
Abstract
Background
Prostate cancer (PCa) is a leading cause of cancer-related death in men. Understanding the proteomic landscape associated with PCa risk can provide insights into its molecular mechanisms and pave the way for potential therapeutic interventions.
Methods
A proteome-wide Mendelian randomization (MR) analysis was employed to determine associations between genetically predicted protein concentrations in plasma and PCa risk. From an initial list of 4,364 proteins, significant associations were identified and validated. Multiple sensitivity analyses were also conducted to enhance the robustness of our findings.
Results
Of the 4,364 genetically predicted proteins, 308 exhibited preliminary associations with PCa risk. After rigorous statistical refinement, genetically predicted concentrations of 14 proteins showed positive associations with PCa risk, with odds ratios spanning from 1.55 (95% CI 1.28–1.87) for ATG4B to 2.67 (95% CI 1.94–3.67) for HCN1. In contrast, genetically predicted concentrations of ATG7, B2M, MSMB, and TMEM108 demonstrated inverse associations with PCa. The replication analysis further substantiated positive associations for MDH1 and LSM1, and a negative one for MSMB with PCa. A meta-analysis harmonizing primary and replication data mirrored these findings. Furthermore, the MVMR analysis pinpointed B2M and MSMB as having significant associations with PCa risk.
Conclusion
The genetic evidence unveils a refined set of proteins associated with PCa risk. The findings underscore the potential of these proteins as molecular markers or therapeutic targets for PCa, calling for deeper mechanistic studies and exploration into their translational relevance.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Prostate cancer (PCa) remains one of the most prevalent forms of cancer in men worldwide, representing a significant public health challenge [1]. Despite advances in diagnostic techniques, surgical interventions, and adjuvant therapies, many patients progress to advanced stages of the disease, underscoring the need for innovative therapeutic strategies [2]. One promising avenue for identifying novel drug targets lies in the intricate molecular landscape of PCa, specifically within its proteome [3, 4]. The proteome, which encompasses the entire complement of proteins expressed by an organism, offers a dynamic view of cellular function, unlike the more static genome [5, 6]. This rich dataset can be leveraged to identify proteins or pathways that play a critical role in PCa pathogenesis and could thus be potential drug targets.
Several epidemiological studies have endeavored to decipher the complex relationship between circulating proteins and the risk of PCa. For instance, elevated levels of prostate-specific antigen (PSA) in the blood have been ubiquitously recognized as a marker for prostate cancer risk, leading to its widespread use as a screening tool [7]. Beyond PSA, studies have indicated that increased circulating levels of insulin-like growth factor-1 (IGF-1) are associated with a heightened risk of PCa [8], but inconsistent findings remained [9]. The prior epidemiological studies, while foundational, have limitations. Their observational nature raises concerns of confounding and reverse causation, potentially skewing true associations between circulating proteins and prostate cancer risk. The Mendelian randomization (MR) approach offers a robust method for establishing causal relationships between exposures and outcomes [10]. MR leverages genetic variants as instrumental variables (IVs) to tease apart cause and effect, thus circumventing some of the confounding and reverse causation issues that typically plague observational studies [11]. In essence, by harnessing the natural experiments conferred by random assortment of alleles during meiosis, MR provides a more reliable window into causality. When applied proteome-wide, such an analysis has the potential to reveal a multitude of proteins causally linked to PCa, offering tantalizing hints for drug discovery [12].
In the current study, by integrating advanced statistical methodologies with large-scale proteomic datasets, we aim to shed light on the potential plasma proteomic biomarkers for PCa, which may open a new avenue for the development of targeted therapies that could revolutionize PCa treatment. As the burden of PCa continues to grow globally, such insights are not just academically interesting but also have the potential to shape the future of therapeutic interventions.
Methods
Study design
In this two-sample MR analysis, we set the plasma proteins as the exposures and the PCa was the outcome. SNPs associated with plasma protein levels were used as the IVs, referred to pQTL. Both cis- and trans-pQTL were considered. To ensure the consistence of genetic background, the exposure and outcome data were collected from participants with European ancestry.
Proteomic data source and pQTLs
We retrieved the genetic summary data of plasma proteins from Ferkingstad et al.’s study [13]. In that study, the genome-wide association study (GWAS) was performed for 4,907 aptamers in 35,559 Icelanders to identify protein-associated SNPs (ie., pQTLs). Briefly, the authors initially adjusted rank-inverse normal transformed levels of each peotein for age, sex and sample age for the deCODE Health study. The residuals were then standardized again using rank-inverse normal transformation and the standardized values were used as phenotypes for GWAS that implementing using BOLT-LMM. A total of 27.2 million variants were tested. Details of the proteome GWAS could refer to the original study [13].
