
Abstract
Background
The precise classification of idiopathic interstitial pneumonia (IIP) is essential for selecting treatment as well as estimating clinical outcomes; however, this is sometimes difficult in clinical practice. Therefore, cluster analysis was used to identify the clinical phenotypes of IIPs, and its usefulness for predicting clinical outcomes was evaluated.
Methods
Cluster analysis was performed using clinical features including patients’ demographics; histories; pulmonary function test data; and laboratory, physical and radiological findings.
Results
In 337 patients with IIPs, four clusters were identified: Cluster I, in which > 80% of the patients had autoimmune features; Cluster II, which had the lowest rate of smoking, the lowest percent predicted forced vital capacity (%FVC) and the lowest body mass index (BMI); Cluster III, which had the highest rate of smoking, the highest rate of dust exposure, the second lowest %FVC and normal BMI; and Cluster IV, which exhibited maintenance of %FVC and normal BMI. Cluster IV had significantly longer overall survival than Clusters II and III. Clusters I and III had significantly longer overall survival than Cluster II. Clusters II and III had a significantly higher cumulative incidence of acute exacerbation than Cluster IV.
Conclusion
Cluster analysis using clinical features identified four clinical phenotypes of IIPs, which may be useful for predicting the risk of acute exacerbation and overall survival.
Similar content being viewed by others
Background
Idiopathic interstitial pneumonias (IIPs) consist of heterogeneous interstitial lung diseases of unknown aetiology. Based on their clinical, radiological and pathologic features, IIPs comprise several disease entities, such as idiopathic pulmonary fibrosis (IPF), nonspecific interstitial pneumonia (NSIP) and cryptogenic organizing pneumonia (COP) [1].
The precise classification of IIPs is essential for selecting treatment and predicting prognosis; however, this is not simple in clinical practice [1]. First, diagnosis at a single point is sometimes difficult, but this can be achieved by monitoring disease behaviour. For example, some patients with IIPs lacking honeycombing experience disease progression over several years, and they develop a usual interstitial pneumonia (UIP) pattern characterized by honeycombing on high-resolution computed tomography (HRCT) that is eventually diagnosed as IPF. In these patients, it is difficult to accurately diagnose the disease in the first examination. Additionally, patients with advanced IIPs other than IPF sometimes develop radiological and pathological honeycombing that mimics IPF [2]. Second, pathological patterns can vary among different sites of the lungs in patients with IIPs [3]. If surgical lung biopsy was not performed for representative lesions, it is difficult to ensure an accurate diagnosis [4, 5]. Furthermore, discordance in diagnosis even among specialists is frequent [6, 7]. Third, surgical lung biopsy, an essential procedure for the diagnosis of IIPs, cannot be performed in all patients [8]. In fact, according to a recent ATS/ERS/JRS/ALAT guideline, surgical lung biopsy is not necessarily required for the diagnosis of IPF in patients with typical radiologic findings of UIP [9]. Transbronchial cryobiopsy has attracted attention as a less invasive procedure than surgical lung biopsy; however, its utility for the diagnosis of IIPs remains a matter of debate [10, 11].
A new trend of classifying interstitial lung diseases (ILDs) using clinical characteristics, regardless of conventional disease entities, has emerged [12]. Recently, the concept of progressive fibrosing ILD (PF-ILD) was proposed, and this concept covers several IIPs featuring self-sustaining fibrosis, a progressive decline in lung function and early mortality [12]. PF-ILD comprises a comprehensive group of ILDs, including IIPs, connective tissue disease-associated ILD (CTD-ILD), sarcoidosis and chronic hypersensitivity pneumonia (CHP). Although the concept of PF-ILD has not been validated, it is expected to play a certain role from a therapeutic perspective [13]. Interstitial pneumonia with autoimmune features (IPAF) is another attempt to identify IIPs with features suggestive of, but not definitive for, CTD-ILD [14]. The diagnostic criteria of IPAF are positivity for two of the following three domains: clinical domain (extra-thoracic symptoms associated with autoimmune diseases), serologic domain (serum autoantibodies) and morphologic domain (HRCT and histopathologic findings and multi-compartment involvement other than IIPs) [14].
