
Abstract
Background
Retinitis pigmentosa (RP) is a heterogeneous group of inherited retinal diseases. However, it is still not well understand about the relationship between PCDH15 variants and RP.
Methods
In this study, we enrolled a Chinese autosomal recessive retinitis pigmentosa (arRP) pedigree and identified the causative gene in the proband by targeted whole exome sequencing (WES). The variants were validated in the family members by Sanger sequencing and co-segregation analysis.
Results
Novel compound heterozygous, Frame shift variants of the PCDH15 gene, NM_001384140.1:c.4368 − 2147_4368-2131del and NM_001384140.1:c exon19:c.2505del: p. T836Lfs*6 were identified in the arRP pedigree, which co-segregated with the clinical RP phenotypes. The PCDH15 protein is highly conserved among species.
Conclusion
This is the first study to identify novel compound heterozygous variants c.4368 − 2147_4368-2131del and c.2505del(p.T836Lfs*6) in the PCDH15 gene which might be disease-causing variants, and extending the variant spectra. All above findings may be contribute to genetic counseling, molecular diagnosis and clinical management of arRP disease.
Similar content being viewed by others

Introduction
Retinitis pigmentosa is one of the most serious inherited retinal disease(IRD), which characterized by progressive degeneration of rod photoreceptors, cone photoreceptors, retinal pigment epithelium(RPE), and remodeling of peripheral vascular [1]. Clinical hallmarks of RP include optic disc pallor, bone-spicule deposits, regression of blood vessels, visual field loss, and diminished or non-recordable electroretinographic(ERG) responses [2].
To date, there are about 270 genes, encoding proteins responsible for various retinal functions, have been reported to cause IRD [3]. The variants of these IRD related genes affect many potential biochemical pathways, including visual cycle regulation, gene transcription, light transduction, ion homeostasis, cellular structure maintenance, ciliary processes, premessenger RNA treatment, and lipid metabolism [4]. The inheritance pattern of RP can follow both Mendelian and non-Mendelian inheritance patterns [5]. Several inheritance models of RP have been reported, autosomal dominan, autosomal recessive and X-linked trait are the most common RP inheritance pattern [6]. Retinitis Pigmentosa (RP) is characterized by remarkable heterogeneity, with genetic, allelic, phenotypic, and clinical variations. This complexity makes research on RP challenging. The disease has both syndromic and non-syndromic forms. Syndromes like Usher syndrome (linked to deafness) and Bardet-Biedl syndrome (involving kidney problems, obesity, extra fingers, and developmental delays) are caused by mutations in 13 and 17 genes respectively. Non-syndromic cases often present with night blindness, progressing to peripheral vision loss initially, followed by central vision decline later in life, sometimes leaving some residual central vision in middle age, but often leading to complete blindness. There are over 50 known gene mutations associated with this phenotype; 23 in autosomal dominant inheritance, 36 in autosomal recessive, and 3 involve X-linked inheritance. These genes do not have a straightforward one-to-one mapping with the disease, indicating that the genetics of RP is highly complex and interconnected [7]. However, rare features such as genes, mitochondria or new variants reported in previous studies may be associated with other complex diseases [8].
Protocadherin 15 (PCDH15) encodes procadherin, a member of a large cadherin superfamily of calcium-dependent cell-cell adhesion molecules, plays an important role in the formation of neural synapses and circuits [9]. Previous studies have confirmed that PCDH15 expression in the brain, inner ear, retinal photoreceptor cell layer and in Müller glia. PCDH15 is essential for the structural maintenance and mechanical transduction function of sensory hair cells in the inner ear of vertebrates. In the retina, PCDH15 protein is important for the morphogenesis and cohesion of stereocilium bundles and retinal photoreceptor cell maintenance or function [10].