To identify suitable genetic IVs from the protein GWAS data, we applied rigorous quality control steps. We began by pinpointing SNPs with genome-wide significance (P < 5 × 10–8). We then clumped SNPs to account for linkage-disequilibrium (LD) using a reference genome panel (window size = 10,000 kb, R2 < 0.01), retaining the SNPs with the lowest P-values in LD pairs exceeding the 0.01 R2 threshold. SNPs with a minor allele frequency (MAF) below 1% were discarded. We matched these selected SNPs against those in the PCa GWAS data (details see the below). If a SNP was missing in the outcome GWAS, we sourced a proxy SNP in strong LD (R2 > 0.8) with the original SNP. Ambiguous or palindromic SNPs with unclear strands were either excluded or adjusted for in the MR analysis.
To validate the strong association between the chosen instrumental SNPs and exposure—a fundamental assumption of MR analysis—we computed the F-statistic using the formula: F = R2(n—k—1)/k(1—R2). Here, R2 denotes the variance proportion of exposure accounted for by the chosen genetic instruments, n is the sample size of the exposure GWAS, and k is the number of IVs chosen. R2 can be calculated using 2 × β2 × EAF × (1-EAF), where β represents the IV’s estimated coefficient in the exposure GWAS and EAF stands for the effect allele frequency. An F-statistic exceeding 10 is deemed satisfactory.
Prostate cancer data source
The GWAS summary data of PCa were retrieved from the study of PRACTICAL Consortium [14], which meta-analyzed genotype data from 79,148 PCa cases and 61,106 controls of European ancestry. Genotypes for ~ 70 M SNPs were imputed for all samples using the October 2014 (Phase 3) release of the 1000 Genomes Project data as the reference panel. After excluding monomorphic SNPs and those with an imputation R2 < 0.3, a total of 21,465,239 SNP across chromosomes 1–22 and chromosome X were included in the GWAS. Per-allele odds ratios and standard errors were generated for the OncoArray and each GWAS, adjusting for principal components and study relevant covariates using logistic regression.
Mendelian randomization analysis
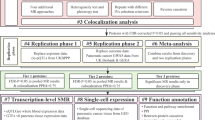
The analytic flowchart is shown in Fig. 1. In our primary analysis, we included 4,364 eligible proteins. To rapidly determine the associations between genetically predicted protein concentrations and PCa risk, we employed the inverse-variance weighted (IVW) method or the Wald ratio test. The IVW method was used when multiple IVs were available, while the Wald ratio test was selected in other instances. We employed the IVW as the primary method for its efficiency and simplicity in estimating the causal effect of plasma proteins on prostate cancer risk, particularly suitable for our large-scale dataset. IVW is robust when the instrumental variables are strong and valid, making it a practical choice for the initial analysis phase. Odds ratio (OR) and the corresponding 95% confidence interval (CI) were calculated to quantify the association. To account for potential biases from multiple testing, we utilized the Bonferroni correction, designating a P value < 1.14e-5 (0.05/4364) as denoting statistical significance.

The study flowchart
We further examined proteins that exhibited significant associations with PCa risk in the primary analysis and possessed multiple IVs. For validation, we employed an array of sensitivity tests, including MR-Egger regression, MR-PRESSO, weighted median, and weighted mode methods. The IVW method gauged between-SNP heterogeneity, while the MR-Egger regression intercept test evaluated the potential presence of horizontal pleiotropy. We also performed HEIDI (Heterogeneity in Dependent Instruments) tests in the SMR software to exploit if the observed association was due to vertical pleiotropy rather than the LD with the causal variant. [15]. A PHEIDI of < 0.05 indicates that the observed association could be due to two distinct genetic variants in high LD with each other rather than pleiotropy.
To corroborate these associations, we retrieved the GWAS summary data for PCa from the FinnGen project’s R9 release, comprising 13,216 cases and 119,948 controls [16]. This data facilitated repeated MR analyses to confirm associations between the pinpointed proteins and PCa. Subsequently, we meta-analyzed the ORs drawn from diverse data sources.