Clinical phenotyping using cluster analysis, which groups subjects according to the similarities and differences of their clinical features, has recently attracted attention for classifying heterogeneous diseases. For example, clinical phenotypes determined via cluster analysis using clinical data reveal distinct clinical outcomes in asthma and COPD [15, 16]. In all ILDs, clinical phenotypes determined using cluster analysis illustrated considerable predictive accuracy for clinical outcomes [17]. However, more than one-third of studied patients had non-IIPs such as CTD-ILD and CHP, both of which exhibit distinct characteristics and different clinical outcomes from IIPs [17].
We hypothesized that the clinical phenotypes of IIPs provide more useful information about clinical outcomes among patients. In this study, we performed cluster analysis using the clinical data of patients with IIPs and evaluated the clinical utility of the phenotypes.
Methods
Study design
This retrospective observational study followed the ethical standards of the Declaration of Helsinki. The study protocol was approved by the Institutional Review Board of Hamamatsu University School of Medicine (Hamamatsu, Japan, approval No. E15-197).
Patients
The medical records of consecutive patients with IIPs who were diagnosed at Hamamatsu Medical University Hospital between September 2004 and August 2018 were retrospectively analysed. The IIP diagnosis followed American Thoracic Society (ATS)/European Respiratory Society (ERS) guidelines [1, 9, 18, 19]. Patients who exhibited acute exacerbation (AE) at the first visit were excluded [20].
Data collection
The following data were collected at the time of IIP diagnosis: age, sex, body mass index (BMI), pack-year smoking history, history of dust exposure, laboratory data (C-reactive protein [CRP], lactate dehydrogenase [LDH], albumin, Krebs von den Lungen-6 [KL-6], surfactant protein D [SP-D]), pulmonary function (percent predicted forced vital capacity [%FVC], percent predicted forced expiratory volume in 1 s [%FEV1]) and the clinical and pathological diagnoses (if performed). The history of dust exposure was based on each patient’s reported history. Dust was classified as organic or inorganic, and the type of exposure was classified as occupational or environmental. The existence of emphysema and honeycombing was evaluated via chest high-resolution computed tomography (HRCT) at the diagnosis. The definitions of emphysema and honeycombing were described elsewhere [21]. Chest HRCT data were reviewed by two independent observers who were masked to patient data. Autoimmune features were recorded according to the IPAF diagnostic criteria [14].
Statistical analysis
Hierarchical clustering was performed using age, sex, BMI, histories of smoking and dust exposure, autoimmune features, laboratory data (CRP, LDH, albumin, KL-6, SP-D), pulmonary function (%FVC, %FEV1) and HRCT findings (emphysema, honeycombing) to identify clinical IIP subtypes. The number of clusters in which the scree plot of the distances between the clusters in a dendrogram of hierarchical clustering rose sharply, indicating different characteristics between clusters, was determined. Overall survival (OS) and the time to the first acute exacerbation (AE) were measured from the IIP diagnosis. The Kaplan–Meier method and log-rank test were used to analyse OS, and Grey’s test was used to analyse the time to the first AE. Wilcoxon’s signed-rank test was used for continuous variables, and Fisher’s exact test was applied for categorical variables. All comparisons among clusters were adjusted using Bonferroni’s correction. Data were expressed as the median (range) unless otherwise indicated. All statistical tests were two-sided, and p < 0.05 indicated significance. All values were analysed using EZR (Saitama Medical Center, Jichi Medical University, Saitama, Japan), which is a graphical user interface for R (The R Foundation for Statistical Computing, Vienna, Austria), except the cluster analysis, which was performed using JMP v13.0.0 (SAS Institute Japan, Tokyo, Japan).