Human PCDH15 consists of 35 exons, including multiple isomers encoding 3–11 exon domains (ECs), a transmembrane domain, and a carboxy-terminal cytoplasmic domain (CD) [9]. PCDH15 susceptible to large deletion and rearrangements since the large intronic and non-coding exonic regions [11]. Ahmed et al. first reported two pathogenic variants in PCDH15 in two DFNB23 families [9, 10]. A genotype–phenotype correlation has been reported for pathogenic variants in PCDH15, with missense variants causing DFNB23 and null variants such as splicing, nonsense, frameshift, and large deletions lead to USH1F [12,13,14]. Diplenic inheritance, copy number variation and premature termination of PCDH15 translation to form truncated proteins are the most common pathogenic variants, and existing murine models like the Pcdh15R250X mice shed light on that disrupted expression of PCDH15 can lead to photoreceptor degeneration, impairing the light transduction cascade and protein localization in the retinal pigment epithelium, resulting in severe retinal dystrophy, a common feature of Retinitis Pigmentosa (RP) [15]. To date, according to Human Gene variant Database (HGMD) and previous reports, a large number of variants including exon copy number variants (CNVs), null variants of single nucleotide variants (SNVs) or insertions and deletions (INDELs) in PCDH15 have been detected in Caucasian USH1F families. However, PCDH15 gene variants and their involvement in RP have not been well studied in the Chinese population [16].
Even though next-generation sequencing (NGS) is the most widely used method in RP genetic diagnosis, there are certain technical limitations in the detection of variation in duplicate regions [17]. Whole exome sequencing(WES) enables researchers to analyze coding regions of the human genome in individuals or small families, including patients who lack a clear genotype-phenotype association [18]. Previous studies have shown that WES offers a promising alternative for molecular diagnosis of Mendelian disorders and genetic identification. In approximately 60% of RP cases, disease-causing variants can be identified by WES [19].
In present study, WES was used to identify the pathogenic variant in a non-consanguineous Chinese family with autosomal recessive RP(arRP). We identified a novel compound heterozygous variation c.4368 − 2147_4368-2131del and c.2505del: p.T836Lfs*6 in PCDH15 gene who presented with arRP. Our findings demonstrated the association between the variant and arRP, thus expands the pathogenic genetic spectrum of PCDH15 in RP.
Method
Subjects
A Chinese family, without any history of consanguineous marriage, was recruited from the Sixth Affiliated Hospital of Kunming Medical University(Yunnan, China). This pedigree includs a total of seven members with one affected patient and six unaffected individuals. The study was approved by the Institutional Review Boards of Sixth Affiliated Hospital of Kunming Medical University and implemented in conformity to the tenets of the Declaration of Helsinki. Written informed consents was obtained from all participants or parents of children prior to their inclusion in the present study.
DNA isolation
Peripheral blood samples were collected in EDTA tubes from the proband(III:3) and his family members. A commercial kit (TIANGEN, China) was used to extract all Genomic DNA samples. NanoDrop ND1000 (Thermo, USA) spectrophotometer and agarose gel electrophoresis was used to analyse the quantity/quality of DNA. DNA samples were stored at -20˚C until use.
WES
The DNA of case no.III:3 (the proband) was analyzed by WES at Shanghai WeHealth Biomedical Technology Co. with a mean read depth of target regions of 300X. xGen Exome Research Panel v1.0 (IDT, USA) and Illumina HiSeq platform (Illumina, USA) were used to Exome capture and 150 bp paired-end sequencing. The sequencing company using the Burrows-Wheeler Aligner (BWA) and SAM tools to aligned the raw reads. variants were classified according to the American College of Medical Genetics guidelines [20].
According to the American College of Medical Genetics and Genomics Guidelines (ACMG), the variant (c.4368 − 2147_4368- 2131del) was determined to be of uncertain significance (PM3_Supporting + PM2_Supporting + PM4) and the evidence are as follows: (1) It was detected in trans with a pathogenic or likely pathogenic variant (PM3_Supporting) (2) It is absent from gnomAD database (PM2_Supporting); (3) The variant is a deletion variant leading to the formation of a truncated protein (PM4). The variant (c.2505del: p.T836Lfs*6) was determined to be of likely pathogenic ( PVS1 + PM2_Supporting) and the evidence are as follows: (1)It is null variant (PVS1); (2) It is absent from gnomAD database (PM2_Supporting).