To comprehensively account for potential horizontal pleiotropy, we executed a multivariable MR (MVMR) analysis. MVMR allows for the adjustment of additional confounding variables and is especially beneficial in dissecting the effects of multiple, possibly interrelated, exposures [17]. This was particularly relevant for our study, where plasma proteins have multifaceted biological roles and could be involved in complex pathways influencing PCa risk. Here, covariates like smoking habits, body mass index (BMI), and alcohol consumption were integrated. Genetic data pertaining to smoking and alcohol was sourced from a GWAS on risk tolerance and associated behaviors, covering over a million participants [18]. Smoking classification was based on individual history (categorized as ever vs. never smokers), while alcohol consumption was quantified by weekly drink intake. Genetic data for BMI was derived from a GWAS examining height and BMI across nearly 700,000 European subjects [19]. In scenarios devoid of between-SNP heterogeneity, the MVMR-IVW method was employed; otherwise, the MVMR-weighted median approach was utilized.
We also performed Gene Ontology (GO) and KEGG enrichment analyses and constructed protein–protein interaction (PPI) network for the PCa-related proteins. The PPI network was developed utilizing the STRING online tool. The interaction data were sourced through diverse methods including textual analysis, experimental results, database records, co-expression analysis, proximity in genomic context, gene fusion evidence, and observed co-occurrence patterns. We incorporated only those proteins into the network that had an interaction confidence score higher than 0.7 with the target protein. Furthermore, we confined our analysis to a maximum of 30 interacting proteins within the immediate vicinity (the first shell) of the PPI network, deliberately excluding any secondary shell interactions for clarity and focus. We compared the mRNA expression levels of the selected proteins between tumor and non-tumor tissues using the TCGA database (52 non-tumor vs. 497 tumor). Furthermore, we evaluated the prognostic values of these proteins based on their mRNA expression levels.
All statistics were performed using R program (v 4.1.3). TwoSampleMR, MendelianRandomization, and MRPRESSO packages were used for MR and MVMR analyses. ClusterProfiler package was utilized for enrichment analysis and STRING database was applied for the construction of PPI network.
Results
In our primary assessment, 4,364 genetically-predicted proteins met the criteria for screening via the IVW method or Wald ratio test. This led to the identification of 308 proteins significantly associated with PCa risk (P value < 0.05; Fig. 2A; Supplementary Table S1). GO analysis demonstrated these proteins were notably enriched in biological processes such as peptidyl-tyrosine modification, peptide hormone response, and the JAK-STAT signaling pathway. Concerning cellular components, these proteins were predominantly located in cytoplasmic vesicle lumens, the collagen-rich extracellular matrix, and the secretory granule lumen. Their molecular functions were significantly tied to receptor ligand activity and cytokine receptor binding (Fig. 2B). Furthermore, KEGG analysis revealed their notable presence in pathways like PI3K-Akt, cytokine-cytokine receptor interaction, HIF-1, and JAK-STAT (Supplementary Figure S1).

The prostate cancer associated plasma proteins and their enriched pathways. A the association between plasma proteins and prostate cancer risk. The blue and red dash lines denote P < 0.05 and P < 1.14e-5, respectively. B the Gene Ontology enrichment analysis of the 308 proteins showing significant association with prostate cancer
From these 308 genetically-predicted proteins, post-multiple testing corrections left 18 with significant associations with PCa (14 positive and 4 negative; P < 1.14e-5; Fig. 2A; Supplementary Table S1). Seventeen of these proteins were functionally annotated using the STRING database, except for MDH1 (Supplementary Table S2). Among them, only B2M, MSMB, and SNX1 had multiple IVs (Table 1; Supplementary Table S3), all with F-statistics > 10. Significant between-SNP heterogeneity was observed for B2M and MSMB, with MSMB also exhibiting horizontal pleiotropy (Table 1). After conducting the HEIDI test, we found that the association between MSMB and PCa might be influenced by pleiotropy, as indicated by a P-value of 0.127. In contrast, for the associations of PCa with the other 17 proteins, no significant evidence of pleiotropy was detected, as detailed in Supplementary Table S3. A constructed PPI network based on these 18 proteins comprised 47 nodes and 137 edges, showcasing an average local clustering coefficient of 0.69 and a highly significant PPI enrichment (P = 2.02e-13; Supplementary Figure S2A). This network notably aligned with immunological pathways such as antigen processing and T-cell-mediated cytotoxicity (Supplementary Figure S2B).