Results
Baseline characteristics
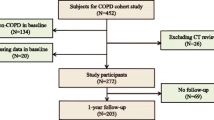
In total, 337 patients with IIPs were screened, and 54 were excluded because of missing data (n = 41) and AE at the time of diagnosis (n = 13), leaving 283 patients (Fig. 1). The clinical characteristics of these patients are presented in Table 1. The study cohort consisted mainly of males, and more than half of the patients had smoking histories. In total, 148 (52.2%) patients had %FVC < 80%, and 134 (47.3%) patients had %FEV1 < 80%. Among 69 (24.3%) patients with histories of dust exposure, organic and inorganic materials were responsible for 2.9 and 97.1% of cases, respectively, and all instances of exposure were associated with occupational exposure. Sixty-eight (24.0%) patients underwent surgical lung biopsy. Additionally, 94 (33.2%), 19 (6.7%), 16 (5.6%), 12 (4.2%) and 4 (1.4%) patients were diagnosed with IPF (clinical, n = 66; pathological, n = 28), COP, NSIP, pleuroparenchymal fibroelastosis (PPFE) and desquamative interstitial pneumonia (DIP)/respiratory bronchiolitis-associated interstitial lung disease (RB-ILD), respectively (Table 2). The remaining 138 patients (48.7%) had unclassifiable IIPs. Thirty-nine (13.7%) patients had autoimmune features, including 6 (17.4%), 39 (100%) and 36 (92.3%) patients in the clinical, serologic and morphologic domains, respectively (Table 2). The median observation time was 39.4 months (range 1.0–165.0 months).

Flow diagram of the study patients. IIP, idiopathic interstitial pneumonia
Clinical characteristics of the clusters
Four clusters were identified in the cluster analysis using clinical data (Additional file 1: Fig. 1).
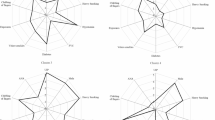
Cluster I (n = 35) was an intermediate-aged cohort (70.7 years) with low proportions of males (62.8%) and smokers (54.2%; Table 1, Fig. 2a). Cluster I featured preserved %FVC (81.8%), the lowest honeycombing rate (14.2%) and the highest rate of autoimmune features (85.7%). Regarding laboratory data, Cluster I had the second highest KL-6 level (1010 U/mL), a slightly increased SP-D level (166 ng/mL) and normal CRP, LDH and albumin levels. In Cluster I, six (17.1%), five (14.2%), three (8.5%) and two (5.7%) patients were diagnosed with IPF (pathological, n = 3; clinical, n = 3), NSIP, COP and DIP, respectively. The remaining 19 (54.2%) patients had unclassifiable IIP (Table 2).

Radar plot of clinical features of the four clusters. a Cluster I, b Cluster II, c Cluster III, and d Cluster IV. Plot scale: 1, < 0.4 SDs below the mean; 2, 0.3–0.4 SDs below the mean; 3, 0.2–0.3 SDs below the mean; 4, 0.1–0.2 SDs below the mean; 5, mean to 0.1 SDs below the mean; 6, mean to 0.1 SDs above the mean; 7, 0.1–0.2 SDs above the mean; 8, 0.2–0.3 SDs above the mean; 9, 0.3–0.4 SDs above the mean; 10, > 0.4 SDs above the mean. BMI, body mass index; CRP, C-reactive protein; IPAF, interstitial pneumonia with autoimmune features; KL-6, Krebs von den Lungen-6; %FVC, percent predicted forced vital capacity; SP-D, pulmonary surfactant protein-D
Cluster II (n = 62) had the oldest population (75.9 years), the smallest percentage of smokers (50.0%), the lowest BMI (20.8 kg/m2) and the lowest %FVC (64.5%; Table 1, Fig. 2b), as well as the second largest male population (79.0%). Emphysema was least frequent in this group (22.5%), but the second highest rate of honeycombing was noted (37.0%). In laboratory findings, Cluster II had the highest CRP level (1.13 mg/dL), the lowest serum albumin level (3.6 g/dL) and slightly increased KL-6 (664 U/mL) and SP-D levels (173 ng/mL). In total, 22 (35.4%), 8 (12.9%) and 8 (12.9%) patients were diagnosed with IPF (clinical, n = 20; pathological, n = 2), PPFE and COP, respectively. The remaining 24 (38.7%) patients had unclassifiable IIP (Table 2).