Variant validation
Novel variants in patients and available family members were confirmed by Polymerase chain reaction(PCR) and Sanger sequencing. The products of PCR were purified (OMEGA, Irving, TX, USA) and sequenced in Shanghai WeHealth Biomedical Technology Co.(Shanghai, China). The detected variants were subjected to cosegregation analysis in the RP families to determine their causalities.
Results
Clinical characteristics of the RP family
The proband had an early onset of night blindness at the age of 6 years old, with subsequent loss of peripheral vision after approximately 20 years. Detailed clinical information of the family is described in Table 1.
Fundus examination exhibited several classic characteristics of RP including attenuated blood vessels, pale optic discs and paravascular bone spicule pigmentation (Fig. 1A). Optical coherence tomography(OCT) showed that the outer nuclear layer was still present in the foveal area, but was markedly thinned in parafoveal regions, the thickness of choroid was significantly reduced (Fig. 1B). Optical coherence tomography angiography(OCTA) images showed that retinal microvessels regress from peripheral to posterior pole, and the posterior polar retinal microvascular density was significantly reduced with peripheral vascular remodeling (Fig. 1C). Full-field ERG showed signifificantly decreased scotopic and photopic responses, with extinguished decrease of a and b waves(Fig. 1D). The visual field test indicated a severe visual field defect ((Fig. 1E)). All above eye examinations suggested classical RP characters. All the members except the proband are normal in fundus despite the genetic variant.

Clinical information for the proband of retinitis pigmentosa. A: Fundus picture of the right and left eyes. B: Optical coherence tomography (OCT) scans of the right and left eyes. C: Optical coherence tomography angiography(OCTA) scans of the right and left eyes. D: Electroretinogram (ERG) results of the right and left eyes. E: Visual fields of the right and left eyes
Variant analysis
To identify the genetic variant causing the RP condition in the family, WES was performed for the proband (subject no. III:3). WES of proband generated a total of 18.79 million clean reads, and the target region with a mean coverage of 99.85% was mapped to the human reference genome. The mean depth of target region was 312.89-fold. Variants filtering strategies based on quality, rareness, deleteriousness, and biological filters. All data called by WES were 90,310 mutations, and after filtering out low-quality data (< 300) 84,924 mutation sites were obtained. By retaining variants located within exonic regions and those associated with splicing, a total of 24,993 variants were identified. The detected variants were filtered with minor allele frequencies (MAFs) of > 0.1% for 1000G, ExAC, GnomAD and GnomAD_EAS databases. There were 574 remaining variants. Variants leading to a premature stop codon, frameshifting, or located at a canonical splicing site, or predicted to be pathogenic by at least one of the 5 following programs: Mutation Taster, Polyphen-2, SIFT, CADD and REVEL (> 0.6). Keep only genes associated with retinitis pigmentosa according to literature or OMIM(Table 2). The filtering resulted in two variants localized in PCDH15 gene (c.4368 − 2147_4368- 2131del and c.2505del: p. T836Lfs*6).
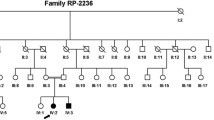
Two compound heterozygous variants in PCDH15 gene, variant c.4368 − 2147_4368-2131del and c.2505del: p.T836Lfs*6 in exon19, were identified in the proband(III:3). The variants c.4368 − 2147_4368-2131del was inherited from his mother, and the c.2505del variant was originated from his father (Fig. 2 A, B). Co-segregation analysis supported that compound heterozygous variants ( c.2505del and c.4368 − 2147_4368-2131del) were responsible for the disease phenotype. The c.4368 − 2147_4368-2131del was classified as “uncertain significance” variant, and the c.2505del was classified as “likely pathogenic” variant, according to the comprehensive analysis of ACMG guidelines. Conservative analysis of the PCDH15 protein from Physeter catodon to human showed that threonine at variant site (p.Thr836) was highly conserved (Fig. 2 C).