The primary analysis suggested that genetically-predicted concentrations of MDH1, HCN1, LSM1, PLS1, TEAD3, SSB, IFNA7, PTK2, CYR61, IFNGR2, UNC51, WFDC11, SNX1, and ATG4B were positively associated with PCa risk, with the OR ranged from 1.55 (95% CI 1.28–1.87) for ATG4B to 2.67 (95% CI 1.94–3.67) for HCN1 (Table 1; Fig. 3). Conversely, proteins like ATG7, B2M, MSMB, and TMEM108 showed negative associations with PCa (Table 1; Fig. 3). Sensitivity analyses of proteins with multiple IVs (B2M, MSMB, SNX1) affirmed these associations across different MR methods (Supplementary Table S4). Compared to the PCa tumor tissue, the expression levels of MDH1, B2M, MSMB, LSM1, TEAD3, CYR61, and TMEM108 were significantly higher, whereas the levels of IFNGR2, UNC5A, and ATG4B were significantly lower in non-tumor tissue (Supplementary Figures S3-18). The differences of mRNA expression levels were mostly inconsistent with the results of MR analyses. Moreover, we found that high levels of HCN1 and ATG4B were significantly associated with a poor prognosis (Supplementary Figures S4 and S17).

The association between eighteen plasma proteins and prostate cancer risk
Replicating the MR analysis using FinnGen’s PCa GWAS summary data reaffirmed positive associations for MDH1 and LSM1 and a negative one for MSMB with PCa (Fig. 3). However, for most proteins, barring HCN1, ATG7, and CYR61, the direction of association mirrored the primary analysis, despite not all reaching statistical significance. Meta-analytic outcomes closely paralleled the primary findings (Fig. 3). MVMR analysis specifically flagged significant associations between PCa risk and both B2M and MSMB (Table 1). Notably, most proteins displayed conditional F-statistics < 10 in this analysis.
Discussion
In this proteome-wide MR analysis aimed at understanding the risk proteins for PCa, our comprehensive approach entailed the meticulous examination of 4,364 genetically-predicted proteins. Such an extensive preliminary assessment underscores the complex proteomic intricacies possibly associated with a prostate precancerous metabolic state, which can serve as a precursor to tumor development and represents an elevated risk of PCa. From this vast assortment, 308 plasma protein concentrations were discernibly associated with PCa risk, a testament to the multitudinous potential molecular interactions linked with this malignancy. However, post stringent statistical rigor, primarily to account for potential false positives inherent in such expansive datasets, a select group of 18 plasma protein concentrations emerged as paramount, meriting further exploration due to their pronounced association profiles with PCa.
Our findings offer a profound deep dive into specific proteins and their associations with PCa risk, opening avenues for both mechanistic understanding and clinical applications. At the forefront is MDH1, a pivotal component of the mitochondrial citric acid cycle [20]. The positive association with PCa risk necessitates an exploration into the metabolic pathways operative within the tumorigenic processes of prostate cells. Given that neoplasias are characterized by distinct metabolic profiles compared to non-tumorous cells [21], the prominence of MDH1 could denote a metabolic aberrancy, indicative of tumorigenic progression, thus heralding its potential as both a biomarker and therapeutic target. The pronounced association of HCN1 with PCa risk is particularly compelling. Traditionally attributed a role in the hyperpolarization-activated cyclic nucleotide-gated channels of neuronal cells [22], its emergence in the context of PCa suggests hitherto uncharted functionalities or interactions. This warrants rigorous investigation into the mechanistic involvement of HCN1 in prostate carcinogenesis, potentially unveiling novel molecular targets. Proteins such as LSM1, PLS1, and TEAD3 further broaden the proteomic spectrum of interest. TEAD3, as an integral constituent of the Hippo signaling pathway, plays a pivotal role in cellular proliferation and apoptotic determinants [23]. Its association with PCa underscores the pathway’s potential dysregulation in this malignancy, emphasizing the translational implications for targeted interventions.