Cluster III (n = 89) had the youngest population (68.9 years), the highest proportion of males (93.2%), the highest BMI (24.9 kg/m2) and the highest frequencies of smoking (88.7%) and dust exposure (44.9%; Table 1, Fig. 2c). This cluster had the second lowest %FVC (73.0%). Cluster III had the highest LDH (241 U/L), KL-6 (1148 U/mL) and SP-D levels (218 ng/mL) and normal CRP and albumin levels. The rates of emphysema and honeycombing were highest in this group (39.3 and 38.2%, respectively). In total, 46 (51.6%), 5 (5.6%) and 3 (3.3%) patients were diagnosed with IPF (clinical, n = 29; pathological, n = 17), NSIP and COP, respectively. The remaining 35 (39.3%) patients had unclassifiable IIP (Table 2).
Cluster IV (n = 97) had the second youngest population (69.2 years), the highest proportion of females (38.2%), normal BMI (22.1 kg/m2) and lower frequencies of smoking (54.6%) and dust exposure (13.4%; Table 1, Fig. 2d). Cluster IV had the highest %FVC (89.1%) and normal CRP, LDH and albumin levels. Cluster IV had the lowest KL-6 (644 U/mL) and SP-D levels (154 ng/mL). The rates of emphysema and honeycombing were 30.9 and 20.6%, respectively. Overall, 20 (20.6%), 6 (6.1%), 5 (5.1%), 4 (4.1%) and 2 patients (2.0%) were diagnosed with IPF (clinical, n = 13; pathological, n = 7), NSIP, and COP, PPFE and DIP, respectively. The remaining 60 (61.8%) patients had unclassifiable IIPs (Table 2).
Differences in clinical outcomes among the clusters
OS was significantly longer in Cluster IV than in Clusters II (p < 0.01) and III (p = 0.01; Fig. 3a). Clusters I and III had significantly longer OS than Cluster II (both p < 0.01). The 5-year OS rates were 87.7%, 79.0%, 70.1% and 32.3% in Clusters IV, I, III and II, respectively, and the 10-year OS rates were 66.6%, 57.6%, 41.4% and 32.3%, respectively.

Overall survival (OS). a OS was significantly longer in Cluster IV (orange line) than in Clusters I (red line, p = 0.53), II (green line, p < 0.01), and III (blue line, p = 0.01). OS was significantly longer in Cluster I than in Clusters II (p < 0.01) and III (p = 0.21). OS was significantly longer in Cluster III than in Cluster II (p < 0.01). b Patients with idiopathic pulmonary fibrosis (IPF, continuous line) had significantly longer OS than those with non-specific interstitial pneumonia (NSIP, dotted line, p = 0.01), whereas there was no significant difference among the other idiopathic interstitial pneumonia groups. COP, cryptogenic organizing pneumonia (short dashed line); DIP/RB-ILD, desquamative interstitial pneumonia/respiratory bronchitis-associated interstitial lung disease (middle dashed line); PPFE, pleuroparenchymal fibroelastosis (long dashed line)
Clusters II and III had significantly higher cumulative incidences of AE than Cluster IV (both p = 0.03; Fig. 4a). The 5-year cumulative incidence rates of AE were 20.6%, 19.9%, 9.7% and 8.0% in Clusters II, III, I, and IV, respectively, and the 10-year cumulative incidence rates were 20.6%, 23.0%, 21.7% and 13.8%, respectively.

Cumulative incidence of acute exacerbation. a Clusters II (green line) and III (blue line) had significantly higher cumulative incidence rates of acute exacerbation than Cluster IV (orange line, both p = 0.03). b Patients with idiopathic pulmonary fibrosis (IPF, continuous line) had a significantly higher cumulative incidence rate of acute exacerbations than those with cryptogenic organizing pneumonia (COP, small dashed line, p = 0.01) and unclassifiable IIPs (long dashed short dashed line, p < 0.01), whereas there was no significant difference among the other idiopathic interstitial pneumonia groups. DIP/RB-ILD, desquamative interstitial pneumonia/respiratory bronchitis-associated interstitial lung disease (middle dashed line); NSIP, non-specific interstitial pneumonia (dotted line); PPFE, pleuroparenchymal fibroelastosis (long dashed line)
Next, OS and AE were evaluated according to the IIP diagnosis. Patients with NSIP had significantly longer OS than those with IPF (p = 0.01), whereas there was no significant difference among the other groups (Fig. 3b). Patients with IPF had a significantly higher cumulative incidence of AE than those with COP (p = 0.01) and unclassifiable IIPs (p < 0.01), whereas there was no significant difference among the other groups (Fig. 4b).