Identification of compound heterozygous variant c.4368 − 2147_4368-2131del and c.2505del in PCDH15 in a Chinese family with arRP. A. Family pedigree. The dark symbol represents the affected individual, and the crossed symbol indicates that the subject has died. The proband patient indicated by the red arrow. B. The heterozygous PCDH15 variants identifified by Sanger sequencing: The sequencing results in III: 1 for PCDH15 compound heterozygous mutants for c.4368 − 2147_4368-2131del and c.2505del, and the sequencing results in a normal for wild-type in both the sites c.4368 − 2147_4368-2131del and c.2505del respectively. The arrows indicate the variants at the nucleotide position NM_001384140.1: M1:c.4368 − 2147_4368-2131del; M2: c.2505del in PCDH15. C. Multiple sequence alignment showing p.Thr836 change inside a conserved region of the protein (yellow highlighted)
Segregation analysis
To confirm the PCDH15 variants and assess the inheritance pattern of RP in the proband. Segregation analysis of this sequence variant by Sanger sequencing was performed in the proband and his family members. (Table 3).
Protein model construction
According to Sorting Intolerant from Tolerant (SIFT) and Polyphen2 prediction showed the variants c.4368 − 2147_4368-2131del may lead to the change of protein length; the variants c.2505del is a heterozygous frameshift deletion, which is the deletion of the 2505 base of cDNA, resulting in the conversion of the 836th codon from encoding threonine to leucine, followed by the frameshift producing a stop codon ahead of time. Cadherins are evolutionary related to the desmogleins which are component of intercellular desmosome junctions involved in the interaction of plaque proteins. Structural modeling showed the conformational change of R-cadherin caused by variants c.2505del (Fig. 3). The conformational change of R-cadherin hinders the transportation of arrestin and transducin between the photoreceptor outer segment (OS) and inner segment (IS) to desensitize or bind to opsin, respectively.

Changes in protein conformation. Protein structures encoded by PCDH15 wild type(A and B) and p.T836Lfs*6(c.2505de)(C and D)
Discussion
In this study, novel compound heterozygous pathogenic variants of the PCDH15 gene, namely c.4368 − 2147_4368-2131del and c.2505del, were successfully identified as the genetic causes for arRP in a Han-Chinese family by WES and Sanger sequencing. The proband appeared to have nyctalopia as an early symptom that later progresses to the classical RP phenotype. Alterations in the c.4368 − 2147_4368-2131del and c.2505del sequences have not been reported in any major public database of human genome variation (HGMD, ClinVar, gnomAD, etc.), the altered residue (p.T836Lfs*6) was super conserved evolutionarily across diferent vertebrate species. The cosegregation analysis demonstrated the novel variants shows a strict genotype–phenotype correlation in the pedigree of the RP family such that the variant was present in a compound heterozygote state in proband; parents and offspring were single heterozygote carriers for the variant and thus remained clinically unaffected. Our findings supported an autosomal recessive inheritance pattern of the disease, as speculated in our family-based analysis, which further confirms several previous studies involving PCDH15 variants in arRP [14, 21, 22]. Finally, our 3D modeling results showed an overall destabilization of the protein upon variant leading to PCDH15 disfunction, which result in abnormal protein localization in the phototransduction cascade and retinoid cycle.
Variants in the PCDH15 gene encoding proto-cadherin-15 are one of the leading causes of Usher syndrome type 1 (USH1F) [8]. Biallelic truncating variants or severe missense variants in PCDH15 result in autosomal recessive deafness type 23(arDT23) or USH1F [23, 24]. To date, our study first reported the pathogenic variation of small deletion of PCDH15 gene and analyzed the genotype–phenotype correlation in which are associated with RP.
At present study, the compound heterozygous PCDH15 variant of c.4368 − 2147_4368-2131del and c.2505del has not been reported previously in RP patients. According to the American College of Medical Genetics and Genomics (ACMG) variant rating, the c.4368 − 2147_4368-2131del variant, whose pathogenicity was unclear, may cause changes in protein length. Thus it is necessary to be elucidated by relevant experiments or segregation analysis was performed on another RP patients with the same variant. Even though c.2505del variant leads to the change protein products and suggest the pathogenesis of photoreceptor degeneration in the current pedigree might be the same as other known RP-causing PCDH15 variants [25], specific molecular mechanism is still unclear.