Conversely, proteins like ATG7, B2M, and MSMB exhibiting inverse associations with PCa proffer alternative avenues of interest. The corroborative association of MSMB, known for its tumor-suppressive attributes [24], reiterates its potential protective role in PCa, rendering it invaluable for both diagnostic and therapeutic paradigms. B2M is a crucial component of the major histocompatibility complex (MHC) class I molecule. Its role in antigen presentation to cytotoxic T lymphocytes directly implicates it in tumor immune interactions. In the current study, an inverse association between B2M and PCa suggests that decreased B2M expression or function might facilitate prostate tumorigenesis by impairing immune surveillance [25]. It’s noteworthy that past research has indicated B2M alterations in advanced PCa stages, with elevated serum levels linked to higher disease severity [26,27,28]. However, tissue expression and serum levels of B2M might convey different implications. From a therapeutic standpoint, the MSMB-PCa and B2M-PCa inverse associations open avenues for potential immunotherapeutic strategies. Enhancing MSMB and B2M expression could potentially augment immune recognition of PCa cells. Furthermore, MSMB and B2M levels might offer utility as a diagnostic or prognostic indicator in PCa.
The replication of our findings using FinnGen’s PCa GWAS summary data reinforces the robustness of our analytical approach. However, the contrary associations in the replicated data for proteins like HCN1, ATG7, and CYR61 elucidate the inherent challenges in cross-cohort validations, underscoring the necessity for even more expansive datasets or collaborative meta-analyses. The congruence of the meta-analytic outcomes with our primary results accentuates the veracity of our findings. Furthermore, the MVMR analysis, which emphasized the associations of B2M and MSMB with PCa, illuminates these proteins’ potential roles in immunity and tumor suppression, suggesting a multifaceted approach to therapeutic intervention.
The GO and KEGG enrichment analyses provided deeper insights into the biological relevance of these proteins. Several of these proteins are involved in pathways like the JAK-STAT and PI3K-Akt signaling pathways, both of which have previously been implicated in various cancers, including PCa [29, 30]. Our results corroborate these known associations, providing further evidence for their pivotal roles in prostate carcinogenesis. The prominence of proteins within these pathways may suggest a centralized node of dysregulation that could be targeted therapeutically.
The vast disparity between the initial protein candidates and the final select cohort is emblematic of the profound molecular complexities associated with PCa pathogenesis. Each pinpointed protein, whether previously documented in PCa pathophysiology or emergent from our study, represents a potential nexus in the intricate web of molecular events driving or deterring prostate carcinogenesis. Such specificity and granularity are indispensable, for they not only spotlight the multifactorial nature of PCa’s molecular underpinnings but also illuminate myriad therapeutic avenues awaiting exploration. As we delve deeper into these findings, our focal point remains firmly on the biological implications of these associations, their translational relevance, and the broader impact on clinical paradigms in PCa management. Due to the absence of suitable proteomic data for PCa tissue, we focused on comparing the mRNA expression levels of the 18 identified proteins between tumor and non-tumor tissues. Our analysis revealed that these results were mostly inconsistent with the MR findings. This discrepancy could be attributed to several factors, including the fundamental differences between blood and tumor tissue composition, as well as the limited sample size available for PCa in the TCGA database. Given these findings, there is a clear need for future research to bridge the gap between blood and tissue proteomes and to further explore the clinical applicability of proteomic data in this context.
Our analysis, while insightful, encounters inherent limitations. The foundational assumptions of MR, such as the absence of horizontal pleiotropy, could potentially introduce biases. Although we have applied several methods to detect and overcome potential pleiotropy, residual pleiotropy cannot be fully ruled out, thus necessitating careful interpretation for our findings. Our results, derived predominantly from specific populations, might lack universal generalizability, emphasizing caution when extrapolating to diverse genetic backgrounds. While associations were established, direct causality remains to be further validated through mechanistic studies. The vast scope of our protein testing, despite stringent statistical corrections, carries the risk of false discoveries. Additionally, the reliance on existing GWAS datasets implies that any biases within them could have influenced our findings. Furthermore, although we validated some findings using FinnGen’s PCa GWAS data, not all associations consistently reached significance. Thus, while our study provides a foundational perspective, the results should be interpreted considering these constraints, warranting further validation and investigation.
In conclusion, our proteome-wide MR analysis has shed light on the intricate molecular underpinnings of PCa. The identified proteins and pathways offer tantalizing hints for drug discovery and therapeutic interventions. While further research is needed to validate these findings and translate them into clinical applications, our study represents a significant stride towards a more comprehensive understanding of PCa and the development of targeted treatments.
Availability of data and materials
The original contributions presented in the study are included in the article/Supplementary material.