Alternative clustering analysis
If we had employed three clusters, Clusters III and IV would have been merged (Additional file 1: Fig. 1). Alternatively, if we had employed five clusters, Cluster II would have been divided into two clusters. One of the new clusters had a significantly lower proportion of males, lower BMI, lower %FVC, lower %FEV1, lower CRP, lower LDH and higher albumin than the other cluster (Additional file 2: Tables 4–5) However, OS and the cumulative incidence of AE did not differ between the clusters (Additional file 1: Figs. 2 and 3).
Discussion
Four IIP clusters with distinct clinical features and outcomes were identified. Cluster I, in which > 80% of the patients had autoimmune features, featured a low risk for AE and good OS. Clusters II and III had the greatest decreases in %FVC and the highest honeycombing rates. More than 80% of patients with IPF belonged to these two clusters. Cluster II had the lowest %FVC, BMI and albumin levels. Cluster III had the second lowest %FVC and normal BMI and albumin levels, as well as the highest LDH, KL-6 and SP-D levels. OS was worst in Cluster II, followed by Cluster III. Both clusters had high risks of AEs. Cluster IV, representing the mildest IIPs with preserved %FVC and almost normal laboratory data, had the best OS and a low risk of AE. In our patients, more than half did not undergo surgical lung biopsies. Additionally, the radiologic findings used in the cluster analysis were simple; namely the existence of honeycombing and emphysema on chest CT. Our clustering of patients with IIPs using clinical data is highly feasible in practice, and this strategy may provide useful information about patients’ clinical outcomes, even without detailed pathological or radiologic findings.
Interestingly, our cluster analysis revealed that the clinical features associated with collagen vascular diseases were important components of the clinical phenotypes of IIPs. A considerable number of patients with IIPs have some features of collagen vascular disease without fulfilling the defined criteria of any collagen vascular diseases. Several classifications have been proposed for such patients, but controversies remain unresolved [22,23,24,25]. The IPAF concept was proposed by ATS/ERS to establish a unified platform for such patients with IIPs [14]. Several studies reported that patients with IPAF have better OS and lower risks of AE than their counterparts [26, 27]. Similarly, Cluster I, in which most patients had autoimmune features, was linked to better clinical outcomes than Clusters II and III. Additionally, patients with IPAF are younger and more commonly female than those without IPAF, coinciding with the characteristics of Cluster I [28,29,30]. The clinical significance and utility of the IPAF concept are poorly validated. Additionally, several studies reported that the IPAF criteria must be revisited. In this context, our simple clustering successfully extracts patients with IIPs and autoimmune features who have distinct outcomes (Cluster I).
Clusters II and III were characterized by advanced fibrosis accompanied by decreased %FVC and honeycombing, which may lead to poor prognoses and high risks of AE. Most study patients with IPF belonged to these clusters. Patients with IPF have higher AE rates and worse OS than those with other IIPs [31]. The high prevalence of IPF might have strong effects on clinical outcomes in Clusters II and III. However, other factors determined the characteristics of these clusters. More than 50% of patients in these clusters had non-IPF IIPs. Specifically, OS was poorest in Cluster II despite its lower proportion of patients with IPF than Cluster III. In this study, we identified age, low serum albumin levels, decreased %FVC and radiologic honeycombing as independent risk factors for OS in all patients with IIP (Additional file 2: Table 1). Additionally, the latter two were also risk factors for AE (Additional file 2: Table 2). Among the two clusters, Cluster II was associated with significantly older patient age, lower serum albumin levels, lower %FVC and higher rates of radiologic honeycombing than Cluster III. These differences may explain the clinical outcomes of Clusters II and III.
A previous study reported the usefulness of four clinical clusters for predicting clinical outcomes in 770 patients with ILDs [17]. The study included non-IIPs including CTD-ILD and CHP and diverse ethnicities (mainly Caucasian and secondarily African American). Therefore, the clusters were not completely consistent with those in the current study. However, the cluster with female-dominant demographics, elevated antinuclear antibody levels, and the best clinical outcomes and another cluster of elderly male smokers with coexisting emphysema and the second worst outcomes were similar to Clusters I and III, respectively, in the current study.
