0601) which is funded by the German Ministry of Education and Research (BMBF, Bonn, Germany).
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
Disclosure of conflict of interest: on behalf of all authors, the corresponding author states that there is no conflict of interest.
Authors’ contributions
WK carried out part of the molecular genetic studies and drafted the manuscript. MvdH and AH carried out the haplotype analysis. EJR, VK and AL carried out the neuropathological analyses. DF and MS participated in the design and the coordination of the study and helped to draft the manuscript. JAP, TK and HHJ conceived of the study, and participated in its design and coordination and helped to draft the manuscript. All authors read and approved the final manuscript. All patients gave their written informed consent for genetic analysis. The study was performed in accordance with the Declaration of Helsinki and approved by the local ethics committee (Kantonale Ethikkommission Zürich).
Authors’ information
Not applicable.
Jens A. Petersen and Thierry Kuntzer contributed equally to this work.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Petersen, J.A., Kuntzer, T., Fischer, D. et al. Dysferlinopathy in Switzerland: clinical phenotypes and potential founder effects. BMC Neurol 15, 182 (2015). https://doi.org/10.1186/s12883-015-0449-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12883-015-0449-3