Abbreviations
- MR:
-
Mendelian randomization
- IVs:
-
Instrumental variables
- GWAS:
-
Genome-wide association studies
- IVW:
-
Inverse-variance weighted
- BMI:
-
Body mass index
- OR:
-
Odds ratio
References
Sung H, Ferlay J, Siegel RL, Laversanne M, Soerjomataram I, Jemal A, Bray F: Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA: a cancer journal for clinicians 2021, 71(3):209–249.
Hodson R. Researchers take on the challenge of prostate cancer. Nature. 2022;609(7927):S31.
Sinha A, Huang V, Livingstone J, Wang J, Fox NS, Kurganovs N, Ignatchenko V, Fritsch K, Donmez N, Heisler LE, et al. The Proteogenomic Landscape of Curable Prostate Cancer. Cancer Cell. 2019;35(3):414-427.e416.
Khoo A, Liu LY, Nyalwidhe JO, Semmes OJ, Vesprini D, Downes MR, Boutros PC, Liu SK, Kislinger T. Proteomic discovery of non-invasive biomarkers of localized prostate cancer using mass spectrometry. Nat Rev Urol. 2021;18(12):707–24.
Ding Z, Wang N, Ji N, Chen ZS. Proteomics technologies for cancer liquid biopsies. Mol Cancer. 2022;21(1):53.
Ku X, Wang J, Li H, Meng C, Yu F, Yu W, Li Z, Zhou Z, Zhang C, Hua Y, et al. Proteomic portrait of human lymphoma reveals protein molecular fingerprint of disease specific subtypes and progression. Phenomics (Cham, Switzerland). 2023;3(2):148–66.
Albertsen PC. Prostate cancer screening and treatment: where have we come from and where are we going? BJU Int. 2020;126(2):218–24.
Wolk A, Mantzoros CS, Andersson SO, Bergström R, Signorello LB, Lagiou P, Adami HO, Trichopoulos D. Insulin-like growth factor 1 and prostate cancer risk: a population-based, case-control study. J Natl Cancer Inst. 1998;90(12):911–5.
Chen C, Lewis SK, Voigt L, Fitzpatrick A, Plymate SR, Weiss NS. Prostate carcinoma incidence in relation to prediagnostic circulating levels of insulin-like growth factor I, insulin-like growth factor binding protein 3, and insulin. Cancer. 2005;103(1):76–84.
Birney E. Mendelian Randomization. Cold Spring Harbor perspectives in medicine. 2021.
Davey Smith G, Hemani G. Mendelian randomization: genetic anchors for causal inference in epidemiological studies. Hum Mol Genet. 2014;23(R1):R89-98.
Zhao H, Rasheed H, Nøst TH, Cho Y, Liu Y, Bhatta L, Bhattacharya A, Hemani G, Davey Smith G, Brumpton BM et al.: Proteome-wide Mendelian randomization in global biobank meta-analysis reveals multi-ancestry drug targets for common diseases. Cell genomics 2022, 2(11):None.
Ferkingstad E, Sulem P, Atlason BA, Sveinbjornsson G, Magnusson MI, Styrmisdottir EL, Gunnarsdottir K, Helgason A, Oddsson A, Halldorsson BV, et al. Large-scale integration of the plasma proteome with genetics and disease. Nat Genet. 2021;53(12):1712–21.
Schumacher FR, Al Olama AA, Berndt SI, Benlloch S, Ahmed M, Saunders EJ, Dadaev T, Leongamornlert D, Anokian E, Cieza-Borrella C, et al. Association analyses of more than 140,000 men identify 63 new prostate cancer susceptibility loci. Nat Genet. 2018;50(7):928–36.
Zhu Z, Zhang F, Hu H, Bakshi A, Robinson MR, Powell JE, Montgomery GW, Goddard ME, Wray NR, Visscher PM, et al. Integration of summary data from GWAS and eQTL studies predicts complex trait gene targets. Nat Genet. 2016;48(5):481–7.
Kurki MI, Karjalainen J, Palta P, Sipilä TP, Kristiansson K, Donner KM, Reeve MP, Laivuori H, Aavikko M, Kaunisto MA, et al. FinnGen provides genetic insights from a well-phenotyped isolated population. Nature. 2023;613(7944):508–18.
Sanderson E. Multivariable mendelian randomization and mediation. Cold Spring Harbor perspectives in medicine. 2021;11(2):a038984.