In this study, the pure-tone audiometry and vestibular examination results showed that the hearing acuity of the proband fell within the normal range, indicating the compound heterozygous PCDH15 variants may cause arRP alone. Our study notably not consist with previous studies that the variant of PCDH15 lead to deafness, vestibular areflexia, and progressive retinal degeneration [26,27,28]. Thus we need to continue to follow-up and analyze the proband’s hearing. What’s more, further studies that investigating the function of the above mutants to understand their roles in the pathogenesis of RP is needed.
In conclusion, novel compound heterozygous variants c.4368 − 2147_4368-2131del and c.2505del(p.T836Lfs*6), were identified as the genetic causes for arRP in a Han-Chinese family by WES and Sanger sequencing. Our study expands the spectrum of the PCDH15 gene variants and may contribute to improved genetic counseling, prenatal diagnosis, and disease management for this family. However, the pathogenesis of variants c.4368 − 2147_4368-2131del still unclear, further construction of site specifific gene-defificient animal models and functional study may help to understand the genetic mechanism of PCDH15-associated arRP, which may further provide a clue to develop target therapy of this complex disorder.
Data availability
No datasets were generated or analysed during the current study.
Abbreviations
- ACMG:
-
American College of Medical Genetics and Genomics
- arRP:
-
Recessive retinitis pigmentosa
- HGMD:
-
Human genome variation
- OCTA:
-
Optical coherence tomography angiography
- PCDH15 :
-
Protocadherin 15
- SIFT:
-
Sorting Intolerant from Tolerant
- WES:
-
Whole exome sequencing
References
Liu W et al. Retinitis pigmentosa: progress in molecular pathology and biotherapeutical strategies. Int J Mol Sci. 2022;23(9).
Kalloniatis M, Fletcher EL. Retinitis pigmentosa: understanding the clinical presentation, mechanisms and treatment options. Clin Exp Optom. 2004;87(2):65–80.
Schneider N, et al. Inherited retinal diseases: linking genes, disease-causing mutations, and relevant therapeutic modalities. Prog Retin Eye Res. 2022;89:101029.
Ben-Yosef T. Inherited retinal diseases. Int J Mol Sci. 2022;23(21).
Daiger SP, Sullivan LS, Bowne SJ. Genes and mutations causing retinitis pigmentosa. Clin Genet. 2013;84(2):132–41.
Newton F, Megaw R. Mechanisms of photoreceptor death in retinitis pigmentosa. Genes (Basel). 2020;11(10):1120.
Birtel J, Gliem M, Mangold E, et al. Next-generation sequencing identifies unexpected genotype-phenotype correlations in patients with retinitis pigmentosa[J]. PLoS ONE. 2018;13(12):e0207958.
Ali MU, et al. Genetic characterization and disease mechanism of retinitis pigmentosa; current scenario. 3 Biotech. 2017;7(4):251.
Murcia CL, Woychik RP. Expression of Pcdh15 in the inner ear, nervous system and various epithelia of the developing embryo. Mech Dev. 2001;105(1–2):163–6. https://doi.org/10.1016/s0925-4773(01)00388-4
Ahmed ZM, et al. Gene structure and mutant alleles of PCDH15: nonsyndromic deafness DFNB23 and type 1 Usher syndrome. Hum Genet. 2008;124(3):215–23.
Ahmed ZM, et al. PCDH15 is expressed in the neurosensory epithelium of the eye and ear and mutant alleles are responsible for both USH1F and DFNB23. Hum Mol Genet. 2003;12(24):3215–23.
Alagramam KN, Miller ND, et al. Promoter, alternative splice forms, and genomic structure of protocadherin 15. Genomics. 2007;90(4):482–92.
Jaffal L, et al. Novel missense and splice site mutations in USH2A, CDH23, PCDH15, and ADGRV1 are associated with Usher syndrome in Lebanon. Front Genet. 2022;13:864228.
Aller E, Jaijo T, et al. Identification of large rearrangements of the PCDH15 gene by combined MLPA and a CGH: large duplications are responsible for Usher syndrome. Invest Ophthalmol Vis Sci. 2010;51(11):5480–5.