The current study had several limitations. First, the numbers of clusters affected the results. In the current study, we also considered the use of three or five clusters. If we had employed three clusters, the distinct clinical features of Clusters III and IV would have been lost. Meanwhile, if we had employed five clusters, the two new clusters derived from Cluster II would have included small numbers of patients (insufficient for reaching statistical significance); thus, they were grouped together. Second, the numbers and/or optimal combination of variables used for the cluster analysis were not validated. We employed clinical variables that could be representative of demographic, historical, physical, laboratory and radiographic information. However, there were limited data available because the study was retrospective. However, there were limited data available because the study was retrospective. For example, a considerable number of study patients lacked data for diffusion capacity of the lung for carbon monoxide, which would have provided important information regarding the outcomes of ILD. The selection of variables can vary according to the purpose of the classification, such as predicting OS, AE risk, therapeutic response. Further studies are warranted to optimize the clinical classifications of IIPs.
Conclusion
Cluster analysis using simple clinical data identified four phenotypes from the heterogeneous group of IIPs. Even without surgical lung biopsy, the four clinical phenotypes were linked to distinct differences in AE risks and OS, which may help to predict clinical outcomes and make treatment decisions.
Availability of data and materials
The datasets used and/or analyzed during the current study are available from the corresponding author on reasonable request.
Abbreviations
- ATS:
-
American Thoracic Society
- BMI:
-
Body mass index
- CHP:
-
Chronic hypersensitivity pneumonia
- COP:
-
Cryptogenic organizing pneumonia
- COPD:
-
Chronic obstructive pulmonary disease
- CPFE:
-
Combined pulmonary fibrosis and emphysema
- CRP:
-
C-reactive protein
- CTD-ILD:
-
Connective tissue disease-associated interstitial lung diseases
- DIP:
-
Desquamative interstitial pneumonia
- ERS:
-
European Respiratory Society
- FEV1 :
-
Forced expiratory volume in 1 s
- FVC:
-
Forced vital capacity
- HRCT:
-
High resolution computed tomography
- KL-6:
-
Krebs von den Lungen-6
- IIP:
-
Idiopathic interstitial pneumonia
- ILD:
-
Interstitial lung disease
- IPAF:
-
Interstitial pneumonia with autoimmune features
- IPF:
-
Idiopathic pulmonary fibrosis
- LDH:
-
Lactate dehydrogenase
- NSIP:
-
Nonspecific interstitial pneumonia
- OS:
-
Overall survival
- PPFE:
-
Pleuroparenchymal fibroelastosis
- SP-D:
-
Surfactant protein D
- %FEV1 :
-
Percent predicted forced expiratory volume in 1 s
- %FVC:
-
Percent predicted forced vital capacity
References
Travis WD, Costabel U, Hansell DM, King TE Jr, Lynch DA, Nicholson AG, et al. An official American Thoracic Society/European Respiratory Society statement: Update of the international multidisciplinary classification of the idiopathic interstitial pneumonias. Am J Respir Crit Care Med. 2013;188(6):733–48.
Watadani T, Sakai F, Johkoh T, Noma S, Akira M, Fujimoto K, et al. Interobserver variability in the CT assessment of honeycombing in the lungs. Radiology. 2013;266(3):936–44.
Barratt SL, Creamer A, Hayton C, Chaudhuri N. Idiopathic pulmonary fibrosis (IPF): an overview. J Clin Med. 2018;7(8):201.
Yagihashi K, Huckleberry J, Colby TV, Tazelaar HD, Zach J, Sundaram B, et al. Radiologic-pathologic discordance in biopsy-proven usual interstitial pneumonia. Eur Respir J. 2016;47(4):1189–97.
Hutchinson JP, Fogarty AW, McKeever TM, Hubbard RB. In-hospital mortality after surgical lung biopsy for interstitial lung disease in the United States 2000 to 2011. Am J Respir Crit Care Med. 2016;193(10):1161–7.