Karlsson Linnér R, Biroli P, Kong E, Meddens SFW, Wedow R, Fontana MA, Lebreton M, Tino SP, Abdellaoui A, Hammerschlag AR, et al. Genome-wide association analyses of risk tolerance and risky behaviors in over 1 million individuals identify hundreds of loci and shared genetic influences. Nat Genet. 2019;51(2):245–57.
Yengo L, Sidorenko J, Kemper KE, Zheng Z, Wood AR, Weedon MN, Frayling TM, Hirschhorn J, Yang J, Visscher PM. Meta-analysis of genome-wide association studies for height and body mass index in ∼700000 individuals of European ancestry. Hum Mol Genet. 2018;27(20):3641–9.
Broeks MH, Shamseldin HE, Alhashem A, Hashem M, Abdulwahab F, Alshedi T, Alobaid I, Zwartkruis F, Westland D, Fuchs S, et al. MDH1 deficiency is a metabolic disorder of the malate-aspartate shuttle associated with early onset severe encephalopathy. Hum Genet. 2019;138(11–12):1247–57.
Vander Heiden MG, Cantley LC, Thompson CB. Understanding the Warburg effect: the metabolic requirements of cell proliferation. Science (New York, NY). 2009;324(5930):1029–33.
He C, Chen F, Li B, Hu Z. Neurophysiology of HCN channels: from cellular functions to multiple regulations. Prog Neurobiol. 2014;112:1–23.
Wang C, Chen S, Li X, Fan L, Zhou Z, Zhang M, Shao Y, Shang Z, Niu Y. TEAD3 inhibits the proliferation and metastasis of prostate cancer via suppressing ADRBK2. Biochem Biophys Res Commun. 2023;654:120–7.
Lou H, Yeager M, Li H, Bosquet JG, Hayes RB, Orr N, Yu K, Hutchinson A, Jacobs KB, Kraft P, et al. Fine mapping and functional analysis of a common variant in MSMB on chromosome 10q11.2 associated with prostate cancer susceptibility. Proceedings of the National Academy of Sciences of the United States of America. 2009;106(19):7933–8.
Wang H, Liu B, Wei J. Beta2-microglobulin(B2M) in cancer immunotherapies: Biological function, resistance and remedy. Cancer Lett. 2021;517:96–104.
Vajda A, Marignol L, Barrett C, Madden SF, Lynch TH, Hollywood D, Perry AS. Gene expression analysis in prostate cancer: the importance of the endogenous control. Prostate. 2013;73(4):382–90.
Abdul M, Hoosein N. Changes in beta-2 microglobulin expression in prostate cancer. Urol Oncol. 2000;5(4):168–72.
Gross M, Top I, Laux I, Katz J, Curran J, Tindell C, Agus D. Beta-2-microglobulin is an androgen-regulated secreted protein elevated in serum of patients with advanced prostate cancer. Clinical cancer research : an official journal of the American Association for Cancer Research. 2007;13(7):1979–86.
Malekan M, Nezamabadi SS, Samami E, Mohebalizadeh M, Saghazadeh A, Rezaei N. BDNF and its signaling in cancer. J Cancer Res Clin Oncol. 2023;149(6):2621–36.
Kim J, Hwang KW, Lee HJ, Kim HS. Systematic analysis of cellular signaling pathways and therapeutic targets for slc45a3: erg fusion-positive prostate cancer. J Personalized Med. 2022;12(11):1818.
Acknowledgements
We thank all the contributors and participants in the genome wide association studies data that used in the current study.
Funding
This study was supported by the National Natural Science Foundation of China (grant number: 82160463), Guangxi Province Self-funded Science and Technology Project (grant number: Z20200131) and the Guilin Scientific Research and Technology Development Project (grant number: 20210227–7-4).
Author information
Authors and Affiliations
Contributions
W.Sun, HL, and WZ designed the research. W.Sun and HL analyzed data, and wrote the original draft. W.Shi, QL, and WZ participated in interpretation of data. WZ and W.Shi revised the draft critically for important intellectual content. WZ had primary responsibility for the final content. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
This study was conducted in accordance with the Helsinki declaration. Ethical approval for GWAS was granted by local agency review board.
Competing interests
The authors declare no potential conflicts of interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Sun, W., Li, H., Shi, W. et al. Associations between genetically predicted concentrations of plasma proteins and the risk of prostate cancer. BMC Cancer 24, 905 (2024). https://doi.org/10.1186/s12885-024-12659-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12885-024-12659-y