Sethna S, Zein WM, Riaz S, Giese AP, Schultz JM, Duncan T, et al. Proposed therapy, developed in a Pcdh15–deficient mouse, for progressive loss of vision in human usher syndrome. Elife. 2021;10:e67361.
Chen DY, et al. Variant in PCDH15 may modify the phenotypic expression of the 7511T > C variant in MT-TS1 in a Chinese Han family with maternally inherited nonsyndromic hearing loss. Int J Pediatr Otorhinolaryngol. 2015;79(10):1654–7.
Jiang L, Liang X, Li Y, et al. Comprehensive molecular diagnosis of 67 Chinese Usher syndrome probands: high rate of ethnicity specific mutations in Chinese USH patients. Orphanet J Rare Dis. 2015;10:110.
Salmaninejad A, et al. Next-generation sequencing and its application in diagnosis of retinitis pigmentosa. Ophthalmic Genet. 2019;40(5):393–402.
Retterer K, et al. Clinical application of whole-exome sequencing across clinical indications. Genet Med. 2016;18(7):696–704.
Dan H, et al. Application of targeted panel sequencing and whole exome sequencing for 76 Chinese families with retinitis pigmentosa. Mol Genet Genomic Med. 2020;8(3):e1131.
Richards S, et al. Standards and guidelines for the interpretation of sequence mutations: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17(5):405–24.
Yang Z, et al. Case report: compound heterozygous nonsense PCDH15 variant and a novel deep-intronic variant in a Chinese child with profound hearing loss. Mol Genet Genomic Med. 2023;11(7):e2193.
Ahmed ZM, S Riazuddin et al. Mutations of the protocadherin gene PCDH15 cause Usher syndrome type 1F. Am J Hum Genet. 2001;69(1):25–34.
Ouyang XM, et al. Characterization of Usher syndrome type I gene mutations in an Usher syndrome patient population. Hum Genet. 2005;116(4):292–9.
Doucette L, et al. Profound, prelingual nonsyndromic deafness maps to chromosome 10q21 and is caused by a novel missense variant in the Usher syndrome type IF gene PCDH15. Eur J Hum Genet. 2009;17(5):554–64.
Le Guédard S, Faugère V, et al. Large genomic rearrangements within the PCDH15 gene are a significant cause of USH1F syndrome. Mol Vis. 2007;13:102–7.
Xu XR, et al. The effect of PCDH15 gene variations on the risk of noise-induced hearing loss in a Chinese population. Biomed Environ Sci. 2017;30(2):143–6.
Schrauwen I, et al. Novel digenic inheritance of PCDH15 and USH1G underlies profound non-syndromic hearing impairment. BMC Med Genet. 2018;19(1):122.
Acknowledgements
We are thankful to the family for their volunteer participation in the study.
Funding
This study was supported by the Natural Science Foundation of Yunnan Province, China (202101AY070001-201).
Author information
Authors and Affiliations
Contributions
Hong Yang: design of the work and write the manuscript. YJZ: recruited patients. LZ: performed clinical evaluations of patients. WYZ: performed all experimental work including DNA extraction, Sanger validation of WES results, and segregation analysis. MYS: collected blood samples. HCZ: prepared the Figures and reviewed the manuscript. WRZ: reviewed the manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
Our study fully adhered to the ARVO statement on the use of human subjects in medical research and our study was conducted following the standards of the Declaration of Helsinki. Formal approval of this study was obtained from the Institutional Review Boards of Sixth Affiliated Hospital of Kunming Medical University(Approval No. 2023kmykdx6f49). Patients and accompanying family members were aware of the purpose of this study and approved their informed consent to participate in this study.
Consent for publication
All participants were willing for their data to be published.
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material

Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

{kind=link}
Cite this article
Yang, H., Zhang, Yj., Zhu, L. et al. A novel compound heterozygous PCDH15 variants is associated with arRP in a Chinese pedigree. BMC Ophthalmol 24, 373 (2024). https://doi.org/10.1186/s12886-024-03640-1
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12886-024-03640-1