Sumikawa H, Johkoh T, Colby TV, Ichikado K, Suga M, Taniguchi H, et al. Computed tomography findings in pathological usual interstitial pneumonia: relationship to survival. Am J Respir Crit Care Med. 2008;177(4):433–9.
Sverzellati N, Wells AU, Tomassetti S, Desai SR, Copley SJ, Aziz ZA, et al. Biopsy-proved idiopathic pulmonary fibrosis: spectrum of nondiagnostic thin-section CT diagnoses. Radiology. 2010;254(3):957–64.
Ryerson CJ, Urbania TH, Richeldi L, Mooney JJ, Lee JS, Jones KD, et al. Prevalence and prognosis of unclassifiable interstitial lung disease. Eur Respir J. 2013;42(3):750–7.
Raghu G, Remy-Jardin M, Myers JL, Richeldi L, Ryerson CJ, Lederer DJ, et al. Diagnosis of idiopathic pulmonary fibrosis An official ATS/ERS/JRS/ALAT clinical practice guideline. Am J Respir Crit Care Med. 2018;198(5):e44–68.
Pajares V, Núñez-Delgado M, Bonet G, Pérez-Pallarés J, Martínez R, Cubero N, et al. Transbronchial biopsy results according to diffuse interstitial lung disease classification Cryobiopsy versus forceps: MULTICRIO study. PLoS ONE. 2020;15(9):e0239114.
Maldonado F, Danoff SK, Wells AU, Colby TV, Ryu JH, Liberman M, et al. Transbronchial cryobiopsy for the diagnosis of interstitial lung diseases: CHEST guideline and expert panel report. Chest. 2020;157(4):1030–42.
Cottin V, Hirani NA, Hotchkin DL, Nambiar AM, Ogura T, Otaola M, et al. Presentation, diagnosis and clinical course of the spectrum of progressive-fibrosing interstitial lung diseases. Eur Respir Rev. 2018;27(150):180076.
Flaherty KR, Wells AU, Cottin V, Devaraj A, Walsh SLF, Inoue Y, et al. Nintedanib in progressive fibrosing interstitial lung diseases. N Engl J Med. 2019;381(18):1718–27.
Fischer A, Antoniou KM, Brown KK, Cadranel J, Corte TJ, du Bois RM, et al. An official European Respiratory Society / American Thoracic Society research statement: interstitial pneumonia with autoimmune features. Eur Respir J. 2015;46(4):976–87.
Moore WC, Meyers DA, Wenzel SE, Teague WG, Li H, Li X, et al. Identification of asthma phenotypes using cluster analysis in the Severe Asthma Research Program. Am J Respir Crit Care Med. 2010;181(4):315–23.
Rennard SI, Locantore N, Delafont B, Tal-Singer R, Silverman EK, Vestbo J, et al. Identification of five chronic obstructive pulmonary disease subgroups with different prognoses in the ECLIPSE cohort using cluster analysis. Ann Am Thorac Soc. 2015;12(3):303–12.
Adegunsoye A, Oldham JM, Chung JH, Montner SM, Lee C, Witt LJ, et al. Phenotypic clusters predict outcomes in a longitudinal interstitial lung disease cohort. Chest. 2018;153(2):349–60.
American Thoracic Society/European. Respiratory Society international multidisciplinary consensus classification of the idiopathic interstitial pneumonias. Am J Respir Crit Care Med. 2002;165(2):277–304.
Raghu G, Collard HR, Egan JJ, Martinez FJ, Behr J, Brown KK, et al. An official ATS/ERS/JRS/ALAT statement: idiopathic pulmonary fibrosis: evidence-based guidelines for diagnosis and management. Am J Respir Crit Care Med. 2011;183(6):788–824.
Collard HR, Ryerson CJ, Corte TJ, Jenkins G, Kondoh Y, Lederer DJ, et al. Acute exacerbation of idiopathic pulmonary fibrosis an international working group report. Am J Respir Crit Care Med. 2016;194(3):265–75.
Hansell DM, Bankier AA, MacMahon H, McLoud TC, Müller NL, Remy J. Fleischner Society: glossary of terms for thoracic imaging. Radiology. 2008;246(3):697–722.
Corte TJ, Copley SJ, Desai SR, Zappala CJ, Hansell DM, Nicholson AG, et al. Significance of connective tissue disease features in idiopathic interstitial pneumonia. Eur Respir J. 2012;39(3):661–8.
Kinder BW, Collard HR, Koth L, Daikh DI, Wolters PJ, Elicker B, et al. Idiopathic nonspecific interstitial pneumonia: lung manifestation of undifferentiated connective tissue disease? Am J Respir Crit Care Med. 2007;176(7):691–7.
Vij R, Noth I, Strek ME. Autoimmune-featured interstitial lung disease: a distinct entity. Chest. 2011;140(5):1292–9.
Omote N, Taniguchi H, Kondoh Y, Watanabe N, Sakamoto K, Kimura T, et al. Lung-dominant connective tissue disease: clinical, radiologic, and histologic features. Chest. 2015;148(6):1438–46.
Yoshimura K, Kono M, Enomoto Y, Nishimoto K, Oyama Y, Yasui H, et al. Distinctive characteristics and prognostic significance of interstitial pneumonia with autoimmune features in patients with chronic fibrosing interstitial pneumonia. Respir Med. 2018;137:167–75.
Lim JU, Gil BM, Kang HS, Oh J, Kim YH, Kwon SS. Interstitial pneumonia with autoimmune features show better survival and less exacerbations compared to idiopathic pulmonary fibrosis. BMC Pulm Med. 2019;19(1):120.
Ahmad K, Barba T, Gamondes D, Ginoux M, Khouatra C, Spagnolo P, et al. Interstitial pneumonia with autoimmune features: Clinical, radiologic, and histological characteristics and outcome in a series of 57 patients. Respir Med. 2017;123:56–62.
Chartrand S, Swigris JJ, Stanchev L, Lee JS, Brown KK, Fischer A. Clinical features and natural history of interstitial pneumonia with autoimmune features: a single center experience. Respir Med. 2016;119:150–4.
Oldham JM, Adegunsoye A, Valenzi E, Lee C, Witt L, Chen L, et al. Characterisation of patients with interstitial pneumonia with autoimmune features. Eur Respir J. 2016;47(6):1767–75.
Leuschner G, Behr J. Acute exacerbation in interstitial lung disease. Front Med. 2017;4:176.
Acknowledgements
We thank Joe Barber Jr., PhD, from Edanz Group (https://en-author-services.edanzgroup.com/ac) for editing a draft of this manuscript.
Funding
This research did not receive any specific grant from funding agencies in the public, commercial, or not-for-profit sectors.
Author information
Authors and Affiliations
Contributions
YA: interpretation of data, writing-original draft, MK: conceptualization, methodology, interpretation of data, writing-review and editing, and supervision. KM: acquisition and analysis, and interpretation of data, YA, HY: acquisition and analysis. HH, YS, KF: conceptualization and supervision. TF, NE, YN, NI: conceptualization, supervision, and writing-review and editing. TS: conceptualization, methodology, writing-review and editing, and supervision. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
The study protocol was approved by the Institutional Review Board of Hamamatsu University School of Medicine (Hamamatsu, Japan, approval No. E15-197). Informed consents were waived because of a retrospective observational study.
Consent for publication
Not applicable.
Competing interests
All authors have no conflict of interest in relation to this manuscript.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1.
Supplementary Figures 1–3: Figure 1. Dendrogram of patients with IIPs using cluster analysis. Figure 2. Overall survival in the new clusters derived from Cluster II. Figure 3. Cumulative incidence of acute exacerbations in the new clusters derived from Cluster II.
Additional file 2. Supplementary Tables 1–4: Table 1
. Cox proportional hazard analysis of overall survival. Table 2. Cox proportional hazard analysis of acute exacerbations. Table 3. Characteristics of the new clusters derived from Cluster II. Table 4. Diagnosis of IIPs in the new clusters derived from Cluster II.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Aoshima, Y., Karayama, M., Horiike, Y. et al. Cluster analysis-based clinical phenotypes of idiopathic interstitial pneumonias: associations with acute exacerbation and overall survival. BMC Pulm Med 21, 63 (2021). https://doi.org/10.1186/s12890-021-01428-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12890-021-01428-3